

Abstract
The Snail gene product is a transcriptional repressor of E-cadherin expression and an inducer of the epithelial-to-mesenchymal transition in several epithelial tumor cell lines. This report presents data indicating that Snail function is controlled by its intracellular location. The cytosolic distribution of Snail depended on export from the nucleus by a CRM1-dependent mechanism, and a nuclear export sequence (NES) was located in the regulatory domain of this protein. Export of Snail was controlled by phosphorylation of a Ser-rich sequence adjacent to this NES. Modification of this sequence released the restriction created by the zinc finger domain and allowed nuclear export of the protein. The phosphorylation and subcellular distribution of Snail are controlled by cell attachment to the extracellular matrix. Suspended cells presented higher levels of phosphorylated Snail and an augmented extranuclear localization with respect to cells attached to the plate. These findings show the existence in tumor cells of an effective and fine-tuning nontranscriptional mechanism of regulation of Snail activity dependent on the extracellular environment.
During gastrulation, the Snail transcription factor is responsible for the transcriptional repression of E-cadherin, thus lowering the level of this protein and allowing cells to detach from their neighbors and migrate (16, 29). This pathway is essential for the success of this process, since mice deficient in the expression of this protein fail to downregulate E-cadherin levels and to complete gastrulation (7). Repression of E-cadherin transcription is also particularly relevant in the late steps of epithelial tumorigenesis, since a causal relationship between loss of expression of this protein and the invasive properties of some tumors has been established (32, 42). Recently, two studies have shown an association of Snail expression with the degree of infiltration of breast carcinomas (4) and with E-cadherin down-regulation in diffuse-type gastric carcinomas (34). According to these results, there is a considerable inverse correlation between E-cadherin and Snail mRNA levels in most epithelial tumor cell lines (2, 6, 8, 19, 33, 44). However, in some cases, E-cadherin and Snail mRNA levels are both detected in the same cell lines (15).
Very little is known about the structure of the Snail protein. Two different regions of this protein have been described: the C-terminal DNA-binding domain (amino acids 152 to 264) and the N-terminal regulatory domain (amino acids 1 to 151) (Fig. 1). The C-terminal part of the protein contains four zinc fingers belonging to the C2H2 subclass. This domain is conserved in other vertebrate and invertebrate homologues of the protein (24, 29) and has the highest affinity for oligonucleotides containing a 5′-CACCTG-3′ sequence (25). It should be noted that the last zinc finger of the Snail protein does not contain the consensus CXXC-HΦR/QS/TH, which is present in the other three fingers, and that the two histidines are separated by only one lysine. The N-terminal domain differs much more among the various Snail homologues. In mammals, a short sequence in the amino terminus, called the SNAG box, is essential for repression (2). Alignment of Snail sequences corresponding to the N-terminal half of known homologues showed that, excluding the SNAG box, the most conserved region corresponds to a subdomain located between residues 82 and 151 in the mouse Snail sequence. This report presents evidence that this subdomain contains a nuclear export sequence (NES). Phosphorylation of an adjacent serine-rich sequence made this NES accessible to the CRM1 transporter and facilitated translocation of the protein to the nucleus. Phosphorylated Snail was much less active as a repressor of E-cadherin transcription or as an activator of the expression of mesenchymal genes.
FIG. 1.

Diagram of Snail protein. Snail domains are highlighted by gray and light blue (N terminal) and dark blue (C terminal). This color code is maintained in the other figures.
MATERIALS AND METHODS
Preparation of DNA constructs and Snail mutants. (i) Snail deletion mutants.
Murine Snail was obtained by reverse transcription (RT)-PCR amplification with specific oligonucleotides as previously described (2). N- and C-terminal halves were prepared from pcDNA3-Snail-HA (1) by using a BglII site present at 455 bp from the ATG site. The 237-to-264 deletion was achieved by high-fidelity PCR amplification of Snail with specific oligonucleotides. Antisense oligonucleotide annealing avoided the last zinc finger and added an EcoRV site used to set the sequence in frame with the hemagglutinin (HA) tag. The oligonucleotide (antisense, 5′-CCGGATATCCGTATCTCTTCACATC-3′; sense, 5′-CCGGATCCACCATGCCGCGCTCCTTCCTGG-3′) encompasses the ATG site (bold) and adds a Kozak sequence (underlined) preceded by a BamHI restriction site. The Snail (152-236) mutant was generated from this construct by BglII digestion to obtain the new C-terminal fragment without the 237-to-264 region. The following specific oligonucleotides were designed to amplify the Snail serine-rich domain (SRD; amino acids 82 to 123): sense oligonucleotide, 5′-GCAATAGATCTTCCACCATGGAGAGCCCCAAGGC-3′ (it adds a Kozak sequence [underlined] and a new ATG start codon [in italics]); antisense, 5′-CGTCTGCAGCTCCAGGGAGGAGGCCG-3′ (this adds a 3′ PstI restriction site). The Snail (82-151) mutant was obtained from an 82-to-264 fragment amplified by PCR; the 152-to-264 sequence was deleted by using the BglII restriction site previously mentioned. Snail (Δ90-120) was made by ligation of two PCR fragments obtained (i) with a sense oligonucleotide corresponding to the ATG site (previously described) and 5′-CAATGAGCTCAGCTCTACGGCCTTGGGGCTCTCCCGCAG-3′ as the antisense oligonucleotide and (ii) with 5′-CCGTTAGAGCTCCGCCGAGGCCTTCATC-3′ as the sense oligonucleotide and an oligonucleotide corresponding to the last amino acids of murine Snail as the antisense oligonucleotide. The SacI site (underlined) was used to ligate both fragments; this ligation introduced two serines instead of the deleted sequence. Deletion of nucleotides 138 to 151 in the N-terminal half was performed with a BstXI restriction site present in the 1-to-151 fragment of the Snail cDNA.
(ii) Snail point mutants.
The Snail Leu139/142→Ala mutant was obtained by site-directed mutagenesis with complementary specific oligonucleotides (sense, 5′-GGCCAACTTCCCAAGCAGGCGGCCAGGGCCTCGGTGGCCAAGGACCCCCAG-3′; antisense, 5′-CTGGGGGTCCTTGGCCACCGAGGCCCTGGCCGCCTGCTTGGGAAGTTGGCC-3′) in accordance with the protocol for the Quick-Change site-directed mutagenesis kit from Stratagene. The bold nucleotides are those mutated from the original sequence. The first codon mutation abolishes a PvuI site present in wild-type Snail and was used to test positive constructs. To mutate all of the serines of the SRD (but not Thr119) in Ser→Ala and Ser→Asp mutants, synthetic oligonucleotides (sense and antisense) corresponding to the 90-to-120 sequence were purchased from Sigma-Genosys. Codons coding for serines were replaced with alanines or aspartates by using codons closest in sequence. 5′ ends were phosphorylated, sense and antisense complementary oligonucleotides were annealed, and blunt-ended inserts were cloned into the Snail (Δ90-120) mutant in the newly created SacI site opened by using the EcoCRI isoschizomer. This means that a new leucine was added to the end of the domain. The Ser→Ala mutation added a new NotI restriction site that was used to test positive constructs. All mutants were confirmed by sequencing.
(iii) Other constructs.
Murine Slug was cloned, by RT-PCR with a One-Step RT-PCR kit with Pfx Taq (Invitrogen), from 500 mg of total RNA from NIH 3T3 cells, in accordance with the manufacturer's procedure, by using oligonucleotides specific for its 5′ and 3′ tails that added a 5′ BamHI restriction site and a 3′ EcoRV restriction site. The EcoRV site was designed to be in frame with an HA tag cloned into the acceptor vector, pcDNA3. Murine SC-35 or U2AF65 was obtained by the same procedure, in these cases by adding a SalI restriction site at the 5′ tail. The absence of mutations in all of these cDNAs was confirmed by sequencing.
cDNAs corresponding to the Snail and Slug cDNAs were cloned into vector pcDNA3 (Invitrogen), peGFP-C1, pDsRed1 (Clontech), pGEX-6P1, pGEX-6P3 (Amersham), or pMAL-c2X (New England BioLabs). SC-35 and U2AF65 were cloned into pDsRed1. pGFP-Tcf-4 and pGFP-ZEB1 were prepared by inserting into peGFP-C1 the full-length cDNAs corresponding to transcriptional factors Tcf-4 and ZEB1, which were previously used in our laboratory (14, 28).
