


Abstract
Genomic DNA of Nostoc commune (Cyanobacteria) became covalently modified during decades of desiccation. Amplification of gene loci from desiccated cells required pretreatment of DNA with N-phenacylthiazolium bromide, a reagent that cleaves DNA- and protein-linked advanced glycosylation end-products. DNA from 13 year desiccated cells did not show any higher levels of the commonly studied oxidatively modified DNA damage biomarkers 8-hydroxyguanine, 8-hydroxyadenine and 5-hydroxyuracil, compared to commercially available calf thymus DNA. Different patterns of amplification products were obtained with DNA from desiccated/rehydrating cells and a liquid culture derived from the dried material, using the same set of primers. In contrast, a reproducible fingerprint was obtained, irrespective of time of rehydration of the DNA, using a primer (5′-GWCWATCGCC-3′) based upon a highly iterated palindromic repeat sequence present in the genome. In vitro, the desiccation of cccDNA led to loss of supercoiling, aggregation, loss of resolution during agarose gel electrophoresis and loss of transformation and transfection efficiency. These changes were minimized when DNA was desiccated and stored in the presence of trehalose, a non-reducing disaccharide present in Nostoc colonies. The response of the N.commune genome to desiccation is different from the response of the genomes of cyanobacteria and Deinococcus radiodurans to ionizing radiation.
INTRODUCTION
The concept of dormant or ‘ancient’ cells is a controversial topic because of largely untested assumptions about the stability of cellular components within dry cytoplasm (1). DNA in growing cells is thought to exist in the fully hydrated B-form and to have properties similar to those measured for DNA in solution (2). The limited chemical stability of DNA under these conditions is considered to be the major determinant of cell survival as well as a factor that must set limits for the recovery of DNA fragments from old cells and fossils (2,3). Yet it is unclear whether desiccated nucleic acids are subject to the same types, degree and rates of damage or modification as observed (or predicted) for hydrated molecules in growing cells. Obviously some cells do tolerate desiccation and they must do so because of an ability to protect vital components of their cellular machinery from damage and/or repair them quickly upon rehydration. The latter strategy, for example, is employed by the ionizing radiation-resistant bacterium Deinococcus radiodurans (4).
One potential modification of nucleic acid and protein occurs through Browning reactions (5,6). The modification is initiated by the spontaneous reaction of reducing sugars with the primary amino groups of proteins and nucleic acids (7). The Browning (Maillard) reaction proceeds from reversible Schiff base and Amadori products to a class of irreversibly bound, structurally heterogeneous products referred to as advanced glycosylation end-products (AGEs) (8). Accumulation of AGEs is implicated in many of the pathophysiological alterations associated with normal aging. Such Maillard products occur in plant and animal remains and are a prominent component of ancient DNA extracts (9–11). Other factors that contribute to DNA modification, and ultimately the killing of cells, include metal-catalyzed Haber–Weiss and Fenton reactions (12) and the presence of reactive oxygen species and free radicals (5,13–15). Five-membered hydantoin rings in DNA originate from oxidative decay of six-membered pyrimidines (3,16,17); their presence is thought to be negatively correlated with the PCR amplification of DNA. Of the free radicals, the highly reactive hydroxyl radical (OH) causes damage to DNA and other biological molecules (18,19). This type of DNA damage is also called ‘oxidative damage to DNA’ and is implicated in mutagenesis, carcinogenesis and aging (20). The occurrence of modified bases is problematic because DNA polymerases are unable to copy these damaged residues. The reagent N-phenacylthiazolium bromide (PTB) disrupts AGE crosslinks and its use made it possible to amplify DNA sequences from ancient samples (11).
There are procedural difficulties when attempting to understand the biochemical properties of cellular components in desiccated cells. Aqueous and organic reagents used in cell extraction and preservation techniques disrupt intracellular microenvironments and the physical properties of dried macromolecules. Non-invasive structural techniques such as Fourier transform infrared spectroscopy and differential scanning calorimetry can provide some information on the physical state of cytoplasmic components (21), but not in sufficient detail to assess the physiological relevance of any modifications. For example, supercoiling of DNA has a profound influence on the regulation of gene expression in whole genomes yet is very difficult to study directly even in fully rehydrated cells (22,23).
To understand how fluctuating water content may influence the integrity of nucleic acids (and ultimately gene expression) within cells of Nostoc commune, we designed assays to obtain empirical data on the effects of long-term desiccation on purified supercoiled and linear DNAs in vitro. Nucleic acids were then obtained from cells following different periods of desiccation and rehydration, in the presence and absence of PTB reagent prior to amplification of selected gene loci, and rapid amplification of polymorphic DNA (RAPD)–PCR assays were used to analyze the in vivo dynamics of DNA in the rehydrated cells.
MATERIALS AND METHODS
Biological materials and growth conditions
Desiccated colonies of N.commune of different ages were collected and stored in the dark, until analysis (Table 1). Genotypic analyses indicated these all belonged to ‘form species’ N.commune (24). Most herbarium specimens were obtained in sealed paper envelopes that were unopened since the time of collection. A liquid culture, N.commune strain DRH1, was derived from a desiccated sample and grown as described (25). Human kidney 293H cells were grown and transfected with plasmid DNA as described (26).
Table 1. Treatment of DNA with PTB and ability to amplify gene loci.
