

Abstract
Abnormal expression of Dnmt1 in vivo induces cellular alterations such as transformation, and an increase in Dnmt1 mRNA plays a causal role in c-fos-, ras- and SV40 large T antigen-induced transformation of fibroblasts in vitro. Here, we have investigated the regulation of Dnmt1 transcription. We identified the promoter region and major transcription start sites of mouse Dnmt1 and found two important cis-elements within the core promoter region. One is an E2F binding site, and the other is a binding site for an as yet unidentified factor. Point mutations in the two cis-elements decreased promoter activity in both non-transformed and transformed cells. Thus, both sites play a critical role in regulation of Dnmt1 transcription in proliferating cells. Treatment with trichostatin A, a specific inhibitor of histone deacetylase, increased Dnmt1 promoter activity in G0/G1-arrested NIH 3T3 cells. Furthermore, the decrease in promoter activity induced by expression of E2F-1 and Rb was reversed by trichostatin A treatment of Saos-2 cells. Taken together, these data indicate that transcription of Dnmt1 is regulated in a complex fashion by E2F and other transcription factors through E2F-Rb-HDAC-dependent and -independent pathways. These findings suggest that Dnmt1 is a target gene of these pathways in cell proliferation, cell transformation and tumorigenesis.
INTRODUCTION
In mammalian cells, genomic DNA is methylated on the fifth carbon position of cytosine in CpG dinucleotides, and the distribution of methylated cytosines is cell and tissue specific (1–4). DNA methylation is correlated with gene silencing and condensation of chromatin structure. Inhibition of transcription by DNA methylation occurs either because methyl groups on cytosine limit the access of transcription factors to DNA binding sites or by the recruitment of histone deacetylase (HDAC) via methyl-CpG binding proteins such as MeCP2 (5).
Patterns of DNA methylation are generated during development by DNA methyltransferases such as Dnmt1 (6,7). Dnmt1 prefers hemi-methylated DNA over non-methylated DNA as a substrate in vitro (8), and expression of Dnmt1 increases during S phase (9,10). Dnmt1 is recruited to replication foci through protein interactions involving the DMAP1 binding region (11), the proliferating cell nuclear antigen (PCNA) binding region (12) and the replication foci targeting sequence (10). We have recently found a direct interaction between Dnmt1 and MeCP2 (13), which may confer accurate maintenance of DNA methylation patterns through stepwise interactions during DNA replication.
Aberrant CpG methylation has been observed in several tumors (14). Some CpG islands are hypermethylated in tumor cells (15,16), suggesting that a fundamental disruption of methylation control has occurred. This aberrant hypermethylation can act in a manner analogous to deletion or mutation of growth inhibitory genes, thereby producing a selective growth advantage (16). Dnmt1 has been shown to act as an oncogene in cultured fibroblasts (17). Further, NIH 3T3 cells transformed by Dnmt1 show enhanced DNA methylation, and these cells form tumors when transplanted into nude mice (17). Conditional expression of c-fos up-regulates expression of Dnmt1, resulting in morphological transformation of rat fibroblasts (18). Interestingly, a low level of Dnmt1 over-expression leads to cell transformation whereas a high level is toxic (17). In addition, s.c. injection of the Dnmt1 inhibitor 5-aza-2′-deoxycytidine decreases polyp number in Min mice that are heterozygous for the multiple intestinal neoplasia mutation of the adenomatosis polyposis coli (Apc) gene (19). Thus Dnmt1 expression must be tightly regulated during normal cell growth.
The mechanisms responsible for regulation of Dnmt1 transcription are poorly understood. Expression of immediate early genes such as c-fos increases drastically within 1 h after serum stimulation (20,21). However, expression of Dnmt1 mRNA varies in a cell cycle-dependent manner reaching a maximum during S phase (9). These findings suggest that Dnmt1 is controlled not only by c-fos but also by other transcription factors. Expression of human DNMT1 is increased by binding of Jun homodimers or Fos–Jun hetrodimers to a region located between exons 1 and 2 (22). Nuclear run-on assays in BALB/c 3T3 and myoblast G8 cells indicated that the transcriptional activity of Dnmt1 is constant throughout the cell cycle and during cellular differentiation, suggesting that mRNA levels may be modulated at the post-transcriptional level (9,23). These contradictory findings stimulated us to investigate the regulatory mechanisms that control expression of Dnmt1.
The expression of many replication-related genes, such as PCNA, cyclin E, polα, E2F-1 and cdc6, is regulated by the E2F family of transcription factors (24). The transcriptional activity of E2F is controlled by binding of Rb family members (24,25). The Rb/E2F complex is disrupted by Rb kinases (cyclin D/cdk4, cdk6 and cyclin E/cdk2) and by simian virus 40 (SV40) large T antigen, resulting in activation of E2F target genes such as cyclin E and E2F-1 and entry into S phase (26,27). The promoters of cyclin E and p107 contain E2F binding sites, and repression of their expression by Rb has been shown to be HDAC-dependent (24). However, the expression of other E2F target genes, PCNA, E2F-1 and b-Myb, is insensitive to treatment with trichostatin A (TSA). (28). For each E2F target gene, the pathways regulating transcription are classified as either HDAC dependent or independent.
In this study, we focus on the transcriptional regulation of mouse Dnmt1. We demonstrate that transcription of Dnmt1 during cell cycle progression is modulated by two cis-elements in the promoter region. We propose that a combination of E2F-Rb-HDAC-dependent and -independent pathways control Dnmt1 transcription during cell growth.
MATERIALS AND METHODS
Cell culture
COS-7 cells, NIH 3T3 cells, 3Y1-B clone 1-6 (RCB 0488), SR-3Y1 (JCRB 0742), HR-3Y1 (JCRB 0743), E1A-3Y1 (JCRB 0738) and SV-3Y1 (JCRB 0735) cells and Saos-2 cells (RCB 0428) were all maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). All culture media were supplemented with 100 U/ml penicillin (Sigma) and 100 µg/ml streptomycin (Sigma).
To bring NIH 3T3 cells to quiescence, 5 × 105 cells were plated on a 150 mm dish and incubated overnight. The next day, the cells were washed three times with phosphate-buffered saline (PBS), and the culture medium was replaced with DMEM containing 0.05% FBS. Cells were incubated for 36 h prior to serum stimulation, then harvested at the indicated time after addition of the complete medium.
Northern blot analysis
Total RNA was isolated from NIH 3T3 cells with TRIzol (Gibco BRL) according to the manufacturer’s instructions. Ten micrograms of total RNA were separated by agarose gel electrophoresis under denaturing conditions and hybridized at high stringency to DIG-labeled Dnmt1 (29), cyclin D1, cyclin E and gapdh antisense RNA probes.
FACScan analysis
Cells were detached by incubation with 0.25% trypsin/1 mM EDTA, washed with ice-cold PBS three times and fixed with ice-cold 75% ethanol. Cells were subsequently washed with PBS three times to remove ethanol and were suspended in PBS containing 50 µg/ml propidium iodide. Cell synchrony was assessed by FACScan (Becton Dickinson) and cell cycle distribution was analyzed by using CellQuest software and Modifit LT software (Becton Dickinson).
Library screening and sequencing
An EcoRI-digested rat Dnmt1 cDNA fragment containing the 5′ flanking region (214 bp) including the somatic translation start codon (29) was used to screen a mouse 129/SvJ male kidney λDashII genomic library (a gift from Dr J. Rossant) under high stringency conditions. The probe has >91% homology with the mouse sequence. Among 5 × 105 phages examined, three positive independent clones containing 25, 16 and 16 kb, respectively, of insert DNA fragment were obtained. A 9.6 kb DNA fragment between BamHI sites from the 25 kb fragment was subcloned into pBluescript SK(–) vector and sequenced using a standard protocol (30) (DDBJ accession no. AB056445).