Analysis of Snail and E-cadherin expression by RT-PCR.
All of the cell lines used in this study were grown in Dulbecco's modified essential medium supplemented with 10% fetal calf serum. Total RNA was obtained and analyzed for Snail and E-cadherin as previously described (14). Briefly, RT-PCR analysis was performed with a SuperScript One-Step kit and Platinum Taq polymerase (Invitrogen) with 1 μg of total RNA and primers corresponding to human Snail (sense, corresponding to nucleotides 104 to 125; antisense, corresponding to nucleotides 290 to 310), human E-cadherin (sense, corresponding to nucleotides 1977 to 2006; antisense, corresponding to nucleotides 2287 to 2316), and human cyclophilin (sense, corresponding to nucleotides 45 to 62; antisense, corresponding to nucleotides 690 to 708). The sense and antisense primers always anneal with different exons. Thirty-five cycles were used for Snail analysis, and 32 were used for E-cadherin or cyclophilin analysis.
Analysis of Snail expression by immunofluorescence assay and Western blotting.
Analysis of endogenous Snail by immunofluorescence assay was performed with an anti-Snail polyclonal antibody obtained in our laboratory by affinity purification of an antiserum raised in a rabbit immunized with a glutathione S-transferase (GST)-Snail fusion protein. Previous use of this antibody has been reported (14). Cells, plated on glass coverslips, were fixed with 4% paraformaldehyde for 30 min and permeabilized by incubation with 1% sodium dodecyl sulfate (SDS) for 10 min or with 70% ethanol for 30 min. Blocking was carried out for 1 h with phosphate-buffered saline (PBS) containing 0.1% saponin and 1% bovine serum albumin (BSA). Affinity-purified rabbit polyclonal anti-Snail antibody was diluted 1:10 in PBS-1% BSA-0.1% saponin and incubated overnight at 4°C (for detection of exogenous Snail) or at room temperature (for detection of endogenous Snail). After washing, binding of primary antibody was detected with anti-rabbit antibodies conjugated to fluorescein isothiocyanate (Dako) or Alexa 488 (Molecular Probes). Controls with irrelevant antibodies (anti-GST [Pharmacia Biotech] or anti-erythropoietin [a kind gift of C. de Bolòs, IMIM, Barcelona, Spain]) confirmed the specificity of labeling. Nuclei were counterstained with propidium iodide. Finally, fluorescence was viewed through a Leica TCS-SP2 confocal microscope. Alternatively, analysis was performed on suspended cells. Trypsinized cells were fixed with paraformaldehyde, permeabilized with ethanol, blocked with PBS plus 3% BSA, and incubated with anti-Snail antibody overnight at 4°C. Cells were centrifuged at 400 × g for 5 min after every incubation.
For Western blot analysis, cell extracts were prepared directly in SDS buffer (25 mM Tris-HCl [pH 7.6], 1 mM EDTA, 1 mM EGTA, 1% SDS) or first in CYT buffer (10 mM HEPES [pH 7.6], 10 mM KCl, 1.5 mM MgCl2, 0.5% NP-40) and centrifuged for 10 min in a Microfuge at 15,000 × g, and the pellet was resuspended in SDS buffer. Samples were separated by SDS-10 or 15% polyacrylamide gel electrophoresis (PAGE). After transference, membranes were analyzed with mouse (Boehringer) or rat (Roche) anti-HA monoclonal antibody (1:1,000; for 1 h) or rabbit polyclonal anti-Snail antibody (1:100; overnight a 4°C).
Cell transfection.
Transient transfections of Snail constructs cloned in plasmid pcDNA3 or pGFP were performed with 0.75 to 1.5 μg of plasmid and Lipofectamine-Plus reagent (Invitrogen) diluted in OptiMEM serum-free medium (Invitrogen) in accordance with the manufacturer's instructions. Cells were plated on glass coverslips unless otherwise indicated. Protein expression was analyzed 16 to 18 h after transfection. To inhibit nuclear export, leptomycin B (LMB; Sigma) was added to the culture medium (5 ng/ml in ethanol) 2 h previous to cell fixation.
Pull-down assays.
GST-Snail and maltose binding protein (MBP)-Snail fusion proteins were prepared from Escherichia coli strain BL21 and purified by chromatography on glutathione-Sepharose (Amersham) or amylose resin (New England BioLabs), respectively. Binding assays were performed with 50 mM Tris-Cl (pH 7.4)-150 mM NaCl-1 mM EDTA-1 mM dithiothreitol (DTT)-1 mM MgCl2-0.5% NP-40 (binding buffer) for 30 min at room temperature with 1 μg of GST-Snail (1-151) (either the wild-type or the mutant form) and MBP-Snail (152-264) (either the wild-type or the 152-to-238 mutant form). Alternatively, RWP-1 cells were transfected with the indicated forms of the C-terminal half of Snail. After 36 h, cell extracts were prepared in binding buffer plus protease inhibitors, and 250 μg of cell lysate was incubated with 1 μg of GST-SNA (1-151). The GST-Snail (1-151) and MBP-Snail (152-264) complexes were precipitated with glutathione-Sepharose or amylose resin, respectively, and proteins bound to the beads were resolved by SDS-PAGE and blotted against rat anti-HA tag or GST (1:2,000; BD Biosciences-Transduction Labs).
Luciferase reporter assays.
Reporter assays were done with RWP-1 cells and 25 ng of the human E-cadherin promoter (−178/+92) (2), 25 ng of the human fibronectin promoter (−637/+105, from the preprofibronectin translation start), or 100 ng of the human LEF1 promoter (−1874/+58, from the translation start), all cloned into the pGL3 basic vector (Promega) with a mutated E box as described elsewhere (2). The human fibronectin promoter was obtained by PCR from HT-29 genomic DNA with high-fidelity Pfx polymerase (Invitrogen) and oligonucleotides 5′-ACACAAGTCCAGCCACTCCC-3′ (−637 to 615) and 5′-TTGCTGAGCCTGCCTGCCTCTTG-3′ (+105 to +83), containing MluI and XhoI sites at the 5′ ends, respectively. The purified PCR product was cloned into the MluI and XhoI restriction sites of pGL3. To keep the fragment from the preproprotein contained in the cloned region and the luciferase gene of the reporter in frame, the construct was linearized with MluI and HindIII, filled in with the Klenow fragment (Promega), religated, and sequenced. The human LEF1 promoter was obtained from the same source with oligonucleotides 5′-CTTGTCTCCAAAGAGCG-3′ (−1874/−1858), containing a KpnI site at the 5′ end, and 5′-TGGCGCAGAGTTCCGG-3′ (+43/+58). The purified PCR product was cloned into the KpnI and SmaI sites of pGL3 and sequenced. Cells were cotransfected with the indicated amounts of reporter plasmids, 1 ng for repression and 25 ng for activation of the wild-type and mutant forms of Snail cloned into pcDNA3 and 0.5 ng of simian virus 40-Renilla luciferase plasmid as a control for transfection efficiency. Expression of the firefly and Renilla luciferases was analyzed in accordance with the manufacturer's instructions.
32P labeling and immunoprecipitation.
RWP-1 or NIH 3T3 cells grown on 60-mm-diameter plates were transfected with pcDNA3-Snail-HA or the different mutants. After 36 h, cells were washed three times in 25 mM Tris HCl (pH 7.5)-140 mm NaCl and incubated for 3 h in Dulbecco's modified essential medium without phosphates but supplemented with 50 mM HEPES (pH 7.5) and 10% dialyzed fetal calf serum. Subsequently, cells were incubated with the same medium but containing 250 μCi of [32P]orthophosphate (Amersham) per ml for an additional 3 h. Afterwards, cells were lysed in immunoprecipitation-kinase assay buffer (50 mM Tris-Cl [pH 7.4], 1 mM EDTA, 150 mM NaCl, 0.2% NP-40, 1% Triton X-100, 1 mM DTT, 5 mM sodium fluoride, 1 mM sodium orthovanadate, 4 mM β-glycerophosphate, protease inhibitors), incubated on rotation for 30 min at 4°C, and centrifuged for 15 min at 20,000 × g. The whole-cell lysates obtained were preabsorbed with protein G-agarose (Roche) for 4 h at 4°C, and supernatants were incubated overnight at 4°C with 1 μg of rat anti-HA monoclonal antibody (Roche). After collection of immunoprecipitates with protein G-agarose beads for 3 h, they were resolved by SDS-PAGE and exposed to Curix (AGFA) film for 24 to 48 h. A parallel plate of cells was used as a control for expression. Cells were transfected with the indicated plasmids, extracts were prepared at the same time but without inclusion of 32P during the labeling period, and anti-HA immunoprecipitates were analyzed by Western blotting with rat anti-HA monoclonal antibody (Roche). Alternatively, the analysis was performed with 32P-labeled MDCK cells constitutively expressing Snail (2).