| Gene amplification material | Age (years) | Viable | tRNALEU+PTB | introna–PTB | tRNALEU+PTB | intronb–PTB | phr+PTB | –PTB | 23S rDNA | sodF | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WH002 | 64 | Yes | + | + | – | – | + | – | + | – | ++ | – |
| WH0014 | 50 | Yes | ++ | ND | +++ | + | + | + | + | + | +++ | + |
| ALD8122 | 29 | Yes | ++ | + | + | + | + | + | + | + | + | + |
| WH010 | ∼120 | – | – | – | – | – | + | + | + | – | – | – |
| WH012 | 118 | – | – | – | + | – | + | + | + | – | – | – |
| WH001 | 75 | ND | + | + | – | – | + | – | + | – | – | – |
| WH004 | 149 | ND | + | + | – | – | + | + | + | + | + | + |
| WH005 | 139 | ND | – | – | – | – | + | + | + | + | – | – |
| WH006 | 120 | ND | + | – | + | – | + | – | ++ | + | – | – |
| WH011 | 138 | ND | – | – | – | – | + | – | + | – | – | – |
| WH013 | 94 | ND | – | – | + | – | + | – | + | – | – | – |
| WH016 | 133 | ND | – | – | + | – | + | + | + | + | – | – |
| MEL1968 | 30 | ND | ++ | + | + | + | ++ | + | ++ | + | +++ | ++ |
| SPH1998 | 26 | ND | + | + | + | + | + | + | + | + | – | – |
| DRH1 | NA | Yes | ND | +++ | ND | + | ND | + | ND | + | ND | + |
| Escherichia coli strains and plasmids | Comments | Source |
|---|---|---|
| Escherichia coli BL21DE3 (pSpsA) | Ampr, spsA of Synechocystis PCC 6803; sucrose 6-phosphate synthesis | (55) |
| Escherichia coli BL21DE3 (pMP005) | Ampr; iphP of Nostoc sp. UTEX 584; phosphomonoesterase activity | (44) |
| Escherichia coli DH10B (pEXPcmvgtβgal) | Ampr; Gateway (Invitrogen) construction; cytomegalovirus promoter; β-galactosidase activity | Fred Bloom |
| Escherichia coli DH10B Electromax™ | Competent cells | Invitrogen |
| Escherichia coli TOP10™ | Competent cells | Invitrogen |
Drying and rehydration of DNA
Escherichia coli strains BL21DE3 (pSpsA), BL21DE3 (pMP005) and DH10B (pEXPcmvgtBgal) were grown overnight in Luria–Bertani (LB) medium containing 200 µg/ml ampicillin. Plasmid DNA was purified using Wizard preparation kits (catalog no. PR-A7510; Promega). Solutions of plasmid and linear DNAs were prepared in 1× TE buffer (1 mM EDTA, 10 mM Tris–HCl, pH 7.5, approximately 1:10, powder:buffer) in the presence or absence of filter-sterilized 100 mM trehalose (d[+] Trehalose T-1901; Sigma). Aliquots of the DNA solutions were used to determine the concentration of DNA, for agarose gel electrophoresis, for field inversion gel electrophoresis and to transform Electromax competent E.coli DH10B (Invitrogen, CA) as described in the figure legends. Aliquots were dried in 1.5 ml microcentrifuge tubes under a stream of sterile air until no liquid remained in the tube (typically overnight). These tubes were then transferred to a glove box and all subsequent manipulations were performed under an atmosphere of nitrogen, under positive pressure. The open tubes were placed on the surface of phosphorus pentoxide powder (P2O5) in 50 ml containers that were later sealed. Storage was continued at room temperature in the light, under an incident photon flux density of 50 µmol photons/m2/s1, or in the dark, for periods from 72 h to 56 days prior to rehydration. In the present experiments the mass ratio of plasmid pMP005 (6.29 kb) DNA to trehalose following drying (50:1711.5 µg) was therefore 1:34.2 (0.0079 × 10–3:0.005 mmol) After a period of storage, replicate tubes were removed and desiccated DNA was rehydrated with sterile distilled water for different periods up to 24 h, in the light, prior to further manipulations.
Genomic DNA purification
Genomic DNA of N.commune DRH1 was purified as described previously (25). Desiccated materials were lyophilized and ground to a powder under liquid nitrogen. The powder was rehydrated with 1× TE. Extraction buffer (1.4 M NaCl, 20 mM EDTA, 1.5% w/v hexadecyl trimethyl ammonium bromide, 1% v/v β-mercaptoethanol, 100 mM Tris–HCl, pH 7.5, approximately 4 times the volume of 1× TE added to the powder) was added and the mixture was incubated at 65°C, for 30 min. The slurry was homogenized, frozen under liquid nitrogen and thawed at 65°C; the freeze–thaw cycle was repeated six times. The mixture was extracted with chloroform:isoamyl alcohol (24:1) and DNA was recovered from the aqueous phase using isopropanol then stored in 1× TE buffer. In some cases DNA was extracted exhaustively (up to six times) with phenol prior to chloroform:isoamyl alcohol extraction and prior to treatment with PTB (see below).
In some cases desiccated colonies were ground to a powder under liquid nitrogen and resuspended in 50 mM EDTA. An equal volume of 1.5% (w/v) molten agarose (type 1-A, low EEO, catalog no. A-0169; Sigma) was added to embed the cell material. The blocks were incubated overnight at 50°C with grade 1 lysozyme (final concentration 2 mg/ml) (Sigma) in 50 mM EDTA and then overnight at 50°C in buffer containing proteinase K (final concentration 2 mg/ml) (catalog no. P6556; Sigma), 0.1 M EDTA, 1% (w/v) N-lauroylsarcosine, 0.05% (w/v) SDS and 10 mM Tris–HCl (pH 8.0). Finally the blocks were equilibrated in 10 mM sodium phosphate buffer, pH 7.0, prior to the addition of PTB at a final concentration of 10 mM. PTB was synthesized as described (8,11). The blocks were incubated in PTB for 10 h at 27°C, and then equilibrated in TAE buffer (2 mM EDTA, 40 mM Tris–acetate). The blocks were transferred to the wells of a 1% (w/v) agarose (catalog no. A-0169; Sigma) gel and the DNA was resolved at 75 mA for 1 h, excised from the gel and purified further using a QIAEX II gel extraction kit (Qiagen, CA).
Highly polymerized calf thymus DNA (ctDNA, type I) was obtained from Sigma Chemical Co. Linear DNA fragments in the size range 75–12 216 bp were obtained from Invitrogen (CA).
Gel electrophoresis
DNA samples were resolved in 1% (w/v) agarose gels (catalog no. A-0169; Sigma) in TAE buffer (1 mM EDTA, 40 mM Tris–acetate). Field inversion gel electrophoresis was in 1.2% (w/v) agarose in TBE buffer (0.44 M Tris–HCl, 0.44 M boric acid, 10 mM EDTA, pH 8.3) at 8°C. Samples were embedded in agarose before transfer to wells of the gel. Separations were performed over 48 h at 150 V, with pulse times that increased from 5 to 120 s over 48 h, using a Gene Navigator system (Pharmacia Biotech). PFG DNA markers (catalog no. 340; New England Biolabs) were used as size standards.
Transformation of E.coli
One microliter (approximately 70–90 ng) of a solution of DNA (diluted where appropriate) in 1× TE buffer or 1× TE buffer + 100 mM trehalose was used for transformation. The 1 µl aliquot was mixed with 20 µl of a 1:1 dilution of Electromax DH10B Competent Cells (catalog no. 18290-015; Invitrogen) and placed in a 0.15 cm LTI Microelectroporation Chamber (catalog no. 11608-31). Electroporations were carried out at 400 V, 330 μF capacitance and 4 Ω using a LTI Cell-Electroporation System (1609-013). Electroporated cells were incubated in SOC (2% w/v bacto tryptone, 0.5% w/v yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, pH 7.5) for 1 h before plating in the presence of 100 µg/ml ampicillin. Plates were incubated overnight, at 37°C, before colonies were counted.
PCR amplification
The loci selected for amplification through PCR assay included the tRNALEU(UAA) group I intron (24), rrn (23S rDNA) (24), phr and sodF (25) of N.commune DRH1. Assays were optimized with each set of primers (Table 2), in multiple trials, prior to experiments. Assays were performed in 50 µl reactions, in pH 9.0 (sodF only) buffer (supplied by the manufacturer; Promega Biotech) or in pH 8.5 (phr, 23S rDNA, group I introns) buffer [60 mM Tris–HCl, 15 mM (NH4)2SO4]. Reactions contained 12.5 nmol of each dNTP (each dNTP at 250 µM), 1.5 (sodF only) or 3.5 mM Mg2+ (phr, 23S rDNA, group I introns) and 2.5 U of Taq polymerase (Promega). Temperature was controlled with a Biometra thermocycler model T-3. All assays began with a denaturation temperature of 95°C for 2 min and ended with an elongation time of 10 min; in each cycle denaturation occurred at 95°C for 1 min and elongation was at 72°C for 90 s. For amplification of tRNALEU group I introns the annealing temperature (60 s) was 66°C in the first cycle and dropped 0.2°C per cycle thereafter for an additional 39 cycles. For amplification of phr and 23S rDNA the annealing temperature was 60°C (60 s) in the first cycle, dropped 0.5°C per cycle for the next 19 cycles (to 50°C) and was kept constant at 50°C for the remaining 19 cycles. For amplification of sodF the annealing temperature was 60°C (90 s) in the first cycle, dropped 1.5°C per cycle for the first 10 cycles (to 45°C) and was kept constant at 45°C for the remaining 30 cycles. Three independent amplifications were performed per sample.