RNase protection assay
Assays were performed by using an RNase Protection kit (Roche) according to the manufacturer’s instructions. Two radiolabeled RNA probes were prepared from mouse Dnmt1 DNA fragments. Probe 380 consisted of 300 bases upstream from the translation start codon and 80 bases downstream, and probe 80 contained the 80 bases downstream of the start codon (Fig. 2B). Both fragments were cloned into pGEM-T easy vector (Promega), from which antisense cRNAs were transcribed with SP6 or T7 RNA polymerase. Ten micrograms of total RNA were added to the labeled cRNA probe. The mixture was denatured at 95°C for 5 min. Hybridization was performed overnight at 41°C. The hybridization reaction was cooled to room temperature and treated with RNase A at 30°C for 30 min. Stop solution (0.5% SDS and 0.27 mg/ml proteinase K) was added and the reaction mixture was incubated at 37°C for 15 min. RNA was extracted with phenol/chloroform/isoamyl alcohol (25:24:1), and the double-stranded RNA was precipitated with ethanol. It subsequently was subjected to electrophoresis in a 4% polyacrylamide/7 M urea gel in Tris–borate buffer. Signal was detected by using a BAS 2000 image analyzer (Fuji film).
Figure 2.
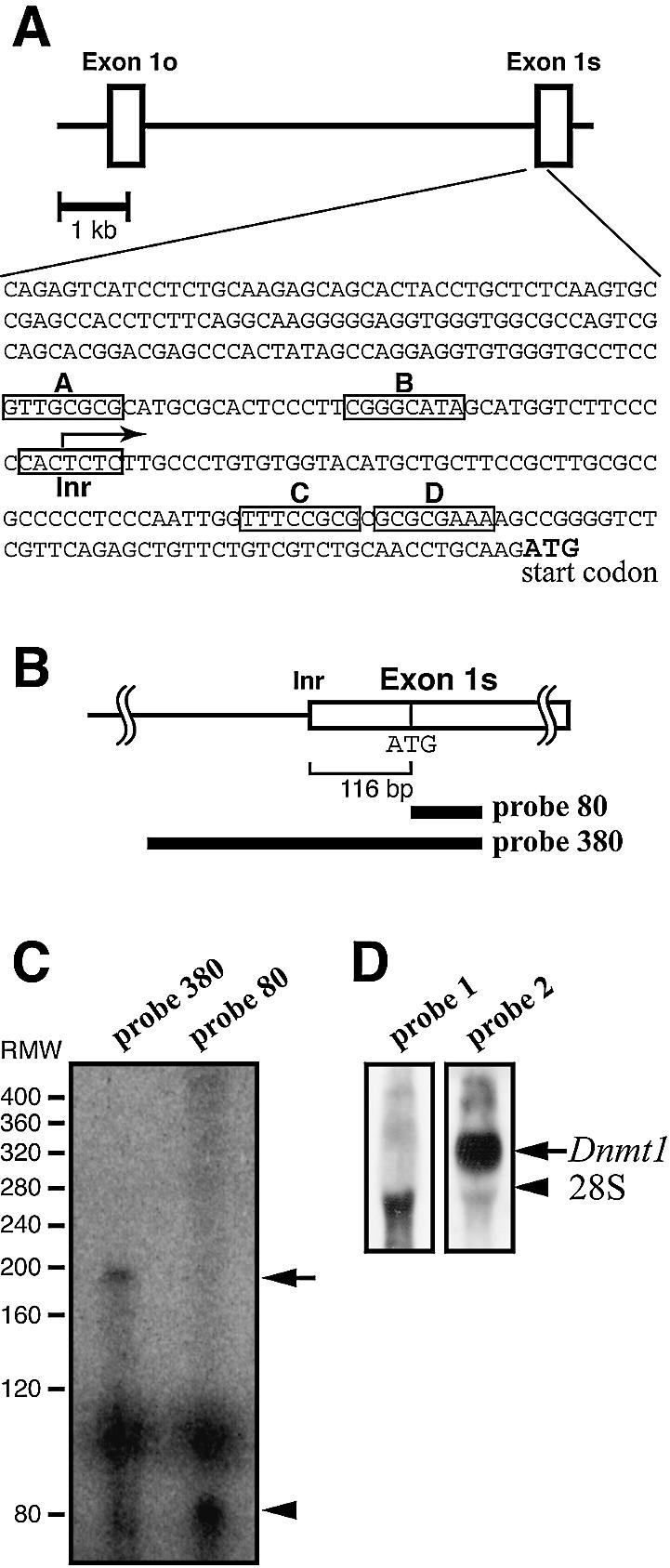
Identification of the transcription start sites of mouse Dnmt1. (A) Nucleotide sequence of the 5′-flanking region and the transcription start site of the mouse Dnmt1 showing the 299 bases upstream from the translation start codon. Boxes A–D indicate putative E2F binding sites. The transcription start site is indicated with an arrow. Exon 1o, oocyte-specific first exon; Exon 1s, somatic first exon; Inr, initiator consensus sequence (34,35). (B) Probes used in RNase protection assay. (C) RNase protection assay. RNA was prepared from NIH 3T3 cells. The major 190 base fragment protected by probe 380 is indicated with an arrow. RMW, size standards with sizes (base pair) corrected for the slower migration of double-stranded RNA based on the migration of the 80 bp double-stranded RNA protected by probe 80 (arrowhead). (D) Northern blot analysis confirmed the transcription of the 5′ region of mouse Dnmt1. Total RNA from NIH 3T3 cells was hybridized to probe 1, located 610 bases upstream from the initiator sequence, or probe 2, which corresponds to the Dnmt1 catalytic domain (33).
Isolation of the 5′ flanking region of the rat Dnmt1
To identify the sequence of the rat Dnmt1 promoter region, rat oocyte-specific short form Dnmt1 cDNA was amplified from ovary-derived total RNA by RT–PCR using the following primer set: forward primer (1o Dnmt1-F3), 5′-AGCCCT CTTGTTTGATGGC-3′; reverse primer (5′ Dnmt1-R), 5′-GCAGGAATTCATGCAGTAAG-3′.
Oocyte-specific exon 1 is located upstream of somatic exon 1 (Fig. 2A) (31). The amplified 194 bp cDNA was subcloned into pGEM-T easy (Promega) and sequenced. Genomic PCR was then performed by using LA-Taq (TaKaRa), genomic DNA derived from rat kidney and the following primer set designed based on sequence of rat oocyte-specific exon 1: forward primer (5′ G-Dnmt1-F), 5′-AGCCCTCTTGTTTG ATGGCCAGGGTGGAGC-3′; reverse primer (3′ G-Dnmt1-R), 5′-TCTTGGGGATCGGTTTGAGTCTGCCATTTC-3′.
The amplified 15 kb DNA fragment was digested with EcoRI, subcloned into pBluescript II SK(+) (Stratagene) and sequenced (DDBJ accession no. AB056446).
Plasmids, transfection and luciferase assay
Different lengths of upstream regions of the mouse Dnmt1 gene with either NheI or SmaI and XhoI sites were generated by a PCR-based method. These fragments were subcloned into PGV-B2 luciferase plasmid (Nippon Gene). Primer sets are as follows: 2061-luc sense, 5′-GCCCGGGAGGATTAAAGGT GTGCACCACTACCGCTG-3′; 500-luc sense, 5′-GGCT AGCTCTGGCTTTTGCATTCTGAG-3′; 300-luc sense, 5′-GGCTAGCGAGCCCACTATAGCCAGGAG-3′; 100-luc sense, 5′-GGCTAGCCATGCTGCTTCCGTTGCGCC-3′; common antisense, 5′-GCTCGAGCTTGCAGGTTGCAG ACGACAGAAC-3′; R756-luc sense, 5′-GCTCGAGCA CTGCGCAGACAAGCCCAC-3′; R756 antisense, 5′-GGCTAGCCTTGCAGGTTGCAGACGACA-3′.