Phosphoamino acid analysis was performed by two-dimensional thin-layer electrophoresis in silica gel plates (35). 32P-labeled Snail was hydrolyzed in 6 M HCl for 1 h at 110°C. After concentration, phosphorylated amino acid standards were added to the sample and separated by electrophoresis at pHs 1.9 (first dimension) and 3.5 (second dimension). The positions of the standards were visualized by staining with ninhydrin, and those of 32P-labeled amino acids were determined by autoradiography.
Nucleotide sequence accession numbers.
The GenBank database accession numbers of the genes studied here are as follows: murine Snail, NM011427; murine Slug, AB38365; human ZEB1, U12170; human Tcf-4, Y11306; mouse SC35, NM011358; mouse U2AF65, NM133671.
RESULTS
Snail is located in the nucleus and cytosol of tumor cell lines.
The presence of the Snail transcription factor was analyzed in different epithelial cell lines by RT-PCR. As has been previously reported (see introduction), there is a good inverse correlation between Snail and E-cadherin in most cell lines, exemplified in these assays by RWP-1 and MDA-231 cells (Fig. 2A). However, some cell lines show high levels of the RNAs corresponding to both genes. Three examples of human cell lines and one of murine cell lines with concomitant expression of Snail and E-cadherin are presented in Fig. 2A. Simultaneous expression of both genes has been previously reported by us and others (9, 14, 15). The expression and intracellular localization of Snail protein were analyzed in a panel of epithelial and mesenchymal cell lines with a polyclonal antiserum prepared against the recombinant protein. As shown in Fig. 2B, this antiserum recognizes ectopically expressed Snail and a protein of NIH 3T3 fibroblasts with a similar molecular mass. By immunofluorescence assay, a signal was detected mainly in the nuclei of cells stably transfected with Snail (2). Most of the cell lines showed endogenous reactivity against our polyclonal antibody not only in the nucleus but also in the cytosol (Fig. 2C). There was a good correlation between the presence of reactivity against our Snail polyclonal antibody and detection of Snail by RT-PCR. For instance, the signal obtained in RWP-1 cells was much weaker than that in NIH 3T3 or MDA-231 cells. Snail was present mostly in the cytosol in some cell lines, such as MCF-7, Caco-2, HCT-116 (Fig. 2C), or EpH4 (not shown); these are the cell lines in which the presence of both E-cadherin and Snail was detected by RT-PCR.
FIG. 2.

Endogenous Snail is distributed not only in the nucleus but also in the cytoplasm of human tumor cells. (A) Coexpression of Snail and E-cadherin in epithelial cell lines. Snail and E-cadherin expression was analyzed by RT-PCR in mouse and human cell lines. Cyclophilin levels were used as a loading control. (B) Western blotting analysis of Snail expression. Cell extracts prepared in SDS buffer (80 μg for exogenous Snail or 100 μg for endogenous Snail) were subjected to SDS-10% PAGE and analyzed by Western blotting with anti-Snail polyclonal antibody. (C and D) Immunofluorescence distribution of anti-Snail reactivity. Cells stably transfected with Snail (MDCK SNA1) or presenting different levels of Snail expression by RT-PCR were analyzed by immunofluorescence assay with the anti-Snail antibody. Cells were stained with propidium iodide to localize nuclei. Note that the staining in panel C was performed at a lower temperature than that in panel D.
In order to better visualize the subcellular location of Snail, and considering the low abundance of the protein (Fig. 2B), the gene was ectopically expressed in different cell lines and the distribution of the protein was analyzed by immunofluorescence assay with our polyclonal antibody against Snail (data not shown) or direct visualization of the subcellular location of the fused green fluorescent protein (GFP) (Fig. 3). The distribution of ectopically expressed Snail was analyzed after 12 to 18 h to avoid excessive accumulation of the protein. Similar results were obtained by both approaches. Ectopically expressed Snail accumulated in the nuclei of some cell lines, such as SW-480 (Fig. 3A). However, in other cell lines, such as MCF-7, Snail protein seemed to be excluded from the nucleus (Fig. 3B). In most of the cases, the pattern was intermediate and cells with a predominant nuclear or cytosolic location coexisted in the same population. In RWP-1 cells, for instance, 72% ± 8% (mean ± standard deviation of three experiments) of the cells transfected with GFP-Snail had nuclear plus cytosolic staining, 18% ± 8% had exclusively nuclear staining, and 10% ± 4% had totally cytosolic staining. Similar proportions were observed in MDCK and NIH 3T3 cells. This pattern of distribution was quite different from that obtained for other transcription factors, such as Tcf-4 and ZEB1 (Fig. 3G and H), which were located only within the nucleus.
FIG. 3.

Exogenous Snail is present in the nucleus and cytoplasm of human tumor cells. Cell lines were transfected with pGFP-Snail. Subcellular location was assessed 16 to 18 h after transfection by analysis of the distribution of GFP in a confocal microscope. Nuclear counterstaining was performed with propidium iodide. Shown are representative stainings of different cell lines. In RWP-1 cells, the most common location was that shown in panel C; less abundant patterns are shown in panels D and E. The distribution in this cell line of other transcription factors (Slug, Tcf-4, and ZEB1 [as fusions with GFP]) is shown in panels F, G, and H.
We analyzed whether the subcellular distribution of Slug was similar to that of Snail. As shown in Fig. 3F, GFP-Slug was detected solely in the nucleus, with a nuclear pattern similar to that shown by Snail. No fluorescence signal in the cytosol was observed in any of the cell lines studied.
The distribution of Snail in the nucleus was not homogeneous. As has been previously described for Slug (17), Snail was present in discrete foci in the nucleus. This dotted staining was detected when the subcellular distribution of GFP-Snail or Snail-HA was determined (Fig. 3 and 4A), especially if nuclear export was blocked (see below). These Snail nuclear structures were not stained with propidium iodide (Fig. 4A), indicating that they corresponded to interchromatin granule clusters (36). Consequently, Snail staining always excluded nucleoli. The identity of these nuclear speckles was studied by cotransfection of GFP-Snail with SC-35 and U2AF65, two proteins that labeled these structures differentially (13, 37). As shown in Fig. 4B, Snail colocalized much more closely with splicing factor U2AF65 than with SC-35, contrary to what has been previously reported for Slug (17).
FIG. 4.

Snail is located in nuclear speckles. RWP-1 cells were transfected with GFP-Snail (A and B) and RFP-SC-35 or RFP-U2AF65 (B). Its subcellular location was assessed 16 to 18 h after transfection by analysis of the distribution of GFP with a confocal microscope. When appropriate, nuclear counterstaining was performed with propidium iodide (A).
Therefore, these results suggest that Snail is distributed differently than Slug in cells and is not present exclusively in the nucleus and prompted us to study the molecular basis of this location.
Distinct domains are responsible for the alternative subcellular location of Snail.
The N- and C-terminal Snail domains were fused to GFP and transiently transfected in several cell lines. In all of the cell lines studied (RWP-1, MDCK, MCF-7, NIH 3T3), the N- and C-terminal domains showed alternative locations (Fig. 5). The C-terminal half, GFP-Snail (152-264), had a restricted nuclear distribution. The pattern was diffuse, with nucleolar exclusion. This contrasted with the speckled pattern frequently obtained with the full-length protein (Fig. 4). On the other hand, the N-terminal half, GFP-Snail (1-151), was strictly cytosolic and thus was probably responsible for the cytosolic location of full-length Snail.
FIG. 5.

A subdomain in the N-terminal half of Snail excludes GFP from the nucleus. The subcellular distribution of GFP fused with distinct domains of Snail is shown. The indicated GFP-SNA mutants were transfected to MDCK cells, and the location of GFP was analyzed 24 h later with a confocal microscope. Shown is the most abundant pattern (in more than 90% of the cells) for each mutant. Although in all cases, cells were counterstained with TOP-Ro or propidium iodide, for the sake of clarity, only the distribution of GFP is shown.