Table 2. Primer sequences used in PCR assays.
| Primer sequence | Forward/reverse | Target | Origin |
|---|---|---|---|
| 5′-TGTGGCGGAATGGTAGACGCTAC-3′ | fa | Bases 6–28 of tRNALEU(UAA) | Anabaena sp. PCC 7120 |
| 5′-GACTTGAACCCACACGAC-3′ | rb | Bases 72–56 of tRNALEU(UAA) | Anabaena sp. PCC 7120 |
| 5′-GGGTGGAGGGACTTGA-3′ | rc | Bases 82–67 of tRNALEU(UAA) | Anabaena sp. PCC 7120 |
| 5′-ACCCTATATACTACCTGCTG-3′ | f | 23S rRNA | Nostoc commune DRH1 |
| 5′-GGTCTTCGCTGATTCACATG-3′ | r | 23S rRNA | Nostoc commune DRH1 |
| 5′-ACATAACCACGCCCGAATGTG-3′ | f | phr | Nostoc commune DRH1 |
| 5′-AAATCACAATGACCGTAAAG-3′ | r | phr | Nostoc commune DRH1 |
| 5′-GAGTATCACTATGGCAAGCA-3′ | f | sodF | Nostoc commune DRH1 |
| 5′-CTAAAGTCAATGTAGTAG-3′ | r | sodF | Nostoc commune DRH1 |
| 5′-GGTGCGGGAA-3′ | f, r | Random 1 | Pharmacia Biotech |
| 5′-GTTTCGCTCC-3′ | f, r | Random 2 | Pharmacia Biotech |
| 5′-GTAGACCCGT-3′ | f, r | Random 3 | Pharmacia Biotech |
| 5′-AAGAGCCCGT-3′ | f, r | Random 4 | Pharmacia Biotech |
| 5′-AACGCGCAAC-3′ | f, r | Random 5 | Pharmacia Biotech |
| 5′-CCCGTCAGCA-3′ | f, r | Random 6 | Pharmacia Biotech |
| 5′-GGCGATCGCC-3′ | f, r | HIP1 | Synechocystis sp. PCC 6803 |
| 5′-AGAGATCGCC-3′ | f, r | HIP1a, random | This study |
| 5′-WGWGATCGCC-3′ | f, r | HIP1b, random | This study |
| 5′-GWCWATCGCC-3′ | f, r | HIP1c, random | This study |
| 5′-GGWGATCGCC-3′ | f, r | HIP1d, random | This study |
RAPD–PCR
Desiccated colonies of N.commune ENG1996 were rehydrated with sterile distilled water for different lengths of time and then ground under liquid nitrogen. Aliquots (1 mm3) were transferred to PCR tubes, mixed with 60 µl of distilled water and heated at 95°C for 1 h. After centrifugation the supernatant fractions were removed, lyophilized and resuspended in 12 µl of distilled water with or without the addition of PTB (12 mM) for 1.5 h. Two types of RAPD–PCR assay were performed. In the first assay, one of six random decameric primers (Pharmacia) was used (Table 2). In the second, the primer (Table 2) was based upon different configurations of the highly iterated palindromic repeat sequence 5′-GCGATCGC-3′ (HIP1). Conditions of PCR assay were as described (above) except that the annealing temperature was 36°C (1 min) and the extension was at 72°C (2 min) for 40 cycles. Amplifications were performed on genomic DNA from N.commune DRH1 for control purposes. PCR reactions were resolved through agarose gel electrophoresis. When necessary bands of interest were purified and subcloned in pCR2.1-TOPO (Invitrogen, CA) and used to transform E.coli TOP10 (Invitrogen, CA) in preparation for DNA sequence analysis.
Measurement of oxidative DNA damage
DNA was extracted by the NaCl method (27–29). Samples were then hydrolyzed and derivatized for gas chromatography/isotope dilution mass spectrometry (GC/IDMS) analysis as previously described (30). Statistical values were derived using the ANOVA single factor test. Briefly, samples were analyzed by GC/IDMS with selected-ion monitoring (SIM) using a gas chromatograph (model 6890 series)–mass spectrometer (model 5973N) system (Agilent Technologies, Rockville, MD) equipped with an automatic sampler. The column was a fused silica capillary column (12.5 m × 0.2 mm i.d.) coated with cross-linked 5% phenylmethylsilicone gum phase (film thickness 0.33 µm) (Agilent Technologies). Ultra-high purity helium was used as the carrier gas. The injection port and the GC/MS interface were kept at 250 and 280°C, respectively. The ion source temperature was 230°C. The column head pressure was 65 kPa. An aliquot of each sample (4 µl) was injected into the injection port of the gas chromatograph using the split mode of injection with a split ratio of 10:1. SIM was performed in the electron ionization mode at 70 eV using the characteristic ions of the trimethylsilyl derivatives of modified bases, guanine and their stable isotope-labeled analogs (31,32). The preparation of aqueous solutions of ctDNA and their exposure to ionizing radiation in a 60Co γ-radiation source at a dose of 5 Gy was performed as described (33). Subsequently, unirradiated, irradiated and treated DNA solutions were dialyzed against water for 18 h. Water outside the dialysis tubes was changed three times during the course of dialysis.
RESULTS
Stability of desiccated DNA in vitro
Native pMP005 and pSpsA DNAs lost supercoiling in a light- and time-dependent manner when they were desiccated and stored over P2O5. The effect was apparent in less than 72 h if the DNA was dried and stored in the absence of trehalose; the DNA formed insoluble aggregates upon rehydration and resolved poorly during conventional agarose gel electrophoresis (Fig. 1b, wells of lanes 2 and 4, and Fig. 1d, well lane 4). Resolution improved considerably if the same desiccated DNA was allowed to rehydrate for 24 h at 4°C prior to electrophoresis (Fig. 1c and e). Trehalose enhanced the short-term stability of desiccated plasmid DNA, it prevented the formation of insoluble aggregates upon rehydration and it diminished the deleterious effects of storage in the light, but it could not do so during longer periods of storage (up to 56 days; Fig. 2). Field inversion gel electrophoresis provided a sensitive assay for the nicking of desiccated supercoiled plasmid DNA, an effect that was apparent after even short-term desiccation despite the presence of trehalose (Fig. 3).
Figure 1.

Conventional gel electrophoresis of pMP005 DNA prepared in TE + 100 mM trehalose (+; lane 2 of a) or TE only (–; lane 3 of a) at 1.0 µg/µl (total volume 50 µl) and desiccated in the light (L) or dark (D) over P2O5, for 72 h (b and c) or 7 days (d and e), prior to rehydration and electrophoresis. Samples were rehydrated with 50 µl of sterile distilled water for ∼10 min (b and d) or 24 h (c and e) prior to loading on gels. Two microliters from each sample were resolved per lane. M, markers (12.2–1.6 kb); oc, open circular pMP005; ccc, covalently closed circular pMP005.