736-luc was generated by insertion of a NheI–XhoI fragment digested from the p2061 luciferase plasmid into the PGV-B2 luciferase plasmid. Point mutations were generated with the QuikChange Site-directed Mutagenesis Kit (Stratagene) as specified in the manufacturer’s instructions. Oligonucleotide sets used for the mutagenesis were as follows (mutated nucleotides are underlined): site A, 5′-GTGCCT CCGTTGCATGCATGCGCACTCCCTTC-3′ and 5′-GAA GGGAGTGCGCATGCATGCAACGGAGGCAC-3′; site B, 5′-CATGCGCACTCCCTTCGATCATAGCATGGTCTTC-3′ and 5′-GAAGACCATGCTATGATCGAAGGGAGTGCG CATG-3′; site C, 5′-CTGCTTCCGCTTGCATCGCCCCCT CCCAATTG-3′ and 5′-CAATTGGGAGGGGGCGATGC AAGCGGAAGCAG-3′; site D, 5′-CTCCCAATTGGT TTCCATGCGCGCGAAAAAGCC-3′ and 5′-GGCTTTTTC GCGCGCATGGAAACCAATTGGGAG-3′.
Further mutagenesis was carried out using the mutated luciferase plasmids as templates and the oligonucleotide sets described above. All clones were verified by sequencing.
For the luciferase assay, 2 × 104 COS-7 cells were seeded in a 24-well plate and cultured overnight. The cells were co-transfected with 0.95 µg of luciferase plasmid and 0.05 µg of Renilla luciferase plasmid pRL-SV40 as an internal control by using a standard DEAE-dextran method (30). Alternatively, NIH 3T3 cells, cultured as above, were co-transfected with a total of 0.2 µg plasmids, which consisted of various luciferase plasmids and expression vector plasmids (pDCE2F1, a kind gift from Dr K. Ohtani, and pME18S SRα mouse Rb, a kind gift from Dr T. Yamamoto) and Renilla luciferase plasmid pRL-SV40 as an internal control. The Lipofectamine PLUS transfection method (Gibco BRL) was used as specified by the manufacturer’s instructions. TSA (a kind gift from Dr M. Yoshida) was added 24 h before harvesting cells (32). The cells were harvested 48 h after transfection, lysed and the degree of transactivation was measured with a PicaGene Dual SeaPansy Luminescence Kit (Nippon Gene) as specified by the manufacturer’s instructions. All assays were repeated at least twice with triplicate determinants.
Electrophoretic mobility shift assay (EMSA)
NIH 3T3 cells were washed twice with ice-cold PBS and scraped into microcentrifuge tubes. Cells were pelleted by centrifugation at 2300 g at 4°C and then pipetted up and down 10 times in 10 vol of lysis buffer [50 mM HEPES, pH 7.9, 250 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 0.1% NP-40, 0.4 mM NaF, 0.4 mM Na3VO4, 10% glycerol, 1 mM dithiothreitol (DTT), 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 2 µg/ml aprotinin, 1 µg/ml pepstatin, 2 µg/ml leupeptin] and were incubated on ice for 30 min. Cleared whole cell extracts were obtained by centrifugation at 20 000 g at 4°C for 30 min. DNA binding assays contained 10 µg of total protein in 10 µl of DNA binding reaction mixture (20 mM HEPES, pH 7.9, 40 mM KCl, 6 mM MgCl2, 1 mM DTT, 1 mM PMSF, 0.1% NP-40, 10% glycerol, 50 µg/ml sonicated salmon sperm DNA) and 0.14 ng of 32P-labeled DNA probe. DNA probes were as follows: E2F oligonucleotide, 5′-TCCGTTTTCGCGCTTAAATTTGAGAAAGGGCGCGAAACTGGA-3′; probe A, 5′-GTGCCTCCGTTGCGCGCA TGCGCAC-3′; probe C, 5′-CCAATTGGTTTCCGCGCGC GCGAAAAAGCCGGGGTCTCGTTC-3′.
The reaction mixtures were incubated at room temperature for 20 min and resolved by electrophoresis on a 5% polyacrylamide gel (acrylamide/bis 80:1) in 0.25× TBEG (12.5 mM Tris–borate, 0.25 mM EGTA) containing 5% glycerol at 4°C. Alternative DNA binding assays were carried out with DNA binding reaction mixture [10 mM Tris, pH 7.5, 50 mM NaCl, 1 mM MgCl2, 0.5 mM EDTA, 50 µg/ml poly(dI–dC)·(dI–dC), 4% glycerol] in TBE (50 mM Tris–borate, 1 mM EDTA) at 4°C.
Western blot analysis
NIH 3T3 cells were harvested and washed three times with ice-cold PBS. The cells were pelleted by centrifugation at 2300 g at 4°C. NP-40 lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP-40, 10% glycerol, 1 mM DTT, 0.5 mM EDTA, 0.5 mM EGTA, 0.5 mM NaF, 1 mM Na3VO4, 1% aprotinin, 5 µg/ml leupeptin, 0.7 µg/ml pepstatin) was added to the cell pellets and placed on ice for 30 min. Cleared whole cell extracts were prepared by centrifugation at 20 000 g for 30 min at 4°C. Protein concentration was measured by using a BCA kit (Pierce) according to the manufacturer’s instructions. Whole cell extracts were subjected to western blot analysis using anti-Dnmt1 (33), anti-p21Waf1 (Santa Cruz) and anti-β-tubulin (Sigma) antibodies.
RESULTS
Dnmt1 mRNA increases at late G1/early S phase
To investigate the cell cycle-dependent Dnmt1 transcription, we examined the expression of three genes in NIH 3T3 cells; Dnmt1, cyclin D1 and cyclin E (Fig. 1A). Analysis of RNA from quiescent and serum-stimulated cells revealed a strong induction of each gene. Dnmt1 transcript started to accumulate at 9 h after serum stimulation and reached maximum levels at 20 h. cyclin D1 mRNA began to accumulate at 6 h and reached maximum levels at 9 h. cyclin E transcript was elevated at 9 h and peaked at 12 h.
Figure 1.
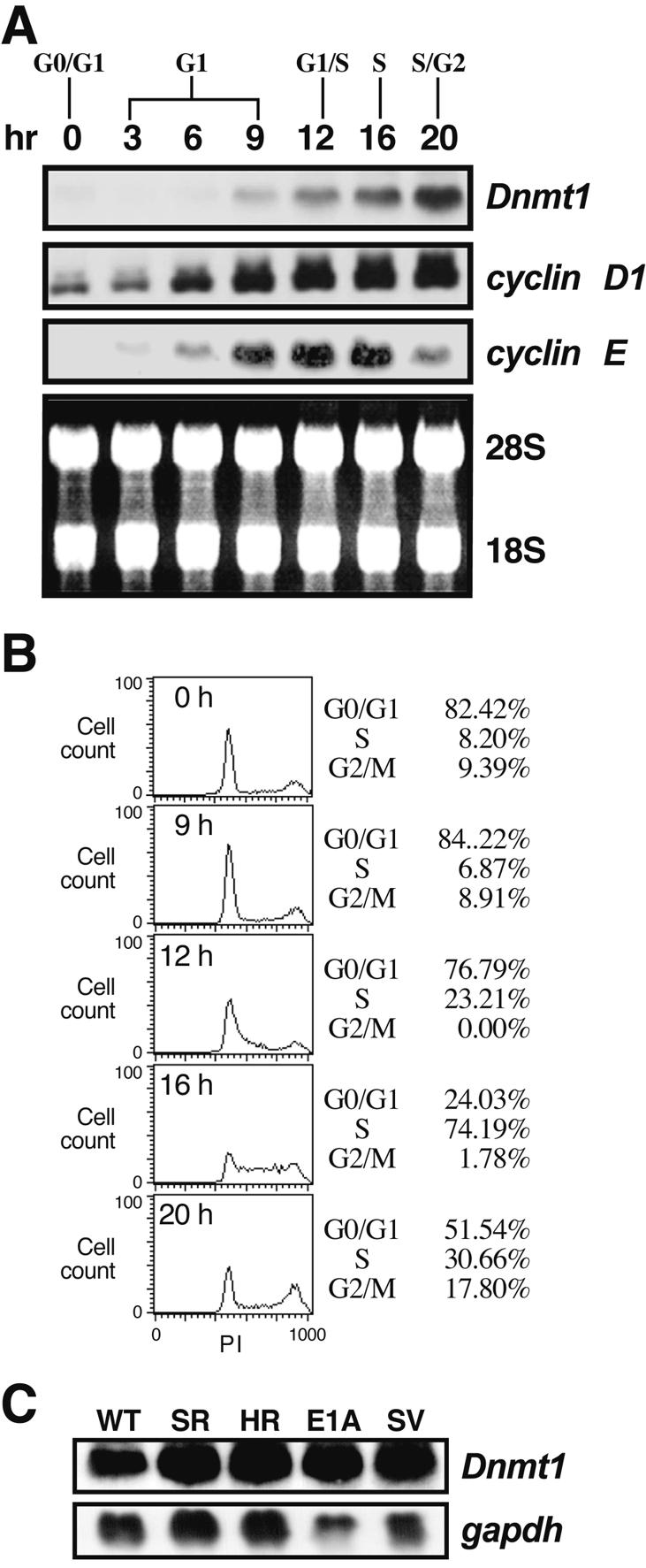
Expression of Dnmt1 mRNA during the cell cycle and in oncogenic transformed cells. (A) Northern blot showing expression of Dnmt1, cyclin D1 and cyclin E during the cell cycle. Quiescent NIH 3T3 cells were stimulated by replacement with complete medium including 10% fetal bovine serum and harvested at the indicated times. (B) Percentage of cells in each stage of the cell cycle at various times after serum stimulation. DNA amounts were measured by propidium iodide (PI) staining and FACScan analysis. The cell cycle distribution is indicated on the right of the histogram. (C) Northern blot showing expression of Dnmt1 in transformed 3Y1 cells, with gapdh as an internal control. WT, parental 3Y1 cells; SR, RSV-transformed 3Y1 cells; HR, Ha-ras transformed 3Y1 cells; E1A, E1A-transformed cells; SV, SV40-transformed 3Y1 cells.