The N-terminal domain was divided into two fragments, 1 to 82 and 82 to 151, fused to GFP and transiently transfected to RWP-1 cells. As shown in Fig. 5, only the 82-to-151 sequence excluded GFP from the nucleus. On the contrary, GFP-Snail (1-82) had the same pattern as control GFP. This result suggested that, at least in part, this 82-to-151 domain of Snail could mediate its cytosolic location.
A leucine-rich signal is involved in CRM1-dependent nuclear export of Snail.
Several different mechanisms can explain the effect of the Snail regulatory domain on the cytosolic location of the protein. This domain may anchor Snail to specific cytosolic proteins, inhibit nuclear import, or mediate its nuclear export. The most common mechanism of nuclear export of proteins in eukaryotic cells is based on CRM1-dependent systems and requires the presence of a hydrophobic Leu-rich sequence (11, 12, 41). We examined the Snail amino acid sequence for a Leu-rich sequence similar to those described for CRM1-mediated nuclear export. The sequence LGQLPKQLARLS (leucines are underlined), between residues 132 and 143 of mouse Snail, almost matches the perfect consensus sequence previously defined for an NES [LX(1-3)LX(2-3)LXL]. Replacement of Leu with Val, as occurs in human Snail (Leu135 to Val), has been observed in some NESs, as well as longer spaces among leucines (21). This putative NES is not present in the Snail relative Slug, whose localization was exclusively nuclear (Fig. 3F). Deletion of part of this sequence (residues 138 to 151, which deletes the last two leucines) from a GFP-N-terminal domain fusion protein was sufficient to impair its exclusion from the nucleus, as shown in Fig. 5.
To check whether CRM1 is responsible for Snail nuclear export, we inhibited the system with LMB. LMB is an antifungal molecule that is able to bind CRM1 and block the formation of the CRM1/Ran-GTP/nuclear protein complex necessary for export (20, 30). Treatment of transfected RWP-1 cells with LMB rapidly relocated Snail protein to the nucleus (Fig. 6A). The effect was analyzed in one of the few cells in this population with cytosolic staining only and in other cell lines where this pattern was more predominant, such as MCF7. LMB caused rapid translocation of Snail from the cytosol to the nucleus as early as 10 min after addition of the drug. Accumulation of the protein in nuclear speckles required some additional time. Similar results were obtained when the effect of LMB on the location of other exclusively cytosolic Snail constructs was studied. In the presence of this drug, both the complete regulatory domain (amino acids 1 to 151) and a fragment containing the NES (82 to 151) were detected mostly in the nucleus (Fig. 6B). Staining in the nucleus was homogeneous without labeling in speckles, indicating that positioning in these structures requires the zinc finger domain.
FIG. 6.

The cytosolic location of Snail depends on its CRM1/NES-mediated nuclear export. (A) LMB rapidly relocates GFP-Snail to the nucleus. RWP-1 cells transfected with pGFP-Snail were treated with LMB (5 ng/ml); the change in the location of GFP in a cell that presented predominant cytosolic staining is shown at different times (in minutes). (B) LMB treatment relocates Snail truncations usually present in the cytosol. This experiment was performed as described for panel A, with the indicated Snail mutants. (C) Mutation of leucines 139 and 142 to Ala prevents nuclear exclusion. Leucines 139 and 142 were mutated to alanines in full-length Snail or the N-terminal domain [GFP-Snail (1-151)]. Mutant constructs were fused to GFP and expressed in RWP-1 cells. The distribution of GFP after an 18-h transfection is shown. WT, wild type.
To further confirm that the 132-to-143 sequence behaves as an NES, we performed a selective mutation of two leucines to alanine, i.e., Leu139,142→Ala, the same residues of the NES eliminated by the 138-to-151 deletion. This mutation almost totally abolished the ability of full-length Snail to be exported and, thus, its cytosolic location. A similar export inhibition was obtained when the distribution of the GFP-SNA (1-151) (Leu139,142→Ala) mutant was determined (Fig. 6C).
We concluded from these results that Snail harbors a functional NES preceding the zinc finger domain. This sequence, which includes residues 132 to 143, is responsible for the efficient nuclear export of Snail and the cytosolic location of this protein in some epithelial cells.
Phosphorylation of the SRD modulates the nuclear export and transcriptional activity of Snail.
The activity of the Snail NES seemed to be variable among the cell lines studied, as Snail preferred a nuclear location in some cell lines, whereas it had a marked preference for an extranuclear location in others (see above). This suggested a possible modulation of the export process. A relevant clue was provided by two independent observations.
First, selective deletion of the SRD of Snail (residues 90 to 120) blocked nuclear export almost entirely. This mutant, GFP-Snail (Δ90-120), was only distributed in the nucleus in all of the cell lines tested (Fig. 7A), including those with a prevailing cytosolic location of wild-type Snail. Therefore, this SRD was a good candidate as a mediator of modulation of the adjacent NES activity. Curiously, the effect of the SRD deletion on nuclear location was dependent on the presence of the zinc finger domain, since GFP-SNA (1-151) (Δ90-120) had the same cytosolic distribution as the wild-type N-terminal domain (Fig. 7A).
FIG. 7.
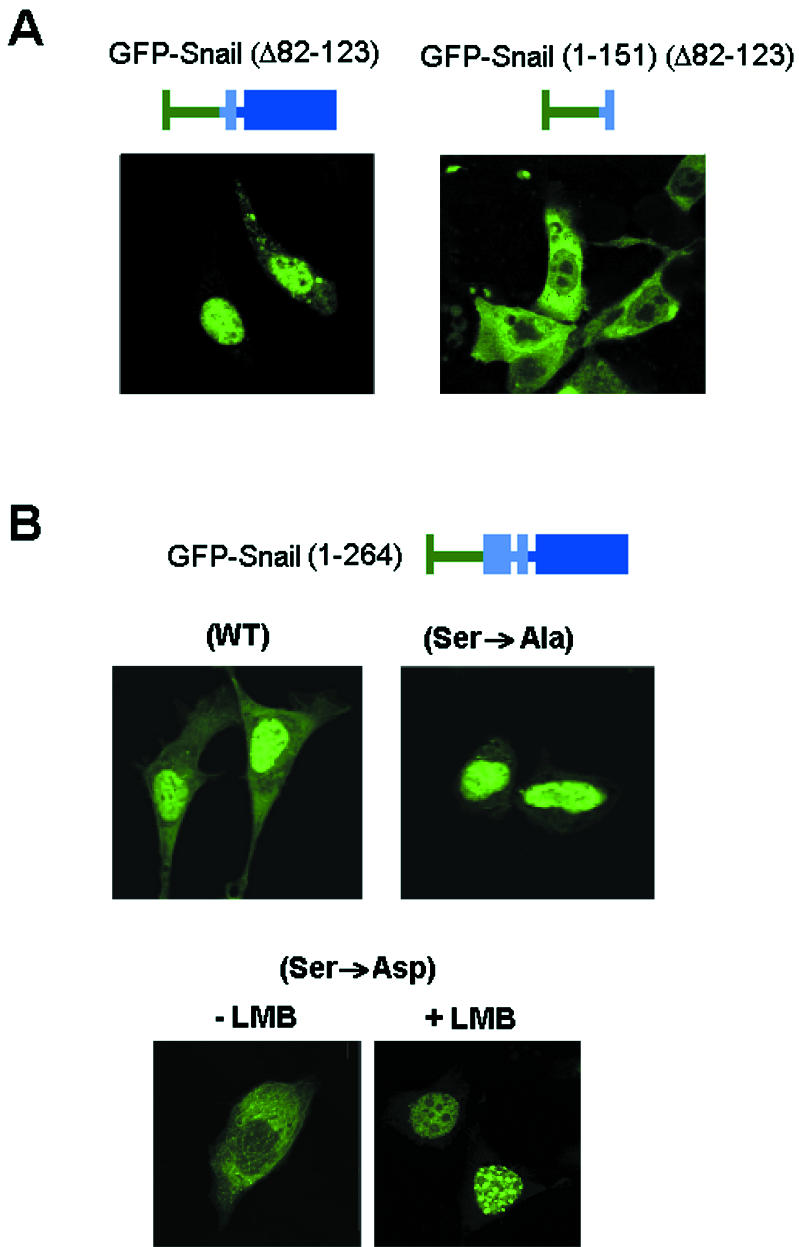
The SRD of Snail modulates its nuclear export. (A) The SRD of Snail determines its nuclear export. The indicated GFP fusions with a deletion comprising amino acids 90 to 120 were expressed in MDCK cells, and their distribution was analyzed in a confocal microscope. (B) Phosphorylation of SRD modifies the subcellular distribution of Snail. This experiment was performed as described for panel A, with the indicated SRD mutant forms of Snail and addition of LMB when appropriate. WT, wild type.