Figure 2.

Conventional gel electrophoresis of samples of pSpsA desiccated for 15 or 56 days prior to rehydration and analysis as described in the legend to Figure 1.
Figure 3.

Field inversion gel electrophoresis of pSpsA plasmid DNA. Samples analyzed prior to desiccation (lanes 2 and 3) or after 7 days desiccation (lanes 4 and 5), in the absence (lanes 2 and 4) or presence (lanes 3 and 5) of trehalose (see legend to Fig. 1). Markers are from 1.018 Mb to 48.5 kb. White arrows indicate ethidium bromide staining material that enters the gel poorly. Black arrows indicate pSpsA with different supercoiling.
There were no discernible effects of either short- or long-term desiccation on the complexity, solubility or resolution of mixtures of linear DNA fragments (∼200–12 500 bp). This was true whether the fragments were desiccated in the light or the dark and irrespective of the presence or absence of trehalose or the time of rehydration prior to electrophoresis.
The data on plasmid stability during desiccation were consistent with further results from transformation assays performed with aliquots of the same samples of DNA (Fig. 4). pSpsA DNA stored in the light in the absence of trehalose gave low transformation efficiencies. Highest transformation efficiencies were achieved with plasmid DNA desiccated and stored in the presence of trehalose. The differences between the transformation efficiencies obtained with light- and dark-stored samples that contained trehalose were negligible (Fig. 4). Also, with human 293H cells, there was a significant increase in Lipofectimine-mediated transfection efficiency (from ∼50 to 75%) if pEXPcmvβgal DNA was mixed with trehalose (100 mM final concentration) immediately after plasmid purification and prior to storage at 4°C (data not shown).
Figure 4.

Electrocompetent E.coli DH10B were transformed with samples of pSpsA DNAs that were stored desiccated in the presence (in light, partial fill; in dark, black fill) or absence of trehalose (in light, open box; in dark, grey shading).
In multiple transformation assays, where samples of pMP005 were exposed to the same times and conditions of desiccation as used for pSpsA, all BL21DE3 (pMP005) transformants had an IphP+ (blue, wild-type) phenotype (from 25 independent trials with >107 transformants scored; data not shown).
DNA modification in desiccated cells
Solutions of genomic DNA from desiccated colonies typically had low A260:A230 and A260:A280 absorbance ratios and retained a dark brown color during exhaustive phenol: chloroform:isoamyl alcohol extraction. Absorbance in the 300–450 nm range was attributed to this pigmentation. After treatment with PTB, and one further round of solvent extraction, the DNA solution lost most of this pigmentation. The effects of PTB treatment on the capacity to amplify defined gene loci was studied to obtain further evidence of DNA modification in vivo. Four selected gene loci [sodF, phr, rrn (23S rRNA) and tRNALEU(UAA) intron] were amplified from colonies of N.commune desiccated for periods of up to 149 years. Products of the expected size were obtained from some of the different samples and their identities were confirmed through DNA sequence analysis. For the majority of the samples amplification of phr and rrn was possible only after pretreatment of the samples with PTB (Fig. 5A). Amplification of tRNALEU(UAA) group I intron and sodF required PTB in one and two samples, respectively. With sample N.commune WH002, amplification of sodF, phr and rrn was possible only after treatment of the DNA with PTB. In contrast, all loci were amplified efficiently without need of PTB using DNA from a liquid culture of N.commune derived from field material (Fig. 5B).
Figure 5.

PCR amplification of gene loci. Numbers in bp; p, primer dimers. (A) DNA from desiccated N.commune WH002 (phr and rrn) or N.commune WH0014 (sodF) with (+) or without (–) prior treatment with PTB. (B) DNA from liquid culture of N.commune with no PTB treatment. Lanes 2, 4 and 6 are control assays (no target DNA in reaction mixture).
Oxidative DNA damage in desiccated cells
Three N.commune samples (GRVE2002, BBC1990 and ENG1996) were analyzed for oxidative DNA damage and compared to commercially available ctDNA using GC/IDMS technology. Biomarkers detected were 8-hydroxyguanine (8-OH-Gua), 8-hydroxyadenine (8-OH-Ade) and 5-hydroxyuracil (5-OH-Ura). No significant level of damage was detected among the samples when compared to control DNA, i.e. unirradiated ctDNA (Table 3). For 8-OH-Gua, the three Nostoc samples were calculated to have between 120 and 154 lesions/106 DNA bases. The control and irradiated ctDNA had levels of 130 and 242 lesions/106 DNA bases, respectively. For 8-OH-Ade, the three Nostoc samples had between 13 and 14 lesions/106 DNA bases, with the ctDNA control and irradiated ctDNA at levels of 13 and 36 lesions/106 DNA bases, respectively. For 5-OH-Ura, the three Nostoc samples had between 1 and 2 lesions/106 DNA bases, with the ctDNA control and irradiated ctDNA at levels of 4 and 12 lesions/106 DNA bases, respectively.
Table 3. Levels of 8-OH-Gua, 8-OH-Ade and 5-OH-Ura in DNA from desiccated N.commune as measured by GC/IDMS.
| Source of DNA | Age (years) | Viable | Modified bases/106 DNA bases | ||
|---|---|---|---|---|---|
| 8-OH-Gua | 8-OH-Ade | 5-OH-Ura | |||
| GRVE2002 | 1 | + | 131.1 ± 10.7a | 13.2 ± 0.9b | 1.3 ± 0.3c |
| BBC1990 | 13 | + | 120.6 ± 28.4a | 12.3 ± 0.7b | 1.5 ± 0.1c |
| ENG1996 | 7 | + | 153.9 ± 12.0a | 14.1 ± 2.2b | 1.8 ± 0.4c |
| ctDNA | NA | NA | 130.6 ± 15.0a | 13.6 ± 2.2b | 3.8 ± 0.8c |
| ctDNA (5 Gy) | NA | NA | 242.8 ± 24.4 | 36.1 ± 1.0 | 12.2 ± 0.3 |
SD error values are based on triplicate measurement analysis (n = 3). NA, not applicable, liquid culture.
a8-OH-Gua column. The values of the ctDNA and Nostoc samples were statistically different from the value of the ctDNA 5 Gy sample (P < 0.05).
b8-OH-Ade column. The values of the ctDNA and Nostoc samples were statistically different from the value of the ctDNA 5 Gy sample (P < 0.001).
c5-OH-Ura column. The values of the ctDNA and Nostoc samples were statistically different from the value of the ctDNA 5 Gy sample (P < 0.001).