To examine the cell cycle distribution of the cells after serum stimulation, we measured DNA content (Fig. 1B). Nine hours after addition of serum, about 7% of cells had entered S phase, whereas after 12 h, 23% of cells had entered S phase. After 16 h, 74% of cells had progressed into S phase, and then passed through S phase at 20 h. Similar results on FACScan analysis were obtained from two independent experiments. At 0 h, the quiescent cells had very low levels of Dnmt1, cyclin D1 and cyclin E mRNA, but expression of all three genes increased 20- to 30-fold following serum stimulation. Dnmt1 and cyclin D1 mRNA remained constant following their initial accumulation, whereas cyclin E mRNA declined as cells proceeded through S phase. cyclin D1 mRNA started to accumulate from mid-G1 phase, while Dnmt1 RNA started to accumulate from late G1 to early S phase. These results indicate that the transcription of Dnmt1 was activated at late G1 in our system.
To examine Dnmt1 expression in transformed cells, total RNA was prepared from rat-derived 3Y1 (WT), RSV-transformed 3Y1 (SR-3Y1), Ha-ras-transformed 3Y1 (HR-3Y1), E1A-transformed 3Y1 (E1A-3Y1) and SV40-transformed 3Y1 cells (SV-3Y1 cells). The transformed cells had approximately 2- to 3-fold more Dnmt1 mRNA than the parental wild-type cells (Fig. 1C), suggesting that a modest increase in Dnmt1 mRNA is associated with cell transformation. Our observations are consistent with the levels of Dnmt1 expression observed in cells transformed by c-fos (18).
Genomic cloning of the 5′ upstream region and identification of the transcription start sites of Dnmt1
To identify the promoter of the mouse Dnmt1 gene, we isolated and sequenced a 9.6 kb fragment containing a complete somatic exon 1 (31) and 8.5 kb upstream from the translation start codon. The 2061 bases immediately upstream from the translation start codon did not contain a TATAA sequence (DDBJ accession no. AB056445).
Dnmt1 codes for three distinct gene products, the somatic form, the oocyte-specific form and the testis-specific form (31). Sequencing showed a possible transcription initiator sequence (Inr) (5′-CCACTCT-3′, PyPyANT/APyPy) (34,35) located at bases –123 to –117 upstream of the translation start codon (Fig. 2A). To confirm this experimentally, RNase protection assays were performed using RNA from NIH 3T3 cells. Probes used were an 80 base fragment directly downstream from the ATG in somatic exon 1 (probe 80) and a 380 base fragment spanning from 300 bases upstream of the ATG to 80 bases downstream of the ATG (probe 380) (Fig. 2B). Probe 80 is a positive control for the molecular weight of hybridized RNAs. As expected, probe 80 detected an 80 bp fragment of somatic exon 1 (arrowhead in Fig. 2C). Probe 380 produced a major signal at ∼190 bp (arrow in Fig. 2C). This result shows that transcription is initiated around 110 bases upstream from the translation start codon ATG. To further confirm the transcription start sites, northern blot analysis was performed using two probes, one of which hybridizes 610 bases upstream from the initiator sequence (probe 1) and the other (probe 2) corresponding to the catalytic domain in the coding sequence of Dnmt1. Probe 1 detected no Dnmt1 mRNA, whereas probe 2 detected a 5.2 kb band, which is the major transcript size of somatic Dnmt1 (Fig. 2D). These data indicate that the major transcription start sites are between –123 and –117.
Two major cis-elements in the core promoter of Dnmt1
To identify the promoter region essential for transcriptional regulation of Dnmt1 during cell proliferation, we transfected COS-7 cells with luciferase reporter constructs containing deletions of the Dnmt1 5′-flanking region. The luciferase activity from plasmids containing 2061, 736, 500 or 300 bp upstream of the translation start codon of Dnmt1 was 25- to 40-fold higher than that of a control plasmid without insert (Fig. 3A). However, the luciferase activity of a plasmid containing 100 bp of 5′ sequence of Dnmt1 was similar to control levels. To confirm that the promoter activity of the upstream sequence was unidirectional, we constructed a luciferase reporter plasmid containing 736 bp of upstream sequence in the reverse orientation (R736). The luciferase activity of R736 was comparable to the control levels. These data indicate that the 300 bases of the 5′ flanking region of Dnmt1 is a core promoter sequence. Additionally, we examined the 1.5 kb region upstream from the oocyte-specific ATG codon using luciferase assays. No luciferase activity was detected in COS-7 cells (data not shown), suggesting that the oocyte-specific form of Dnmt1 may be regulated by an oocyte-specific regulatory mechanism.
Figure 3.

Identification of the core promoter region of mouse Dnmt1 and two critical binding sites. (A) COS-7 cells were transfected with several promoter fragments linked to a luciferase reporter construct. Results are shown as relative luciferase activity after 48 h. Bars indicate standard deviations (SD). COS-7 cells were transfected with serial deletion constructs of the upstream region: 2061-luc, 736-luc, 500-luc, 300-luc, 100-luc, R736-luc (the 736 bp upstream region in reverse orientation) and No insert-luc (PGV-B2) constructs. (B) COS-7 cells were transiently transfected with constructs containing the first 300 bases upstream of the start codon either with wild-type or mutated (crosses) putative E2F binding sites, or with No insert-luc (PGV-B2). Luciferase activity of the wild-type 300-luc (column 1) was defined to be 100%. (C) As in (B), except that NIH 3T3 cells were used instead of COS-7 cells.
As shown in Figure 1, Dnmt1 is expressed in a cell cycle-dependent manner. The core promoter region of Dnmt1 contained four putative E2F binding sites (sites A–D, Fig. 2A). To test the possibility that transcription of Dnmt1 may also be regulated by E2F transcription factors, we analyzed the luciferase activity of the core promoter sequence of Dnmt1 by creating point mutations in the putative E2F binding sites. Relative to wild-type sequence (Fig. 3B, column 1), point mutations at either site A or site C decreased the promoter activity by 40–50% (Fig. 3B, columns 2 and 4). However, mutation in either site B or site D reduced the promoter activity only slightly (Fig. 3B, columns 3 and 5). The combination of several point mutations showed that mutations of both site A and site C further decreased the promoter activity by ∼60% (Fig. 3B, columns 7, 8 and 10). Other plasmids containing mutations in either site A or site C produced 60% of the promoter activity (Fig. 3B, columns 6 and 9). These findings showed that the putative E2F binding site A and site C in the core promoter play a significant role in the activation of Dnmt1 transcription.