Moreover, analysis of the Snail protein in stable transfectants (2) showed a doublet, which is indicative of a covalent modification of the protein (Fig. 8A). This doublet was more evident when the Western blot assay was performed after electrophoresis in high-percentage polyacrylamide gels. Incubation of the cell extracts with recombinant alkaline phosphatase led to the disappearance of the upper band, suggesting that this band corresponded to the phosphorylated protein (Fig. 8B).
FIG. 8.

Snail is phosphorylated at the SRD. (A and B) Detection of Snail protein as a doublet in cell lines. (A) Extracts were prepared from MDCK or HT-29 M6 cells stably transfected with Snail (1) by lysis of the cells in 25 mM Tris HCl (pH 7.5)-1 mM EDTA-1 mM EGTA-1% SDS and analyzed by Western blotting after electrophoresis in 15% polyacrylamide gels. (B) Cell extracts were diluted 1:10 in 25 mM Tris HCl (pH 7.9)-10 mM MgCl2-1 mM DTT-100 mM NaCl-1% Triton X-100 and incubated in a final volume of 350 μl without (−CIP) or with (+CIP) 20 U of calf intestinal phosphatase (New England BioLabs) for 6 h at 37°C. Migration of the Snail band was analyzed by Western blotting as described above. (C) Immunoprecipitation of HA-tagged Snail after 32P labeling. The indicated forms of Snail, tagged with the HA epitope, were inserted into plasmid pcDNA3 and transfected into RWP-1 cells. At 24 h after transfection, cells were labeled for 3 h with 32P, cell extracts were prepared, and Snail was immunoprecipitated with anti-HA antibody. A representative result of four experiments performed is shown. No radioactivity was observed in the Snail band when cells were transfected with an empty plasmid. A parallel plate of cells transfected with the indicated plasmids was used as a control for expression of the mutants; extracts were prepared without labeling, immunoprecipitated, and analyzed by Western blotting with anti-HA antibody. (D) Phosphoamino acid analysis of 32P-labeled Snail. An immunoprecipitate from the experiment whose results are shown in panel C was hydrolyzed and analyzed by two-dimensional thin-layer electrophoresis as indicated in Materials and Methods. The migration of the phosphoamino acid standards is indicated. PTyr, phosphotyrosine; PSer, phosphoserine; PThr, phosphothreonine; WT, wild type.
To assess more definitively whether Snail was being phosphorylated in vivo, the protein was immunoprecipitated from transiently transfected RWP-1 cells labeled with 32P. Snail was consistently labeled with this isotope, either in these cells (Fig. 8C) or in NIH 3T3 or MCF-7 cells (not shown). This phosphorylation was mainly due to serine modification, as confirmed by phosphoamino acid analysis, although a minor spot corresponding to PThr was also detected (Fig. 8D). Furthermore, phosphorylation occurred within the SRD, as a mutant Snail protein without this subdomain [deletion mutant Snail (Δ90-120)] was not labeled with 32P (Fig. 8C).
We also checked whether phosphorylation of the SRD can affect NES function. As described for other factors (31, 39), we considered the possibility that a charge accumulation in this region may unmask a sequence relevant to its nuclear location. This idea is supported by the fact that 14 out of the 18 serines predicted to be phosphorylated in the protein are concentrated within this domain. To test this hypothesis, we replaced all of the serine residues in the SRD with alanines (Ser→Ala mutant) or aspartates (Ser→Asp mutant). These changes gave an unphosphorylated domain and mimicked a phosphorylated form, respectively. Both the Ser→Ala and Ser→Asp mutants were not phosphorylated in 32P-labeled cells (Fig. 8C), confirming that these serines were the target of the phosphorylation in vivo. After transfection, the GFP fusion constructs of these two mutants disclosed a stunning inversion of their subcellular location patterns: whereas the Ser→Ala mutant was mainly nuclear, the Ser→Asp mutant was almost exclusively cytosolic (Fig. 7B). Moreover, LMB totally reversed the Ser→Asp mutant extranuclear distribution and translocated it to the nucleus (Fig. 7B), thus confirming that the Ser→Asp mutant was mostly cytosolic, not because of its inability to travel to the nucleus but because of its increased nuclear export. According to these findings, we concluded that phosphorylation of the SRD was responsible for the modulation of Snail nuclear export. When inside the nucleus, the two mutants were located differently: whereas the Snail Ser→Asp mutant accumulated in speckles, the Snail Ser→Ala mutant (either in the presence of LMB or not in the presence of LMB) was distributed homogeneously apart from nucleoli.
As has been previously reported, Snail directly binds to the E-cadherin promoter and represses its activity (2, 6). Nuclear location of Snail protein should be essential for its transcriptional function. Therefore, we predicted that cytosolic Snail would not repress the E-cadherin promoter. Similarly, those deletion mutants with impaired or absent nuclear location would have inefficient repressor activity. To assess this, we analyzed the repressive activity of the Snail mutants on a 300-bp proximal human E-cadherin promoter, which includes the three E boxes described for Snail binding (2). As shown in Fig. 9A, mutants with greater export efficiency, such as the Snail Ser→Asp mutant, presented lower repression activity than the wild-type protein or mutants deficient in export. This effect was not due to impaired binding to DNA, since the Snail Ser→Asp mutant bound an oligonucleotide probe containing the CACCTG sequence just as efficiently as the wild-type protein did (data not shown).
FIG. 9.

Phosphorylation of the SRD of Snail modulates its transcriptional activity. Effects of Snail mutations on repression of the E-cadherin promoter and activation of the fibronectin or LEF1 promoter are shown. Promoter activity was assessed by using luciferase reporter assays, as described in Materials and Methods, after transfection of the indicated Snail forms inserted into the vector pcDNA3. The results shown are the average ± the standard deviation of three (fibronectin or LEF1 promoter) or four (E-cadherin promoter) experiments performed in triplicate. WT, wild type.
Snail expression also causes induction of the mesenchymal LEF-1 and fibronectin genes (14). As shown in Fig. 9B and C, Snail induces a significant increase in the activity of the LEF-1 or fibronectin promoter. A P2A Snail mutant, unable to repress E-cadherin (2), did not induce these promoters, although its cellular distribution was identical to that of the wild-type protein, indicating that the SNAG box is essential for Snail transcriptional effects independently of the nuclear localization. As expected, Snail mutants that were retained better in the nucleus, such as the Snail Ser→Ala and Leu139,142→Ala mutants, caused greater activation of these promoters than did the wild-type form, whereas Snail (Ser→Asp) was consistently less active (Fig. 9B and C).
Phosphorylation of the serine-rich sequence modulates the interaction between the amino- and carboxy-terminal domains of Snail.
Thus, phosphorylation of the SRD modulates the function of the NES of Snail. One possible reason for this result is that accumulation of charges in this region may promote conformational changes in the protein that cause unfolding of the protein and exposure of the NES, which is normally not accessible to the CRM1 transporter. Data obtained with several Snail deletion mutants seemed to favor this hypothesis. First, as mentioned above, deletion of the SRD in GFP-Snail (1-151) did not impair its nuclear export, as it did when this deletion was performed on the full-length protein (Fig. 7A). This indicated that the presence of the zinc finger domain is necessary for the SRD effects on NES function and suggested the existence of an interrelationship between these two domains.
Moreover, a mutant lacking the last zinc finger, Snail (1-236), was excluded from the nucleus (Fig. 10A). The cytosolic location of the Snail (1-236) mutant protein was reversed by LMB treatment, indicating that this mutant had not lost its ability to travel to the nucleus but that it was rapidly exported (Fig. 10A). This mutation did not modify the nuclear distribution of the C-terminal domain, since Snail (152-236), a fragment unable to be exported because it lacked the NES, was detected in the nucleus (Fig. 10A). Hence, we deduced that the last C-terminal amino acids may preclude the accessibility of CRM1 to the NES in concert with the SRD.
FIG. 10.