Stability of desiccated DNA in vivo
Any modifications or changes to the structure of genomic DNA in vivo must be rectified upon rehydration if cells are to remain viable. Genomic DNA may thus undergo dynamic changes during the rehydration process. To test this hypothesis, colonies of N.commune ENG1996 were rehydrated (with distilled water) for different time periods before isolation of the DNA for RAPD–PCR assay. In repeat trials with several of the primers the pattern of PCR products differed according to the time of hydration of the target cells (see for example Fig. 6A, primers 1–3). In contrast, other primers gave a consistent pattern with the same samples of DNA (primers 4 and 6). The capacity to generate products with a given sample was strictly primer dependent (compare the amplification pattern from the 24 h rehydrated sample when using primers 1 and 4). For primers 1–4 the greatest variability in the pattern of products occurred with the 3 h and/or 24 h rehydration time point samples. With the exception of assays using primers 4 and 5 the general trend was decreasing numbers and amounts of products with increasing time of rehydration of the cells. When PTB-treated as opposed to non-PTB-treated DNA was used in assays the amplification products tended to be (i) in greater quantity and of higher quality and (ii) formed more consistent patterns for the different rehydrated samples (compare for example Fig. 6B with A, panel 3, which is representative of the improved consistency and resolution achieved with PTB). All primers showed improved resolution with PTB; only the results for primer 3 are shown as with non-PTB-treated DNA the greatest variation between samples was noted with this primer (Fig. 6A). The most effective primers with PTB-treated samples were those that provided the most consistent patterns with non-treated samples (primers 4 and 6). DNA from a liquid culture of N.commune derived from the desiccated material generated quite different genomic fingerprints using the same six primers as tested on the desiccated/rehydrated samples (Fig. 6C).
Figure 6.

RAPD–PCR assay. (A) DNA from N.commune ENG1996 colonies desiccated for 4 years at time of assay or rehydrated (lanes from left to right) for 5 or 30 min or 1, 3 or 24 h; redundant primers 1–6 (see Table 1). (B) As (A) but DNA treated with PTB; primer 3. (C) DNA from liquid culture of N.commune derived from desiccated material with the same primers (lanes 1–6, primers 1–6, respectively). Lane M, molecular size markers (12.2 kb to 520 bp).
HIP sequence amplification
Results of RAPD–PCR assays performed with redundant HIP primers were more reproducible and consistent in multiple trials than those obtained with the set of non-redundant decameric primers. HIP primer 4 (5′-GWCWATCGCC-3′) generated the most bands with greatest size distribution, and was used for further analysis. A very consistent pattern of products was obtained with the same series of rehydrated samples of N.commune ENG1996 as described above (Fig. 7). The largest fragments amplified in multiple assays were no greater than ∼1200 bp (compare with DNA from the liquid culture control, Fig. 7, lane C). In repeat trials one band showed marked enrichment from the 24 h rehydration sample; the DNA was isolated, cloned and sequenced. The end of the fragment amplified with the primer (5′-GWCWATCGCC-3′) was, in each case, 5′-GACAATCGCC-3′. The fragment contained an ORF of 96 codons stretching from the 5′ end of the fragment to an internal stop codon. The partial ORF shared 65% amino acid sequence identity with the C-terminus of an AtoS-like protein (polyamine biosynthesis and osmoregulation) in Nostoc punctiforme ATCC 29133 (NPU2424 gi), as well as a number of other putative sensory transduction histidine kinases.
Figure 7.
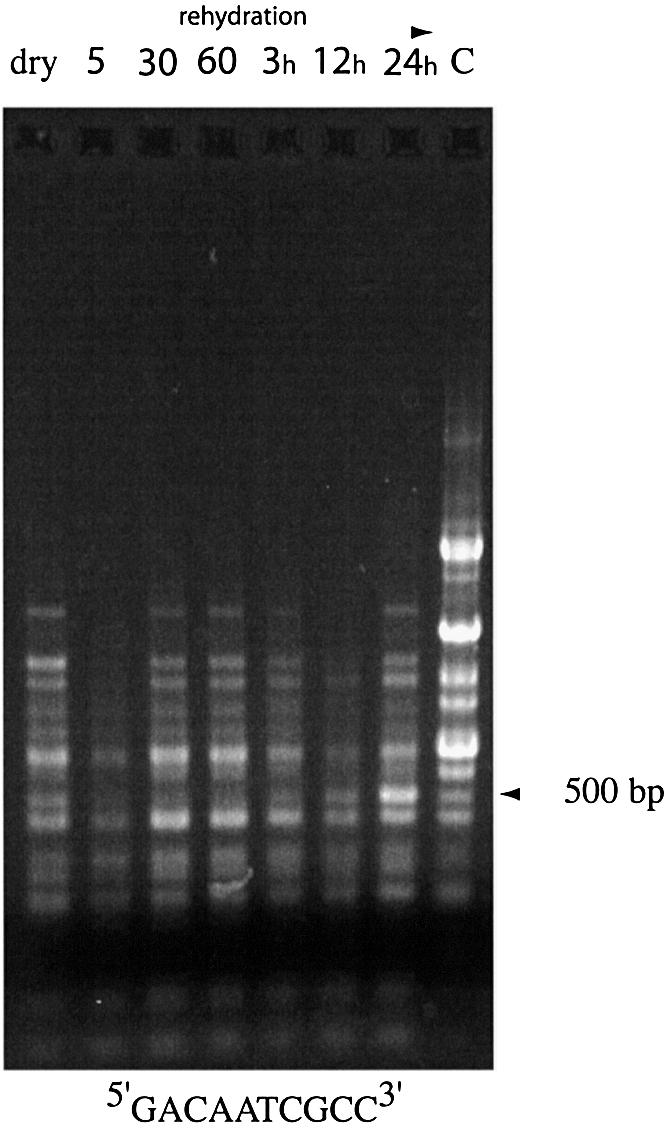
See the legend to Figure 6A but with time points at 0, 5 or 30 min or 1, 3, 12 or 24 h; assays performed with HIP-1 derivative primer 5′-GACAATCGCC-3′. C, control RAPD–PCR using purified N.commune DRH1 genomic DNA. Arrow indicates abundant product at 24 h.
Viability of desiccated Nostoc colonies
In cases where sufficient material was available dried colonies were rehydrated on moist filter paper followed by incubation at 25°C under subdued illumination. Colonies of N.commune TOP1993 (9 years dry), ENG1996 (6 years dry), WH002 (63 years dry) and WH0014 (55 years dry) all contained material that grew after rehydration. However, WH010 and WH012 (120 and 118 years old, respectively) failed to grow and failed to generate two of the markers despite treatment with PTB (Table 1).
DISCUSSION
Despite the fundamental role of water in the structure and properties of DNA, much of our understanding of the form and organization of the bacterial nucleoid is only theoretical in nature (34). In vivo, the hydrated DNA double helices of plasmids and the bacterial chromosome are untwisted or underwound in a left-handed fashion, causing the helices to be negatively supercoiled. This supercoiling is thought to play a crucial role in the regulation of replication, transcription and gene expression. There is virtually nothing known about the structure and properties of genomes in desiccated cells with water contents of around 0.03 g H2O/g cell solids (5,35). Marked changes in appearance of the nucleoid accompanied rewetting of desiccated N.commune and some of these are consistent with a rehydration of condensed material (36). These included increases in the density of staining, polydispersion, microstructure and numbers of peripheral ribosomes and the appearance of a large central polyphosphate body within 24 h (see fig. 4 in 36).
A feature of N.commune desiccated for short (weeks) or long (centuries) periods was the brown pigmentation of purified DNA that was recalcitrant to solvent extraction. Our data confirm that the DNA was covalently modified, presumably through Maillard reactions (11). The differences in RAPD–PCR fingerprints observed during rehydration of DNA may reflect changes in accessibility of priming sites due to interaction with proteins or carbohydrates and/or conformational changes determined by the water content and rehydration status of the cytosol. The changes may also reflect repair processes that must accompany rehydration and recovery of the cells since there was no evidence of modification in DNA from a liquid culture that was derived from desiccated material.