To confirm that these decreases in the promoter activity of Dnmt1 are not specific to COS-7 cells, we also analyzed Dnmt1 promoter activity of site A and site C in NIH 3T3 cells (Fig. 3C). The decrease in promoter activity in NIH 3T3 cells was similar to that observed in COS-7 cells, indicating that these two cis-elements are crucial for the transcriptional regulation of Dnmt1 in proliferating cells.
Dnmt1 core promoter activity in oncogenic transformed cells
As shown in Figure 1C, Dnmt1 mRNA increased approximately 2-fold in transformed cells. To examine whether this accumulation of Dnmt1 mRNA reflects altered promoter activity, we transfected the luciferase reporter construct containing the Dnmt1 core promoter (300-luc in Fig. 3A) into transformed 3Y1 cells. All transformed cells showed at least 30 times more transctiptional activity than parental 3Y1 cells (Fig. 4A), indicating that the promoter activity is increased in the transformed cells.
Figure 4.

Promoter activity of the core promoter region of mouse Dnmt1 in transformed 3Y1 cells. (A) Several cell lines were transiently transfected with the 300-luc construct and luciferase activity was measured as in Figure 3. WT, parental 3Y1 cells; SR, RSV-transformed 3Y1 cells; HR, Ha-ras transformed 3Y1 cells; E1A, E1A-transformed cells; SV, SV40-transformed 3Y1 cells. (B) SV-3Y1 cells were transfected with the 300-luc construct, a construct with mutated site A (300 A), a construct with mutated site C (300 C) or a construct in which both sites A and C were mutated (300 AC). Numbers shown are fold induction relative to luciferase activity of 300-luc. (C) Comparison of the core promoter region of Dnmt1 between mouse and rat. Inr, initiator sequence: sites A and C correspond to those shown in Figure 2A.
E2F trans-activity is inactivated by binding to Rb, but it is activated by releasing Rb on phosphorylation of Rb by Rb kinases (24). The Rb-E2F complex is also disrupted by SV40 large T antigen or expression of a pocket domain mutant form of Rb which cannot bind to E2F (24). To determine whether up-regulation of Dnmt1 mRNA is dependent on E2F binding to site A and site C in the promoter, we measured luciferase activity of mutated Dnmt1 core promoters in SV-3Y1 cells which have been transformed with SV40 (Fig. 4B). In SV-3Y1 cells, the wild-type Dnmt1 promoter had 30-fold greater promoter activity than parental 3Y1 cells (Fig. 4A). The constructs containing mutations in site A (300A), site C (300C) or in both (300AC) showed lower promoter activity than the wild-type construct (300) in SV-3Y1 cells (Fig. 4B). These results indicate that Dnmt1 transcription may be activated by SV40 large T antigen through sites A and C. Therefore, transcription of Dnmt1 is likely activated by E2F transcription factors binding to sites A and C in the promoter, and both putative E2F binding sites A and C are required to enhance promoter activity in SV-3Y1 cells. These observations are unlikely to result from differences between species, because the core promoter region is highly conserved across mouse and rat Dnmt1 (Fig. 4C).
Interestingly, the sequence of site C in mouse Dnmt1 matches exactly with that of site C in rat, but the sequence of site A differs between mouse and rat. Taken together, our findings suggest that Dnmt1 transcription is controlled through two cis-elements in both normal and transformed cells. It is possible, however, that factors other than E2F are responsible for activation of Dnmt1 expression through these cis-elements.
Two distinct transcription factors bind to two cis-elements
To examine whether E2F binds to sites A and C in the promoter of Dnmt1, we performed EMSA using oligonucleotides corresponding to site A or site C (Fig. 4C, probes A and C) and to the E2F consensus binding sequence (36). Binding activity to probe A was detectable (Fig. 5A, lane 2), and the DNA–protein complexes disappeared when unlabeled probe A was used as a competitor (Fig. 5A, lane 3). However, binding was present when the sequence of site A was mutated in the competitor (Fig. 5A, lane 6), consistent with the luciferase assays using the point mutated constructs shown in Figure 3. Moreover, neither the E2F consensus oligonucleotide nor the site C oligonucleotide was able to compete against probe A (Fig. 5B, lanes 4 and 5), indicating that E2F does not bind site A. To confirm this, we repeated EMSA under another condition in which poly(dI–dC) was used as a blocking reagent. In this condition, the major shifted band was competed off by probe A but not by mutated probe A, E2F consensus oligonucleotide or Sp-1 consensus oligonucleotide (Fig. 5, lanes 3–6). EMSA with mutant probe A showed no detectable specific shift (Fig. 5B, lane 7), findings that indicate that a transcription factor other than E2F binds to site A. To examine this further, we analyzed whole cell extracts from NIH 3T3 cells by EMSA, using the site A and E2F consensus oligonucleotides as probes (Fig. 5C). The mobility shift of the DNA–protein complexes using site A differed from that of those using the E2F consensus oligonucleotide (Fig. 5C, lanes 1 and 2). These results indicate that E2F transcription factors are unable to bind to site A. The oligonucleotides corresponding to site A differ from the Sp-1 consensus sequence (G/T)GGGCGGG(GGC/AAT) and Sp-1/3 consensus sequence (GGCAAGGGGGAGGTG), which lie upstream of site A (37), and also from the AP-1 consensus sequence TGACTCA. As expected, the protein bound to site A was not competed off by the Sp-1 consensus sequence (Fig. 5B, lane 6). EMSA using the Sp-1/3 binding element previously detected three major bands (37). Therefore, our data indicate that the protein bound to site A is a factor other than E2F, Sp-1 or AP-1. On the other hand, site C oligonucleotide specifically inhibited formation of E2F consensus oligonucleotide–protein complexes (Fig. 5C, lane 4) and vice versa (Fig. 5D, lane 4). These data collectively suggest that the Dnmt1 promoter may be activated by both E2F at site C and another unidentified transcription factor at site A.
Figure 5.

EMSAs to identify molecules bound to site A and site C in the core promoter region of mouse Dnmt1. (A) Complex formation of 10 µg of whole cell extracts (WCE) from NIH 3T3 cells and 32P-labeled site A oligonucleotides (probe A) analyzed under E2F gel shift conditions. Excess unlabeled oligonucleotides (1:150 molar) were used as competitors; A, site A oligonucleotide; E2F, E2F binding site consensus oligonucleotide; C, site C oligonucleotide; mA, mutated site A oligonucleotide. An arrow indicates the probe A–protein complex. (B) Complex formation as above analyzed under alternative gel shift conditions (see Materials and Methods) with 8 µg of WCE. Excess unlabeled oligonucleotides (1:75 molar) were used as competitors; Sp, Sp-1 consensus oligonucleotide; others as in (A). An arrow indicates the probe A–protein complex. (C) Different patterns of mobility shift in sites A and C. The assays were carried out as in (A). An arrow indicates the probe A–protein complex. (D) Complex formation on 32P-labeled site C oligonucleotides. Conditions and competitors are the same as in (A). The upper bracket indicates probe C–protein complexes, representative of E2F bound to site C.
E2F-1 activates the Dnmt1 promoter
The family of E2F transcription factors consists of E2F-1, E2F-2, E2F-3, E2F-4 and E2F-5, all of which possess DNA binding activity, and E2F-6, which does not have a transcriptional activation domain (24). Individual E2F members recognize specific binding sequences of target genes in vivo (38). E2F-1 possesses both oncogenic and tumor suppressor properties (27,39). Ectopic expression of Dnmt1 in NIH 3T3 cells causes cellular transformation and tumorigenesis (17). Considering these observations and the apparent E2F binding properties of site C in the mouse Dnmt1 promoter, Dnmt1 is potentially a target gene of E2F-1. To examine this, we co-transfected the core promoter reporter plasmids along with an E2F-1 expression vector into NIH 3T3 cells (Fig. 6A). With the wild-type promoter, co-transfected E2F-1 increased the promoter activity (Fig. 6A, column 2). The mutation in site C decreased luciferase activity of E2F-1 by half (Fig. 6A, column 4), whereas the mutation in site A decreased luciferase activity only slightly (Fig. 6A, column 3). These results indicate that activation of the Dnmt1 core promoter by E2F-1 depends upon site C but not site A. Remarkably, double mutation at sites A and C reduced the promoter activity further than the site C single mutation (Fig. 6A, column 5). These data indicate that Dnmt1 is activated by E2F-1 mainly through site C; however, a molecule bound to site A works cooperatively with E2F-dependent transcriptional activity.