Interaction between the N and C domains controls the intracellular location of Snail. (A) Deletion of the last 28 amino acids modifies the subcellular distribution of Snail. Mutant forms of Snail were expressed as GFP fusions in RWP-1 cells for 24 h before their distribution was determined. When indicated, LMB (5 ng/ml) was added and the mixture was incubated for 2 h. (B) The C-terminal domain of Snail shifts the cellular distribution of the N domain. GFP-Snail (1-151) was cotransfected into RWP-1 cells with the RFP-Snail (152-264) fusion, and the subcellular location of both fluorescent proteins was examined by confocal microscopy. WT, wild type.
This effect could be explained by a direct interaction between the N domain and the last zinc finger of the C-terminal tail. To test this hypothesis, we cotransfected the two domains as fusions with fluorescent proteins: Snail (1-151) was fused to GFP, and Snail (152-264) was fused to red fluorescent protein (RFP) (Fig. 10B). As expected, the C-terminal domain was expressed in the nucleus. Only in cells expressing the RFP-C domain was the above-described cytosolic location of the N domain not observed. In these cells, and not in those without expression of the C domain, the pattern observed for GFP-Snail (1-151) was similar to that observed for the complete protein. This confirmed that the C domain restricted the export ability of the N domain.
The association between the two domains was verified by pull-down assays. The Snail N- and C-terminal domains were fused to GST and MBP, respectively, and purified. As shown in Fig. 11A and B, direct binding of the two domains was evidenced by the copurification of both in either glutathione-Sepharose or amylose resin. An MBP-C-terminal domain fusion lacking the fourth zinc finger, Snail (152-236), interacted with GST-Snail (1-151) significantly less than the whole zinc finger domain, Snail (152-264) (Fig. 11A and B). The interaction was also detected when the C-terminal domain was transiently expressed in RWP-1 cells. Very little association was observed when the N-terminal domain was limited to the 1-to-82 sequence (Fig. 11C), suggesting that the 82-to-151 fragment, which included the SRD and the NES, was necessary for the association.
FIG. 11.

Phosphorylation of the SRD regulates the interaction between the Snail N and C regions. (A and B) GST-Snail (1-151) was incubated with MBP fusions containing the 152-to-264 and 152-to-236 C-terminal fragments of Snail, both tagged with the HA epitope. Complexes were bound to glutathione-Sepharose (A) or amylose resin (B) and analyzed by Western blotting (WB) with the indicated antibodies. As a control, the same GST and MBP fusions were incubated with similar amounts of MBP and GST, respectively. (C and D) The N-terminal half of Snail (1 to 151) and the corresponding mutant forms were fused to GST and incubated with cell extracts from RWP-1 cells expressing the C-terminal domain of Snail (152 to 264) tagged with the HA epitope. Complexes bound to glutathione-Sepharose beads were analyzed by Western blotting against HA or GST as a control. The molecular masses of the fusion proteins containing the different fragments of Snail are indicated. WT, wild type.
The interaction between the two domains was impaired when the serines present in the SRD were mutated to Asp, thus mimicking its phosphorylation (Fig. 11D). As expected, no significant differences were observed between the binding of the C-terminal domain to the GST-SNA (1-151) wild type or the Ser→Ala mutant (data not shown). These results explain why phosphorylation of SRD facilitates export: it hinders the interaction between the two domains, exposing the NES.
The same conclusion was reached when the ability of the N- and C-terminal domains to interact was analyzed in vivo with different fluorescent proteins (Fig. 11B). The ability of the RFP-Snail (152-264) fusion to translocate GFP-Snail (1-151) to the nucleus was not detected when the Ser→Asp N-terminal mutant protein was used, confirming that the presence of a negative charge in this sequence prevents the interaction of the two domains.
Snail phosphorylation and subcellular distribution are controlled by cellular attachment to the ECM.
We looked for conditions that could regulate Snail phosphorylation and function. The extracellular matrix (ECM) microenvironment has been reported to affect the characteristics of epithelial cells (22). Acting through integrin activation, ECM proteins have been shown to modulate E-cadherin expression (23, 26, 43). Therefore, we checked whether integrin adhesion modulates Snail phosphorylation. As shown in Fig. 12A and B, detachment of cells from the plate caused an increase in the phosphorylation of the Snail protein present in the MDCK SNA1 cell line. This increase was observed following the shift in the molecular weight of the Snail protein or the extent of 32P incorporation into the protein. In both cases, a significant increase in the phosphorylation of Snail was observed in cells that were trypsinized before lysis, in contrast to the cells directly scraped into homogenization buffer (Fig. 12A and B). Trypsinization also affected the cellular distribution of Snail protein. As shown in Fig. 12C, Snail was observed mostly in the nuclei in adherent MDCK SNA1 cells. However, resuspended cells presented a different pattern, with a diffuse reactivity throughout the cell. Vertical sections evidenced nuclei that were not predominantly stained and, in some cases, seemed to be excluded. Therefore, Snail phosphorylation and distribution were altered by loss of cell attachment.
FIG. 12.

Attachment to the ECM regulates the phosphorylation, subcellular localization, and activity of Snail. (A) Cell attachment modifies the molecular weight of the Snail protein doublet. MDCK SNA1 cell total extracts were prepared from cells homogenized in lysis buffer (adherent [ADH]) or previously resuspended and treated with trypsin for 20 min (suspended [SUSP]). Snail protein was analyzed by SDS-15% PAGE and Western blotting with anti-HA antibody. (B) Cell suspension increases Snail phosphorylation. Transfected MDCK SNA1 cells were labeled with 32P and treated as described for panel A, with trypsin when indicated (suspended cells). Extracts were prepared, and Snail was immunoprecipitated with HA antibody. (C) Localization of Snail is modified by cell attachment. MDCK SNA1 clones grown on a glass coverslip (adherent) or released with trypsin as described above (suspended) were analyzed by immunofluorescence assay with anti-Snail polyclonal antibody. Nuclei were localized by counterstaining with propidium iodide. Shown are representative sections (xy and xz) of the suspended cells obtained in a confocal microscope.
DISCUSSION
There is increasing evidence that regulation of the activity of some transcription factors can be achieved by determining their ability to reach the nucleus or stay in it (3, 27, 38, 47). Some of these transcription factors, such as p53, have NESs that direct their export through the CRM1 transporter (11, 12). In some cases, nuclear export of a factor can be regulated by phosphorylation on Ser/Thr residues (18, 31, 46). Thus, a mechanism based on phosphorylation can control the transcriptional activity of these factors by modulating their location. As has been reported for these factors (10, 46), this way of regulation permits rapid responses to extracellular stimuli or other signals and results in a powerful modulation of the system, as it can be switched on or off almost immediately.
In this report, we present data indicating that the transcription factor Snail cycles between the nucleus and the cytosol of cells and that the higher activity of a NES present in the protein is responsible for its exclusion from the nucleus. This NES has been allocated to amino acids 132 to 143 in mouse Snail, a sequence that is conserved among all known mammalian Snail homologues (human, mouse, dog, pig, and cow). However, it is worth noting that this sequence is not present in other family members, such as Slug. This raises the possibility that only mammalian Snail can be exported from the nucleus. It would be relevant to assess whether other previously unrecognized sequences can perform a similar function in other homologues or if the cytosolic location is exclusive to mammalian Snail. Our results obtained with mutant forms that are preferentially cytosolic indicate that nuclear export controls the transcriptional activity of Snail, as could be foreseen. However, we cannot totally discard the possibility that cytosolic Snail plays some positive role in the acquisition of migratory abilities by tumor cells.
Although the activity of the NES seems to be responsible for the cytosolic location of Snail, we have also considered that enhanced nuclear import or retention in the cytosol can also contribute to this location. Although the mechanism responsible for Snail import into the nucleus is still unknown, the similar and rapid import of the Ser→Asp mutant and wild-type forms of Snail after addition of LMB does not support this possibility.
The distribution of Snail in the nucleus is not homogeneous. As has been previously described for Slug (17), and as also reported here, Snail is present in discrete foci in the nucleus. These nuclear speckles have been identified as sites of active RNA splicing on the basis of their costaining with the factor U2AF65 (13). Snail accumulation in nuclear speckles was better observed when nuclear export was inhibited with LMB, suggesting that its presence in these structures precedes its export. The presence of Snail in these nuclear foci was not detected with the N and C domains of the protein in the presence of LMB, which indicates that the entire protein is required. Moreover, the Ser→Ala mutant does not accumulate in speckles, suggesting that phosphorylation may be a requisite for translocation to these structures. Other Snail mutants that present an unfolded conformation, such as Snail (1-236) or Snail (Ser→Asp), show a remarkable dotted staining when nuclear export is inhibited with LMB, indicating that the open conformation of the protein (see below) is required for association with nuclear speckles.