Nostoc commune colonizes habitats that are rarely dry for longer than 6 months, however, in those cases where sufficient material was available, it was possible to demonstrate viability and recovery of samples desiccated for at least 60 years but not in samples desiccated for >100 years. Clearly, DNA modification is a natural consequence of desiccation in situ and one that the cells can tolerate for considerable periods, but not indefinitely. To do so, the cells presumably require one or more enzymes to remove such modification at the time of rehydration; this could account in part for some of the differences observed in genome fingerprints.
Data from experiments with supercoiled DNA in vitro suggest that the potential for disruption of genome structure during changes in water content is significant. The significance of the role of water in the higher order structure of DNA is emphasized since no such degradation or aggregation was detected when linear DNAs were subjected to identical treatments using the same reagents. Robinson et al. (37) suggest that HIP1 elements may promote recombination repair following desiccation and/or exposure to other factors that cause DNA damage. The fact that consistent and reproducible genome fingerprints were obtained with the redundant HIP primer (as opposed to the other primers) may suggest that HIP priming sites are always readily accessible despite the degree of hydration or condensation of the DNA. This may promote reorganization of the genome after long-term desiccation. It has also been observed that HIP sequences participate in gene amplification in response to environmental cues. For example, the number of copies of the smt (metal toxicity) locus in Synechocystis PCC 6301 increased in response to cadmium addition to the medium (38).
Oxidative DNA damage occurs in cells as a consequence of normal aerobic metabolism and by generation of oxygen-derived free radicals from exposure to ionizing radiation and other DNA-damaging agents (16). This type of DNA damage appears to be one of the main causes of aging and gene-related diseases (39,40–43). Evaluation of the levels of the three biomarkers of oxidative DNA damage used in this study, 8-OH-Gua, 8-OH-Ade and 5-OH-Ura, showed that free radical damage was not a significant event during the prolonged desiccation of dormant N.commune cells. The fact that the Nostoc DNA lesion levels were similar to that obtained with the ctDNA control indicates that free radical DNA damage was not a significant event during desiccation. These results indicate that when dry, the DNA is protected from free radical damage. In this regard it can be noted that a highly active and abundant Fe-SOD was identified in cells of N.commune that were stored desiccated for 13 years (25). It should be noted that while the chemical composition of the brown pigmentation on the isolated DNA is still not clear, it appeared to have hindered the obtaining of reliable mass spectrometry data for an additional seven biomarkers of oxidative DNA damage that were screened (data not shown).
While desiccation of plasmid DNA had marked effects on plasmid structure, stability, solubility and transformation efficiency, there was no evidence following numerous trials with pMP005 that any of these changes become manifest as mutations following rehydration of the plasmid DNA and transformation of E.coli. Plasmid pMP005 carries the iphP gene of Nostoc sp. UTEX 584. The phosphatase is expressed constitutively in E.coli from its own promoter and encodes a triple specificity Ser/Thr/Tyr protein phosphatase (44). IphP (indole phosphate hydrolase) is secreted by E.coli and causes a blue colony phenotype if cells are plated in the presence of 5-bromo-4-chloro-3-indolylphosphate (BCIP) (45). Previous work demonstrated that loss of enzyme activity through deletions (44,45) or point mutations (unpublished results) within the coding or control regions of iphP leads to a white colony phenotype in the presence of BCIP.
Trehalose is hypothesized to replace water molecules that are lost from biological systems following desiccation (46). Although not of physiological relevance, it may have been possible to demonstrate greater protection of plasmid DNA in this study by using ultrapure trehalose, free of trace metals. In the present experiments the mass ratio of plasmid pMP005 (6.29 kb) DNA:trehalose following drying (50:1711.5 µg) was 1:34.2 (0.0079 × 10–3:0.005 mmol). However, the relevant calculation is the ratio of base pairs to trehalose molecules since the protective effect of the disaccharide is thought to be the replacement of water molecules [bp:water molecules = 1:10 in hydrated B-DNA (5)] during drying. Assuming the molecular weight of one base pair as 6.6 × 102 Da, 50 µg of pMP005 represents 4.6 × 1016 bp and these were in contact with 3 × 1018 molecules of trehalose (1711 µg, mol. wt 342.3). The ratio of base pairs to trehalose molecules used was therefore 1:100. In view of the other structural waters lost from the DNA upon drying the ratio used can be considered to be of physiological relevance. Several observations can be made to assess the potential protective effect of trehalose in vivo. Calculations indicate that an average cell of E.coli B/r contains 3.1% DNA, i.e. 31 mg DNA/g dry cells (35), and concentrations of trehalose in some anhydrobiotic organisms have been reported to be as high as 20% of the dry weight, i.e. 200 mg trehalose/g dry cells (5). Thus the possibility that desiccated cells may have a DNA:trehalose mass ratio of 31:200 (1:6.5) is not unreasonable. In fact the levels of trehalose measured in N.commune colonies were around 1 mg/g dry weight of colony (47). Because some 95% of the dry weight of Nostoc colonies is a complex extracellular polysaccharide (47), the level of trehalose per cell is calculated as ∼1 mg/50 mg/dry cell material (20 mg/g dried cells), with an estimated DNA:trehalose mass ratio of around 31:20. If one assumes a genome complexity of 9 × 106 base pairs (http://www.jgi.doe.gov/JGI_microbial/html/nostoc/nostoc_homepage.html), then 31 mg of Nostoc genomic DNA is equivalent to 0.028 × 1021 bp. As 20 mg of trehalose is equivalent to 0.00035 × 1023 molecules, the ratio of base pairs to trehalose molecules is calculated as 1:1. Although this estimated ratio is lower than that used experimentally, the protective effect of trehalose may be enhanced with condensed chromosomal DNA in vivo, despite the presence of Fenton-active metals, due to ‘depletion’ caused by crowding in the cell compartment and increased protein–DNA contacts upon removal of water (34).
Desiccated colonies of N.commune recover their capacities for respiration, photosynthesis and nitrogen fixation in a step-wise, orderly fashion following rehydration (48). The onset of respiration and some processes such as lipid biosynthesis appears to be instantaneous (49), while the recovery of the capacity for nitrogen fixation may require days of rehydration (48). Physical uptake of water is mediated by copious amounts of an extracellular glycan biopolymer with unusual rheological properties (50), and complex ultrastructural changes were observed in the glycan following rehydration (51).
In earlier studies there was no evidence of extensive degradation of DNA in cells of N.commune following decades of desiccation (52), and the same cells showed striking preservation of cellular structure (51). Some cells remain viable long after the time that solution theory predicts their DNA to be fully depurinated, and there are well-documented reports of DNA being extracted from ancient sources (1). Desiccation and rehydration thus effect both physical and chemical changes within genomic DNA, and these changes accompany marked fluxes in pools of mRNA (25,53), as well as possible amplification of DNA sequences that may be involved in osmoregulation.