Figure 6.
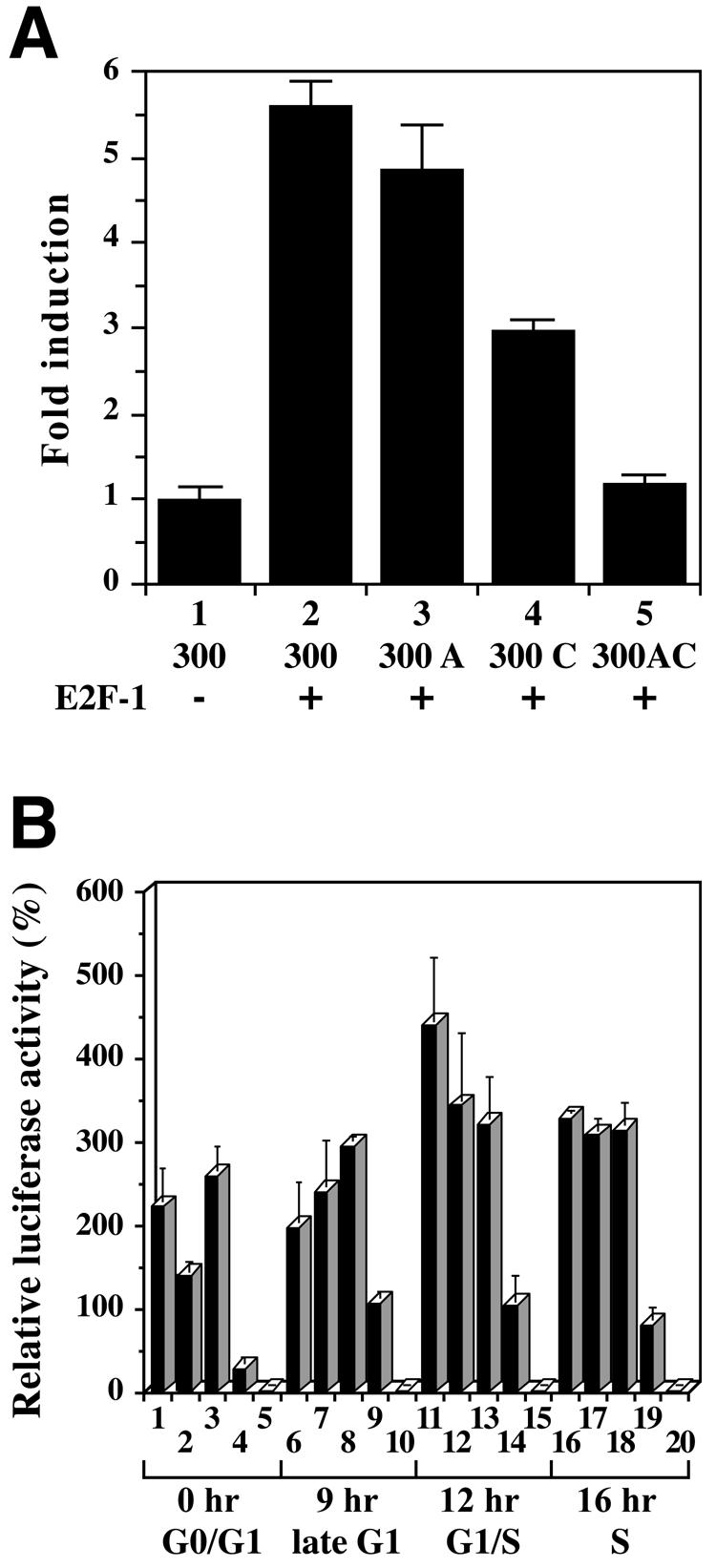
Promoter activity of two cis-elements enhanced by E2F-1 during the cell cycle. (A) Effect of E2F-1 on the activity of the Dnmt1 core promoter region in NIH 3T3 cells co-transfected with the E2F-1 expression vector pDCE2F-1 and the Dnmt1 promoter constructs. NIH 3T3 cells were co-transfected with either the 300-luc construct (300), the construct with mutated site A (300 A), the construct with mutated site C (300 C) or a construct with mutated sites A and C (300 AC). The ratio of luciferase reporter plasmid, E2F-1 expression vector and pRL-SV internal control vector is 10:10:1. Luciferase activity was determined as described in Figure 3A. Numbers shown are fold induction relative to luciferase activity of 300-luc in wild-type, parental 3Y1 cells. (B) Activity of intact and mutated Dnmt1 promoters at various stages in the cell cycle. NIH 3T3 cells were transfected with the 300-luc (columns 1, 6, 11 and 16), 300 A-luc (columns 2, 7, 12 and 17), 300 C-luc (columns 3, 8, 13 and 18), 300 AC-luc (columns 4, 9, 14 and 19) and No insert-luc (columns 5, 10, 15 and 20). After 24 h, the cells were arrested by serum starvation, and 36 h later were stimulated by replacement with complete medium. The cells were harvested at the indicated times after serum stimulation for luciferase activity analysis. The luciferase activities are normalized to Renilla luciferase activity. Numbers indicated are luciferase activity relative to the No insert-luc at each time point.
Distinct roles of two cis-elements during the cell cycle
To examine which cis-element contributes to the regulation of transcriptional activity of Dnmt1 during the cell cycle, we transfected NIH 3T3 cells with luciferase reporter plasmids containing the wild-type Dnmt1 core promoter or the promoter containing point mutations in site A and site C. Luciferase activities were measured in quiescent cells by serum starvation (G0/G1 phase) and 9 (late-G1 phase), 12 (G1/S phase) and 16 h (S phase) after serum stimulation of these cells (Fig. 6B, cf. Fig. 1B). Overall, plasmids in which site C remained intact showed cell cycle-dependent variation in luciferase activity, whereas plasmids with mutated site C showed far less variation.
In G0/G1 phase, luciferase activity was detectable, illustrating that the Dnmt1 promoter had basal Dnmt1 promoter activity in resting cells (Fig. 6B, column 1), consistent with previous reports (9,40). The plasmids with mutated site A had decreased luciferase activity whereas those with mutated site C showed increased activity (Fig. 6B, columns 2 and 3). These results suggest that transcriptional activity of Dnmt1 is repressed by the binding of E2F-Rb complex to site C, and that a nuclear factor which activates Dnmt1 expression through site A exists in G0/G1 phase cells.
In late G1 phase, transcriptional activity was repressed through the E2F binding site (site C) (Fig. 6B, compare columns 6 and 8). Unlike in G0/G1 phase cells, the plasmid with mutated site A showed no decrease in luciferase activity (Fig. 6B, columns 2 and 7). The site A-binding nuclear factors may act differently between G0/G1 and mid/late-G1 phase. Dnmt1 promoter activity peaked in G1/S phase (Fig. 6B, column 11), and declined slightly at S phase (Fig. 6B, column 16).
Promoter activity of reporter plasmids with the mutation at site C did not vary during the cell cycle (Fig. 6B, columns 3, 8, 13 and 18), whereas in each phase transcription from the double mutant plasmids was lower than transcription in the wild type (Fig. 6B, columns 4, 9, 14 and 19). These data suggest that these two cis-elements play distinct and important roles in the regulation of Dnmt1 expression. During the cell cycle, basal transcriptional levels of Dnmt1 appear to be maintained through site A while site C (E2F site) modulates Dnmt1 transcriptional levels both positively and negatively in a cell cycle-specific manner.