The activity of the NES is regulated by phosphorylation in an adjacent SRD. Although we do not know which kinase(s) modifies Snail, a significant amount of the protein seems to be modified in vivo. Analysis by Western blot assay indicates that Snail migrates as a doublet with the two bands, corresponding to the phosphorylated and unphosphorylated forms, present at almost equimolecular levels. This result indicates that approximately 50% of the Snail protein in the cells studied is modified. Furthermore, phosphorylation is relevant to the structure of Snail: introduction of negative charges into this SRD hinders the interaction of this region with the C-terminal zinc finger domain and induces a conformational change that, presumably, makes the NES more accessible to the CRM1 transporter. Our data also suggest an important role in the interaction with the N domain for the last zinc finger, since a C-terminal domain lacking this structure interacts inefficiently with the N terminus. However, it is also possible that this atypical zinc finger does not interact directly with the N domain but coordinates the structure of the rest of the fingers in the zinc finger domain. Anyhow, these results indicate for the first time the role of the SRD, a domain conserved in all Snail family members. Since only unfolded Snail protein is located in the nuclear speckles, it is also possible that phosphorylation, through changes in the structure of Snail, may modulate other Snail actions, even in the absence of export. This possibility would require that its presence in the different nuclear structures affects the activity of Snail.
Our results show that a substantial amount of Snail is phosphorylated in epithelial cells and that the modified protein is much less active as a repressor of E-cadherin or as an inducer of the mesenchymal genes. However, when phosphorylated, it retains the ability to bind DNA. We think that, rather than inactivating the transcriptional repressor activity of Snail, phosphorylation probably just shifts the protein out of the nucleus, blocking its access to the target promoters. Therefore, in the absence of other changes, Snail promotes its effects on the epithelial phenotype when its cellular levels increase above a certain threshold, it cannot be totally phosphorylated and exported, and it stays in the nucleus. Accordingly, Snail overexpression has been reported to repress epithelium-specific genes and to induce mesenchymal genes in several cellular systems (2, 6, 45). However, as this threshold does not have to be the same in different epithelial cell lines, the total levels of Snail protein are not fully indicative of its activity. These results explain why, in some cell lines, Snail and E-cadherin RNAs have been detected simultaneously (15).
We also present data indicating that Snail phosphorylation, intracellular distribution, and activity are controlled by cell attachment to the ECM. There is a rapidly growing body of evidence indicating that the extracellular environment affects the phenotypic characteristics of many epithelial cells (22). Effects of the ECM have also been attributed to tumor progression, in particular, to the acquisition of invasive properties by the cells situated at the edge of the tumor (5; discussed in reference 1). We have previously reported that the ECM, acting through the activation of the integrin-linked kinase, can promote activation of Snail transcription and a mesenchymal transition (40). The results presented here add additional complexity to this system, since Snail is also activated posttranslationally by attachment to the extracellular environment. Experiments need to be performed to further characterize the signal pathway involved in the phosphorylation of Snail and to identify the Snail kinase. The possible involvement of kinases related to integrin activation is currently being studied in the laboratory. In any case, these results suggest that cells with low levels of Snail present the ability to respond to changes in the microenvironment with a rapid modulation of Snail-sensitive genes. The possibility that Snail also plays a role in certain properties of epithelial cells, for instance, in cell migration, also deserves to be investigated.
Acknowledgments
We greatly appreciate the advice of Francesc Posas and Jose Aramburu and the help of Arrate Mallabiabarrena with the confocal microscopy. We thank Gabriel Gil, Mercè Martín, and Mireia Duñach for the generous gift of plasmids. The technical assistance of M. Carmen Torns, Marta Garrido, and Francisco Sánchez-Aguilera is greatly appreciated.
This study was supported by grants awarded to A.G.H. by the Ministerio de Ciencia y Tecnología (PM99-0132), the Fundación Científica de la Asociación Española Contra el Cáncer, and the Direcció General de Recerca of the Generalitat de Catalunya (2001/SSSGR410). B.M.-S. and S. G. were recipients of predoctoral fellowships from the Ministerio de Ciencia y Tecnología. D.D., A.V.-S., and I.P. were recipients of predoctoral fellowships from the Instituto Carlos III, the Universitat Pompeu Fabra, and the Ministerio de Educación, respectively.
REFERENCES
- 1.Barker, N., and H. Clevers. 2001. Tumor environment: a potent driving force in colorectal cancer? Trends Mol. Med. 7:535-537. [DOI] [PubMed] [Google Scholar]
- 2.Batlle, E., E. Sancho, C. Francí, D. Domínguez, M. Monfar, J. Baulida, and A. García de Herreros. 2000. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumor cells. Nat. Cell Biol. 2:84-89. [DOI] [PubMed] [Google Scholar]
- 3.Beals, C. R., N. A. Clipstone, S. N. Ho, and G. R. Crabtree. 1997. Nuclear localization of NFA-Tc by a calcineurin-dependent, cyclosporin-sensitive intramolecular interaction. Genes Dev. 11:824-834. [DOI] [PubMed] [Google Scholar]
- 4.Blanco, M. J., G. Moreno-Bueno, D. Sarrio, A. Locascio, A. Cano, J. Palacios, and M. A. Nieto. 2002. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene 21:3241-3246. [DOI] [PubMed] [Google Scholar]
- 5.Brabletz, T., A. Jung, S. Reu, M. Pozner, F. Hulbek, L. A. Kunz-Schugart, R. Knuechtel, and T. Kirchner. 2001. Variable β-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. USA 98:10356-10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cano, A., M. A. Pérez-Moreno, I. Rodrigo, A. Locascio, M. J. Blanco, M. G. del Barrio, F. Portillo, and M. A. Nieto. 2000. The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2:78-83. [DOI] [PubMed] [Google Scholar]
- 7.Carver, E. A., J. Rulang, Y. Lan, K. F. Oram, and T. Gridley. 2001. The mouse Snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol. Cell. Biol. 21:8184-8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng, C. W., P. E. Wu, J. C. Yu, C. S. Huang, C. T. Yue, C. W. Wu, and C. Y. Shen. 2001. Mechanisms of inactivation of E-cadherin in breast carcinoma: modification of the two-hit hypothesis of tumor suppressor gene. Oncogene 20:3814-3823. [DOI] [PubMed] [Google Scholar]
- 9.Comijn, J., G. Berx, P. Vermassen, K. Verschuren, L. van Grunsven, E. Bruyneel, M. Mareel, D. Huylebroek, and F. van Roy. 2001. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 7:1267-1278. [DOI] [PubMed] [Google Scholar]
- 10.Engel, K., A. Kotlyarov, and M. Gaestel. 1998. Leptomycin B-sensitive nuclear export of MAPKAP kinase 2 is regulated by phosphorylation. EMBO J. 17:3363-3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fornerod, M., M. Ohno, M. Yoshida, and I. W. Mattaj. 1997. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell 90:1051-1060. [DOI] [PubMed] [Google Scholar]
- 12.Fukuda, M., S. Asano, T. Nakamura, M. Adachi, M. Yoshida, M. Yanagida, and E. Nishida. 1997. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature 390:308-310. [DOI] [PubMed] [Google Scholar]