Long-term desiccation of N.commune for up to 60 years leads to modification of DNA, without degradation or oxidative damage or loss of viability of cells, and complex structural changes (sequence-dependent) accompany rehydration, including the removal of DNA modification. This is fundamentally different to the response of D.radiodurans, where desiccation leads to massive damage and degradation of the genome; this damage is repaired rapidly and faithfully during rehydration and recovery of the cells, apparently through the same pathway that repairs damage by 60Co γ-radiation (4). The response of the N.commune genome to water stress is therefore distinct, because N.commune, and other cyanobacteria, are equally resistant to 60Co γ-radiation as D.radiodurans (6,54). Whether the capacity for radiation tolerance is a consequence of the evolution of a capacity for DNA repair following desiccation is, therefore, questionable.
Acknowledgments
ACKNOWLEDGEMENTS
We thank two anonymous reviewers for helpful comments. This work was supported by a Multi University Research Initiative (MURI) infrastructure award (N00014-01-1-0852) and contracts through the Defense Advanced Research Programs Agency (DARPA) (N00173-98-1-G005-P00004 and N00173-02-1-G016).
REFERENCES
- 1.Potts M. (2001) Desiccation tolerance: a simple process? Trends Microbiol., 9, 553–558. [DOI] [PubMed] [Google Scholar]
- 2.Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature, 362, 709–715. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl T. (1997) Facts and artifacts of ancient DNA. Cell, 90, 1–3. [DOI] [PubMed] [Google Scholar]
- 4.Battista J.R,, Earl,A.M. and Park,M.J. (1999) Why is Deinococcus radiodurans so resistant to ionizing radiation? Trends Microbiol., 7, 362–365. [DOI] [PubMed] [Google Scholar]
- 5.Potts M. (1994) Desiccation tolerance of prokaryotes. Microbiol. Rev., 58, 755–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Potts M. (1999) Mechanisms of desiccation tolerance in cyanobacteria. Eur. J. Phycol., 34, 319–328. [Google Scholar]
- 7.Papoulis A., Al-Abed,Y. and Bucala,R. (1995) Identification of N2-(1-carboxyethyl)guanine (CEG) as a guanine advanced glycosylation end product. Biochemistry, 34, 648–655. [DOI] [PubMed] [Google Scholar]
- 8.Vasan S., Zhang,X., Zhang,X.N., Kapurniotu,A., Bernhagen,J., Teichberg,S., Basgen,J., Wagle,D., Shih,D., Terlecky,I., Bucala,R., Cerami,A., Egan,J. and Ulrich,P. (1996) An agent cleaving glucose-derived protein crosslinks in vitro and in vivo. Nature, 382, 275–278. [DOI] [PubMed] [Google Scholar]
- 9.Evershed R.P., Bland,H.A., vanBergen,P.F., Carter,J.F., Horton,M.C. and Rowley Conwy,P.A. (1997) Volatile compounds in archaeological plant remains and the Maillard reaction during decay of organic matter. Science, 278, 432–433. [Google Scholar]
- 10.VanBergen P.F., Bland,H.A., Horton,M.C. and Evershed,R.P. (1997) Chemical and morphological changes in archaeological seeds and fruits during preservation by desiccation. Geochim. Cosmochim. Acta, 61, 1919–1930. [Google Scholar]
- 11.Poinar H.N., Hofreiter,M., Spaulding,W.G., Martin,P.S., Stankiewicz,B.A., Bland,H., Evershed,R.P., Possnert,G. and Paabo,S. (1998) Molecular coproscopy: dung and diet of the extinct ground sloth Nothrotheriops shastensis. Science, 281, 402–406. [DOI] [PubMed] [Google Scholar]
- 12.Buyuksonmez F., Hess,T.F., Crawford,R.L., Paszczynski,A. and Watts,R.J. (1999) Optimization of simultaneous chemical and biological mineralization of perchloroethylene. Appl. Environ. Microbiol., 65, 2784–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Billi D. and Potts,M. (2000) Life without water: responses of prokaryotes to desiccation. In Storey,K.B. and Storey,J. (eds), Environmental Stressors and Gene Responses. Elsevier Science, St Louis, MO, pp. 181–192.
- 14.Lindahl T. and Wood,R.D. (1999) Quality control by DNA repair. Science, 286, 1897–1905. [DOI] [PubMed] [Google Scholar]
- 15.Beckaman K.B. and Ames,B.N. (1997) Oxidative decay of DNA. J. Biol. Chem., 272, 19633–19636. [DOI] [PubMed] [Google Scholar]
- 16.Dizdaroglu M., Jaruga,P., Birincioglu,M. and Rodriguez,H. (2002) Free radical-induced damage to DNA:mechanisms and measurement. Free Radic. Biol. Med., 32, 1102–1115. [DOI] [PubMed] [Google Scholar]
- 17.Höss M., Jaruga,P., Zastawny,T.H., Dizdaroglu,M. and Pääbo,S. (1996) DNA damage and DNA sequence retrieval from ancient tissues. Nucleic Acids Res., 24, 1304–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dizdaroglu M. (1992) Oxidative damage to DNA in mammalian chromatin. Mutat. Res., 275, 331–342. [DOI] [PubMed] [Google Scholar]
- 19.Breen A.P. and Murphy,J.A. (1995) Reactions of oxyl radicals with DNA. Free Radic. Biol. Med., 18, 1033–1077. [DOI] [PubMed] [Google Scholar]
- 20.Cutler R.G. and Rodriguez,H. (2003) Critical Reviews of Oxidative Stress and Aging: Advances in Basic Science, Diagnostics and Intervention. World Scientific Publishing, River Edge, NJ.
- 21.Buitink J., Leprince,O., Hemminga,M.A. and Hoekstra,F.A. (2000) Molecular mobility in the cytoplasm: an approach to describe and predict lifespan of dry germplasm. Proc. Natl Acad. Sci. USA, 97, 2385–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.St Jean A. (1999) Local genetic context, supercoiling and gene expression. In Charlebois,R.L. (ed.), Organization of the Prokaryotic Genome. ASM Press, Washington, DC, pp. 203–215.
- 23.Higgins N.P. (1999) DNA supercoiling and its consequences for chromosome structure and function. In Charlebois,R.L. (ed.), Organization of the Prokaryotic Genome. ASM Press, Washington, DC, pp. 189–202.