TSA activates the Dnmt1 promoter
Since it has been shown that E2F-Rb complexes can repress transcription by recruiting HDAC (25), we examined expression of Dnmt1 in proliferating NIH 3T3 cells after treatment with TSA, a HDAC-specific inhibitor. The amount of Dnmt1 mRNA was reduced in TSA-treated cells (Fig. 7A). TSA can also activate the promoter activity of p21Waf1, a cdk inhibitor (41) which arrests cells at G1 or G2 phase (42). We confirmed an increase in p21Waf1 protein level that occurs in a dose-dependent manner in response to TSA treatment (Fig. 7B). Dnmt1 protein level at 50 nM TSA was decreased, in agreement with measured mRNA levels.
Figure 7.

HDAC-dependent repression of Dnmt1 transcription. (A) Northern blot showing expression of Dnmt1 mRNA in NIH 3T3 cells 24 h after addition of TSA at the indicated concentrations. Ten micrograms of total RNA were analyzed. (B) Western blot analysis reveals protein levels in NIH 3T3 cells treated with TSA. Twenty micrograms of WCE were subjected to western blot analysis with the indicated antibodies. (C) Cell cycle distribution of NIH 3T3 cells 24 h after addition of TSA at indicated concentrations. DNA content was measured by flow cytometry. (D) Effect of TSA on the activity of the Dnmt1 core promoter region at G0/G1 phase. NIH 3T3 cells were transfected with 300-luc (columns 1 and 6), 300 A-luc (columns 2 and 7), 300 C-luc (columns 3 and 8), 300 AC-luc (columns 4 and 9) or No insert-luc (columns 5 and 10). Twenty-four hours later, cells were growth-arrested by serum starvation for 36 h. Cells were subsequently treated with or without TSA (200 nM) for 24 h before luciferase activity was determined. Luciferase activity is normalized to Renilla luciferase activity and is shown relative to No insert-luc transfections (columns 5 and 10). (E) Effect of TSA on growth-arrested cells. Prior to flow cytometry, NIH 3T3 cells were serum starved for 36 h, and then treated with (+TSA) or without (–TSA) 200 nM TSA for 24 h. Asyn, asynchronized NIH 3T3 cells.
To investigate the cell cycle distribution of cells after addition of TSA, we measured the DNA content by FACScan analysis (Fig. 7C). A low dose of TSA (50 nM) arrested the cells in G1 or G2 phase, confirming that Dnmt1 mRNA declined in G1 and G2 phases. However, the S phase distribution of the cells treated with higher doses of TSA increased slightly (Fig. 7C), and Dnmt1 mRNA levels in TSA-treated cells appeared to increase in a TSA dose-dependent fashion (Fig. 7A).
It is known that supercoiled plasmid DNA introduced into mammalian cell lines or Xenopus oocytes forms nucleosome-containing minichromosomes in the nucleus after 16 h (43,44). We examined Dnmt1 promoter activity in cells at G0/G1 phase by luciferase assay to see if there is any HDAC-dependent repression. NIH 3T3 cells transfected with luciferase reporter plasmids were arrested at G0/G1 and treated with 200 nM TSA for 24 h. In all cases, the luciferase activity was higher in TSA-treated cells than in untreated cells (Fig. 7D). The luciferase activity of plasmids with a mutation in site A increased to a greater extent than that of the control plasmids (Fig. 7D, columns 6 and 7), implying that the binding of E2F to site C resulted in the formation of E2F-Rb-HDAC complex. When 200 nM TSA was added to serum-starved NIH 3T3 cells, the distribution of the cell cycle was unchanged (Fig. 7E). The luciferase activity of the plasmids with the mutated E2F site (site C) increased 3-fold after TSA treatment (Fig. 7D, columns 3 and 8), suggesting that the site A-binding complex possesses some HDAC activity. The luciferase activity of plasmids with a mutation in site A is dependent upon promoter activity of site C. Plasmid 300A showed a 7-fold increase in luciferase activity following TSA treatment in G0/G1 arrested cells (Fig. 7D, columns 2 and 7, and 7E). As expected, the double mutated promoter activity did not increase significantly when TSA was added (Fig. 7D, columns 4 and 9). These data indicate that the Dnmt1 core promoter is regulated through dual pathways dependent upon sites A and C, and these sites are associated with HDAC activity.
Dnmt1 promoter activity is negatively regulated by E2F-Rb-HDAC
To investigate whether negative regulation of Dnmt1 promoter activity is associated with the formation of E2F-Rb-HDAC complexes, we co-transfected luciferase reporter plasmids and E2F-1 and Rb expression vectors into Saos-2 osteosarcoma cells which lack functional Rb and express E2F target genes at high levels (45). TSA was added to the cells 24 h after transfection, and the cells were harvested after 24 h in culture (Fig. 8). As expected, the core promoter activity of Dnmt1 when co-transfected with the E2F-1 expression vector increased by 50% (Fig. 8, column 2); however, co-transfection with E2F-1 and Rb expression vectors decreased luciferase activity by 50% as compared to co-transfection with E2F-1 expression vector alone (Fig. 8, columns 2 and 3). These results indicate that the formation of an E2F-1-Rb complex repressed the Dnmt1 core promoter activity. The decrease in luciferase activity caused by Rb expression was reversed by addition of 100 or 200 nM TSA (Fig. 8, columns 4 and 5). We performed luciferase assays with the reporter plasmids containing a mutation in site A and obtained similar results (Fig. 8, columns 6–10). Our data indicate that repression of Dnmt1 core promoter activity by E2F-1-Rb complexes is dependent upon HDAC and occurs through site C. Taken together, these findings show that the Dnmt1 promoter activity is both positively and negatively regulated by E2F complexes.
Figure 8.

The E2F-Rb-HDAC complex decreases the promoter activity of mouse Dnmt1. Saos-2 cells were co-transfected with the plasmids indicated in the figure. The ratio of luciferase reporter plasmid, E2F-1 expression vector, Rb expression vector and pRL-SV40 internal control vector was 10:10:60:1. TSA was added 24 h after transfection. TSA (+) and TSA (++) indicate treatment with concentrations of 100 and 200 nM, respectively. Luciferase activity was determined 48 h post-transfection. The luciferase counts were normalized to Renilla luciferase activity. The standard luciferase activity (100%) was defined as that of cells transfected with the 300-luc and empty expression vectors (column 1).
DISCUSSION
In the present study, we demonstrate that there are two major cis-elements in the promoter region of mouse Dnmt1 that control transcriptional levels during the cell cycle. Dual regulatory pathways, one involving unknown transcription factors and the other involving E2F-Rb-HDAC, play a pivotal role in cell cycle-dependent regulation of Dnmt1 expression. Both pathways may be necessary for controlling gene expression by methylation; however, further investigation of growth-sensitive methylation checkpoints will be required in both normal cell growth and tumor development.
It has been thought that DNA methylation in vivo is maintained by activation of Dnmt1, whereas demethylation is achieved passively by inactivation of Dnmt1. However, over-expression of Dnmt1 has been reported in several cancer cells (46,47), and an intimate relationship between aberrant methylation of genomic DNA and tumorigenesis has been documented (16,17). To clarify how cells produce the appropriate amount of Dnmt1, we investigated the regulation of Dnmt1 expression. First, we identified the promoter region and the major transcript start sites of mouse Dnmt1. Expression of Dnmt1 was regulated through at least two distinct cis-elements (sites A and C). Site C binds E2F and site A binds factors yet to be identified. Our data show that the E2F binding site regulates transcriptional activity of Dnmt1 both positively and negatively in a cell cycle-dependent manner. A model of this complex regulation is proposed in Figure 9. The E2F binding site activates transcription at late G1/S phase and represses it to basal levels at G0/G1 phase by recruiting HDAC through the formation of E2F-Rb-HDAC complexes (Fig. 9). Since Dnmt1 transcription occurs in resting cells, it appears that Dnmt1 functions outside of the cell cycle in quiescent and terminally differentiated cells.
Figure 9.