- 13.Gama-Carvalho, M., R. D. Krauss, L. Chiang, J. Valcárcel, M. R. Green, and M. Carmo-Fonseca. 1997. Targeting U2AF65 to sites of active splicing in the nucleus. J. Cell Biol. 197:975-987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guaita, S., I. Puig, C. Franci, M. Garrido, D. Dominguez, E. Batlle, E. Sancho, S. Dedhar, A. Garcia de Herreros, and J. Baulida. 2002. Snail induction of epithelial-to-mesenchymal transition in tumor cells is accompanied by MUC-1 repression and ZEB1 expression. J. Biol. Chem. 277:30209-39216. [DOI] [PubMed] [Google Scholar]
- 15.Hajra, K. M., D. Y. Chen, and E. R. Fearon. 2002. The SLUG zinc finger protein represses E-cadherin in breast. Cancer Res. 62:1613-1618. [PubMed] [Google Scholar]
- 16.Hemavathy, K., S. I. Ashraf, and Y. T. Ip. 2000. Snail/Slug family of repressors: slowly going into the fast lane of development and cancer. Gene 257:1-12. [DOI] [PubMed] [Google Scholar]
- 17.Hemavathy, K., S. C. Guru, J. Harris, J. D. Chen, and Y. T. Ip. 2000. Human Slug is a repressor that localizes to sites of active transcription. Mol. Cell. Biol. 26:5087-5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishida, N., T. Hara, T. Kamura, M. Yoshida, K. Nakayama, and K. I. Nakayama. 2002. Phosphorylation of p27kip1 on serine 10 is required for its binding to CRM1 and nuclear export. J. Biol. Chem. 277:14355-14358. [DOI] [PubMed] [Google Scholar]
- 19.Jiao, W., K. Miyazaki, and Y. Kitajima. 2002. Inverse correlation between E-cadherin and Snail expression in hepatocellular carcinoma cell lines in vitro and in vivo. Br. J. Cancer 86:98-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kudo, N., B. Wolff, T. Sekimoto, E. P. Schreiner, Y. Yoneda, M. Yanagida, S. Horinouchi, and M. Yoshida. 1998. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp. Cell Res. 242:540-547. [DOI] [PubMed] [Google Scholar]
- 21.Lischka, P., O. Rosorius, E. Trommer, and T. Stamminger. 2001. A novel transferable nuclear export signal mediates CRM1-independent nucleocytoplasmic shuttling of the human cytomegalovirus transactivator protein pUL69. EMBO J. 20:7271-7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lochter, A., and M. J. Bissell. 1995. Involvement of extracellular matrix in breast cancer. Semin. Cancer Biol. 6:165-173. [DOI] [PubMed] [Google Scholar]
- 23.Lochter, A., S. Galosy, J. Muschler, N. Freedman, Z. Werb, and M. J. Bissell. 1997. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J. Cell Biol. 139:1861-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manzanares, M., A. Locascio, and M. A. Nieto. 2001. The increasing complexity of the Snail gene superfamily in metazoan evolution. Trends Gen. 17:178-181. [DOI] [PubMed] [Google Scholar]
- 25.Mauhin, V., Y. Lutz, C. Dennefeld, and A. Auberga. 1993. Definition of the DNA-binding site repertoire for the Drosophila transcription factor SNAIL. Nucleic Acids Res. 21:3951-3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Menke, A., C. Philippi, R. Vogelmann, B. Seidel, M. P. Lutz, G. Adler, and D. Wedlich. 2001. Down-regulation of E-cadherin gene expression by collagen type I and type III in pancreatic cancer cell lines. Cancer Res. 61:3508-3517. [PubMed] [Google Scholar]
- 27.Middeler, G., K. Zerf, S. Jenovai, A. Thulig, M. Tschodrich-Rotter, U. Kubitscheck, and R. Peters. 1997. The tumor suppressor p53 is subject to both nuclear import and export, and both are fast, energy-dependent and lectin-inhibited. Oncogene 14:1407-1417. [DOI] [PubMed] [Google Scholar]
- 28.Miravet, S., J. Piedra, F. Miró, E. Itarte, A. García de Herreros, and M. Duñach. 2002. The transcriptional factor Tcf-4 contains different binding sites for β-catenin and plakoglobin. J. Biol. Chem. 277:1884-1891. [DOI] [PubMed] [Google Scholar]
- 29.Nieto, M. A. 2002. The Snail superfamily of Zinc finger transcription factors. Nat. Rev. Mol. Cell. Biol. 3:155-166. [DOI] [PubMed] [Google Scholar]
- 30.Nishi, K., M. Yoshida, D. Fujiwara, M. Nishikawa, S. Horinouchi, and T. Beppu. 1994. Leptomycin B targets a regulatory cascade of CRM1, a fission yeast nuclear protein, involved in control of higher order chromosome structure and gene expression. J. Biol. Chem. 269:6320-6324. [PubMed] [Google Scholar]
- 31.Okamura, H., J. Aramburu, C. García-Rodríguez, J. P. B. Viola, A. Raghavan, M. Tahiliani, X. Zhang, J. Qin, P. G. Hogan, and A. Rao. 2000. Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol. Cell 6:539-550. [DOI] [PubMed] [Google Scholar]
- 32.Perl, A. K., P. Wilgenbus, U. Dahl, H. Semb, and G. Christofori. 1998. A causal role for E-cadherin in the translation from adenoma to carcinoma. Nature 392:190-193. [DOI] [PubMed] [Google Scholar]
- 33.Poser, I., D. Dominguez, A. García de Herreros, A. Varnai, R. Buettner, and A. K. Bosserhoff. 2001. Loss of E-cadherin expression in melanoma cells involves upregulation of the transcriptional factor Snail. J. Biol. Chem. 276:24661-24666. [DOI] [PubMed] [Google Scholar]
- 34.Rosivatz, E., I. Becker, K. Specht, E. Fricke, B. Luber, R. Busche, H. Hofler, and K. F. Becker. 2002. Different expression of the epithelial-mesenchymal transition regulators Snail, SIP1, and Twist in gastric cancer. Am. J. Pathol. 161:1881-1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sefton, B. M. 1995. Phosphoamino acid analysis, p. 13.3.1-13.3.8. In J. E. Coligan, B. M. Dunn, H. L. Ploegh, D. W. Speicher, and P. T. Wingfield (ed.), Current protocols in protein science. John Wiley & Sons, Inc., New York, N.Y.
- 36.Spector, D. L. 2001. Nuclear domains. J. Cell Sci. 114:2891-2893. [DOI] [PubMed] [Google Scholar]
- 37.Spector, D. L., X. D. Fu, and T. Maniatis. 1991. Association between distinct pre-mRNA splicing components and the cell nucleus. EMBO J. 10:3467-3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stommel, J. M., N. D. Marchenko, G. S. Jimenez, U. M. Moll, T. J. Hope, and G. M. Wahl. 1999. A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J. 18:1660-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sussman, J., D. Stokoe, N. Ossina, and E. Shtivelman. 2001. Protein kinase B phosphorylates AHNAK and regulates its subcellular location. J. Cell Biol. 154:1019-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan, C., P. Costello, J. Sanghera, D. Domínguez, J. Baulida, A. García de Herreros, and S. Dedhar. 2001. Inhibition of integrin linked kinase (ILK) suppresses β-catenin-Lef-1/Tcf-dependent transcription and expression of the E-cadherin repressor, snail, in APC −/− human colon carcinoma cells. Oncogene 20:133-140. [DOI] [PubMed] [Google Scholar]
- 41.Ullman, K. S., M. A. Powers, and D. J. Forbes. 1997. Nuclear export receptors: from importin to exportin. Cell 90:967-970. [DOI] [PubMed] [Google Scholar]
- 42.Vleminckx, K., L. Vakaet, M. Mareel, W. Fiers, and F. Van Roy. 1991. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell 77:1605-1613. [DOI] [PubMed] [Google Scholar]
- 43.Wu, C., S. Y. Keightley, C. Leung-Hagesteijn, G. Radeva, M. Coppolino, S. Goicoechea, J. A. McDonald, and S. Dedhar. 1998. Integrin-linked kinase regulates fibronectin matrix assembly, E-cadherin expression, and tumorigenicity. J. Biol. Chem. 273:528-536. [DOI] [PubMed] [Google Scholar]
- 44.Yokoyama, K., N. Kamata, E. Hayashi, T. Hoteiya, N. Ueda, R. Fujimoto, and M. Nagayama. 2001. Reverse correlation of E-cadherin and Snail expression in oral squamous cell carcinoma cells in vitro. Oral Oncol. 37:65-71. [DOI] [PubMed] [Google Scholar]
- 45.Yokoyama, K., N. Kamata, R. Fujimoto, S. Tsutsumi, M. Tomomari, M. Takai, H. Hoshokawa, and M. Nagayama. 2003. Increased invasion and matrix metalloproteinase-2 expression by Snail-induced mesenchymal transition in squamous cell carcinomas. Int. J. Oncol. 22:891-898. [PubMed] [Google Scholar]
- 46.Zhang, Y., and Y. Xiong. 2001. A p53 amino-terminal nuclear export signal inhibited by DNA damage-induced phosphorylation. Science 292:1910-1915. [DOI] [PubMed] [Google Scholar]
- 47.Zhu, J., and F. McKeon. 1999. NF-AT activation requires suppression of CRM1-dependent export by calcineurin. Nature 398:256-260. [DOI] [PubMed] [Google Scholar]