- 24.Wright D., Prickett,T., Helm,R.F. and Potts,M. (2001) Form species Nostoc commune (Cyanobacteria). Int. J. Syst. Evol. Microbiol., 51, 1839–1852. [DOI] [PubMed] [Google Scholar]
- 25.Shirkey B., Kovarcik,D.P., Wright,D.J., Wilmoth,G., Prickett,T.F., Helm,R.F., Gregory,E.M. and Potts,M. (2000) Active Fe-SOD and abundant sodF mRNA in Nostoc commune (Cyanobacteria) after years of desiccation. J. Bacteriol., 182, 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bloom F., Price,P., Lao,G., Xia,J.L., Crowe,J.H., Battista,J.R., Helm,R.F., Slaughter,S. and Potts,M. (2001) Engineering mammalian cells for solid-state sensor applications. Biosensors Bioelectronics, 16, 603–608. [DOI] [PubMed] [Google Scholar]
- 27.Balajee A.S., DeSantis,L.P., Brosh,R.M.,Jr, Selzer,R. and Bohr,V.A., (2000) Role of the ATPase domain of the Cockayne syndrome group B protein in UV induced apoptosis. Oncogene, 19, 477–489. [DOI] [PubMed] [Google Scholar]
- 28.Miller S.A., Dykes,D.D. and Polesky,H.F. (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res., 16, 1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tuo J., Muftuoglu,M., Chen,C., Jaruga,P., Selzer,R.R., Brosh,R.M.,Jr, Rodriguez,H., Dizdaroglu,M. and Bohr,V.A. (2001) The Cockayne Syndrome group B gene product is involved in general genome base excision repair of 8-hydroxyguanine in DNA. J. Biol. Chem., 276, 45772–45779. [DOI] [PubMed] [Google Scholar]
- 30.Dizdaroglu M., Jaruga,P. and Rodriguez,H. (2001) Measurement of 8-hydroxy-2′-deoxyguanosine in DNA by high-performance liquid chromatography-mass spectrometry: comparison with measurement by gas chromatography-mass spectrometry. Nucleic Acids Res., 29, e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dizdaroglu M. (1994) Chemical determination of oxidative DNA-damage by gas chromatography mass-spectrometry Methods Enzymol., 234, 3–16. [DOI] [PubMed] [Google Scholar]
- 32.Dizdaroglu M. (1991) Chemical determination of free radical-induced damage to DNA. Free Radic. Biol. Med., 10, 225–242. [DOI] [PubMed] [Google Scholar]
- 33.Dizdaroglu M., Bauche,C., Rodriguez,H. and Laval,J. (2000) Novel substrates of Escherichia coli Nth protein and its kinetics for excision of modified bases from DNA damaged by free radicals. Biochemistry, 39, 5586–5592. [DOI] [PubMed] [Google Scholar]
- 34.Woldringh C.L. and Odijk,T. (1999) Structure of DNA within the bacterial cell: physics and physiology. In Charlebois,R.L. (ed.), Organization of the Prokaryotic Genome. ASM Press, Washington, DC, pp. 171–187.
- 35.Niedhardt F.C. (1987) 2. Chemical composition of Escherichia coli. In Neidhardt,F.C. (ed.), Escherichia coli and Salmonella typhimurium. ASM Press, Washington, DC, pp. 3–6.
- 36.Potts M. (2000) Nostoc. In Whitton,B.A. and Potts,M. (eds), The Ecology of Cyanobacteria: Their Diversity in Time and Space. Kluwer Academic, Dordrecht, The Netherlands. pp. 465–504.
- 37.Robinson N.J., Rutherford,J.C., Pocock,M.R. and Cavet,J.S. (2000) Metal metabolism and toxicity: repetitive DNA. In Whitton,B.A. and Potts,M. (eds), The Ecology of Cyanobacteria: Their Diversity in Time and Space. Kluwer Academic, Dordrecht, The Netherlands, pp. 443–463.
- 38.Gupta A., Whitton,B.A., Morby,A.P., Huckle,J.W. and Robinson,N.J. (1992) Amplification and rearrangement of a prokaryotic metallothionein locus smt in Synechococcus PCC 6301 selected for tolerance to cadmium. Proc. R. Soc. Lond. B, 248, 273–281. [DOI] [PubMed] [Google Scholar]
- 39.Halliwell B. and Gutteridge,J.M. (1990) Role of free radicals and catalytic metal ions in human disease: an overview. Methods Enzymol., 186, 1–85. [DOI] [PubMed] [Google Scholar]
- 40.Wallace S.S. (2002) Biological consequences of free radical-damaged DNA bases. Free Radic. Biol. Med., 33, 1–14. [DOI] [PubMed] [Google Scholar]
- 41.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC, pp. 42–43.
- 42.Lindahl T., Karran,P. and Wood,R.D. (1997) DNA excision repair pathways. Curr. Opin. Genet. Dev., 7, 158–169. [DOI] [PubMed] [Google Scholar]
- 43.Wood R.D. (1996) DNA repair in eukaryotes. Annu. Rev. Biochem., 65, 135–167. [DOI] [PubMed] [Google Scholar]
- 44.Potts M., Sun,H., Mockaitis,K., Kennelly,P., Reed,D. and Tonks,N.K. (1993) A protein-tyrosine/serine phosphatase encoded by the genome of the cyanobacterium Nostoc commune UTEX 584. J. Biol. Chem., 268, 7632–7635. [PubMed] [Google Scholar]
- 45.Xie W.-Q., Whitton,B.A., Simon,J.W., Jäger,K., Reed,D. and Potts,M. (1989) Nostoc commune UTEX 584 gene expressing indole phosphate hydrolase activity in Escherichia coli. J. Bacteriol., 171, 708–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crowe J.H., Hoekstra,F.A. and Crowe,L.M. (1992) Anhydrobiosis. Annu. Rev. Physiol., 54, 579–599. [DOI] [PubMed] [Google Scholar]
- 47.Hill D.R., Peat,A. and Potts,M. (1994) Biochemistry and structure of the glycan secreted by desiccation-tolerant Nostoc commune (Cyanobacteria). Protoplasma, 182, 126–148. [Google Scholar]
- 48.Scherer S., Ernst,A., Chen,T.-W. and Böger,P. (1984) Rewetting of drought-resistant blue-green algae: time course of water uptake and reappearance of respiration, photosynthesis and nitrogen fixation. Oecologia, 62, 418–423. [DOI] [PubMed] [Google Scholar]
- 49.Taranto P.A., Keenan,T.W. and Potts,M. (1993) Rehydration induces rapid onset of lipid biosynthesis in desiccated Nostoc commune (Cyanobacteria). Biochim. Biophys. Acta, 1168, 228–237. [DOI] [PubMed] [Google Scholar]
- 50.Helm R.F., Huang,Z., Leeson,H., Edwards,D., Peery,W. and Potts,M. (2000) Structural characterization of the released polysaccharide of desiccation-tolerant Nostoc commune DRH-1. J. Bacteriol., 182, 974–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hill D.R., Keenan,T.W., Helm,R.F., Potts,M., Crowe,L.M. and Crowe,J.H. (1997) Extracellular polysaccharide of Nostoc commune (Cyanobacteria) inhibits fusion of membrane vesicles during desiccation and freeze-drying. J. Appl. Phycol., 9, 237–248. [Google Scholar]
- 52.Jäger K. and Potts,M. (1988) Distinct fractions of genomic DNA from the cyanobacterium Nostoc commune that differ in the degree of methylation. Gene, 74, 197–201. [DOI] [PubMed] [Google Scholar]
- 53.Xie W.-Q., Tice,D. and Potts,M. (1995) Cell water deficit regulates expression of rpoC1C2 (RNA polymerase) at the level of mRNA in desiccation-tolerant Nostoc commune UTEX 584 (Cyanobacteria). FEMS Microbiol. Lett., 126, 159–164. [Google Scholar]
- 54.Billi D., Friedmann,E.I., Hofer,K.G., Caiola,M.G. and Ocampo-Friedmann,R. (2000) Ionizing-radiation resistance in the desiccation-tolerant cyanobacterium Chroococcidiopsis. Appl. Environ. Microbiol., 66, 1489–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Billi D., Wright,D.J., Helm,R.F., Potts,M. and Crowe,J.H. (2000) Engineering desiccation tolerance in Escherichia coli. Appl. Environ. Microbiol., 66, 1680–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]