A proposed model for the regulatory mechanisms of mouse Dnmt1 transcription. At all stages of the cell cycle, there is basal level expression of Dnmt1 driven by site A. The E2F site alters transcription by different means, depending on the phase of the cell cycle. At G0/G1 phase Rb can bind to E2F, recruiting HDAC to the complex, resulting in repression of transcription of Dnmt1. A transcription factor bound to site A (TFBA) maintains low transcriptional levels of Dnmt1. At G1/S phase, the inhibition of Dnmt1 transcription mediated by the E2F site is released, and E2F and TFBA now work cooperatively to increase the expression levels in a regulated manner.
Loss of E2F-1, E2F-2, E2F-3, E2F-4 and E2F-5 gene function results in a diversity of phenotypes (39,48–51). In particular, mice lacking E2F-1 develop tumors (39), implying that E2F-1 functions as a tumor suppressor in vivo. Additionally, the Rb-E2F pathway is responsible for control of cell growth (33). Our data suggest that disruption of the E2F-1-Rb-HDAC regulatory complex formed on the Dnmt1 promoter leads to ectopic expression of Dnmt1, potentially contributing to tumor progression. The different patterns of methylation observed in cancer cells suggest that this E2F-Dnmt1 pathway might accelerate aberrant methylation in tumor malignancy. Rb-E2F pathways may be involved in Dnmt1-associated tumorigenesis at multiple levels. Recently, mice have been made with disruptions in Rb family genes, eliminating the possibility for compensation (52,53). Embryonic fibroblasts derived from these mice are immortal and exhibit properties of transformed cells (52,53). Colony formation was completely inhibited by reintroduction of Rb into these cells (52). Considering these results, our findings strongly suggest that disruption of the Rb-E2F-Dnmt1 regulatory pathway may play a critical role in tumorigenesis.
Robertson et al. have reported that Dnmt1 directly interacts with Rb, thereby reducing transcriptional activity of E2F-responsive genes via histone deacetylation (54). Dnmt1 also interacts directly with E2F-1 and HDAC (11,55), implying that Dnmt1 works not only through maintenance of genome-wide methylation, but also through histone deacetylation. Indeed, recent studies have shown that the inhibition of Dnmt1 results in enhanced promoter activity of tumor suppressor genes such as p16Ink4A, p19Arf and p21Waf1 (54,56,57). In the case of p21Waf1, although its promoter activity is elevated by decreases in Dnmt1, the CpG sequences in the promoter region are completely unmethylated even though Dnmt1 is expressed normally (57). This suggests that Dnmt1 itself may participate in a transcriptional regulatory mechanism in addition to its role in maintenance methylation. Since Dnmt1 is E2F-responsive, Dnmt1 may regulate its own transcription through a negative feedback system, through the formation of complexes such as E2F-Rb-Dnmt1-HDAC. Ectopic expression of Dnmt1 in higher levels than levels in transformed cells leads to cell death (6), while most transformed cells express apporoximately 2-fold more Dnmt1 mRNA than normal cells (6,18). Therefore, Dnmt1 may be required as part of a restrictive regulatory system to continue normal cell growth.
Inappropriate levels of Dnmt1 may trigger tumor initiation or progression at multiple steps by disturbing tightly regulated molecular interactions. In SV40-transformed cells, increased levels of Dnmt1 interfere with the molecular interaction between p21Waf1 and PCNA (12), because the binding domain of PCNA for p21Waf1 is the same as that for Dnmt1. Thus, increased Dnmt1 may enhance tumor progression by inappropriate interaction with cell cycle- or DNA repair-related molecules such as Rb (54) and PCNA (12). Importantly, down-regulation of Dnmt1 decreases or arrests cell growth (56,58). Dnmt1-deficient ES cells grow normally, but they die when induced to differentiate (6). Furthermore, Min mice crossed with Dnmt1S/+ mice, which completely lack Dnmt1 activity on one allele, showed 50% fewer intestinal adenomas without large-scale demethylation of genomic DNA (19). A recent study has reported that Min mice crossed to Dnmt1N/+ mice, which partially retain Dnmt1 activity on one allele, also show fewer polyps (59). The mutant Dnmt1N/N ES cells have slightly reduced levels of Dnmt1 enzymatic activity relative to the parental ES cells, yet they retain nearly normal genomic CpG methylation levels (60). Although both monoallelic expression of Dnmt1 and slightly reduced levels of Dnmt1 activity seem to be sufficient for the maintenance of genome-wide methylation and cell viability, these subtle differences affect tumor development significantly. These reports strongly suggest that maintenance of absolute levels of Dnmt1 is critical for both normal and abnormal cell growth. Both methyltransferase activity of Dnmt1 and its ability to interact with other molecules may play important roles in cell proliferation.
Although site A of Dnmt1 resembles a putative E2F binding sequence, the molecule bound to site A appears to be quite different from E2F transcription factors. Site A mainly functions as a positive regulator of Dnmt1 during the cell cycle. The coordination of the two regulatory sites controls basal and functional activation of the Dnmt1 promoter in proliferating cells (Fig. 9). The transcriptional activity mediated by site A can explain the constant transcriptional rate in G0 phase cells and differentiated cells, compared to proliferating cells (9,29,40). The sequence of the mouse site A is not completely conserved in rat, but proteins derived from rat act on mouse site A (Fig. 4A and B). These findings indicate that the molecule bound to site A recognizes a longer sequence motif than the putative E2F binding site. Further study is required to identify the molecule that binds site A, and to understand the complex molecular mechanisms of Dnmt1 expression regulation in cell growth.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs J. Rossant, M. Yoshida and T. Yamamoto for reagents and Drs K. Ohtani, H. Yamaguchi and K. Imakawa for reagents and helpful advice on EMSA. We also thank Dr Jared Ordway for helpful comments and appreciate proofreading by David Benhayon. This work was supported by the Program for Promotion of Basic Research Activities for Innovative Biosciences (PROBRAIN) (K.S.) and the Japan Society for the Promotion of Science (H.K.).
DDBJ/EMBL/GenBank accession nos AB056445, AB056446
REFERENCES
- 1.Razin A. and Szyf,M. (1984) DNA methylation patterns. Formation and function. Biochim. Biophys. Acta, 782, 331–342. [DOI] [PubMed] [Google Scholar]
- 2.Ohgane J., Wakayama,T., Kogo,Y., Senda,S., Hattori,N., Tanaka,S., Yanagimachi,R. and Shiota,K. (2001) DNA methylation variation in cloned mice. Genesis, 30, 45–50. [DOI] [PubMed] [Google Scholar]
- 3.Cho J.H., Kimura,H., Minami,T., Ohgane,J., Hattori,N., Tanaka,S. and Shiota,K. (2001) DNA methylation regulates placental lactogen I gene expression. Endocrinology, 142, 3389–3396. [DOI] [PubMed] [Google Scholar]
- 4.Shiota K., Kogo,Y., Ohgane,J., Imamura,T., Urano,A., Nishino,K., Tanaka,S. and Hattori,N. (2002) Epigenetic marks by DNA methylation specific to stem, germ and somatic cells in mice. Genes Cells, 7, 961–969. [DOI] [PubMed] [Google Scholar]
- 5.Bird A.P. and Wolffe,A.P. (1999) Methylation-induced repression—belts, braces and chromatin. Cell, 99, 451–454. [DOI] [PubMed] [Google Scholar]
- 6.Li E., Bestor,T.H. and Jaenisch,R. (1992) Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell, 69, 915–926. [DOI] [PubMed] [Google Scholar]
- 7.Okano M., Bell,D.W., Haber,D.A. and Li,E. (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell, 99, 247–257. [DOI] [PubMed] [Google Scholar]
- 8.Okano M., Xie,S. and Li,E. (1998) Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nature Genet., 19, 219–220. [DOI] [PubMed] [Google Scholar]
- 9.Szyf M., Bozovic,V. and Tanigawa,G. (1991) Growth regulation of mouse DNA methyltransferase gene expression. J. Biol. Chem., 266, 10027–10030. [PubMed] [Google Scholar]
- 10.Leonhardt H., Page,A.W., Weier,H.U. and Bestor,T.H. (1992) A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell, 71, 865–873. [DOI] [PubMed] [Google Scholar]
- 11.Rountree M.R., Bachman,K.E. and Baylin,S.B. (2000) DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nature Genet., 25, 269–277. [DOI] [PubMed] [Google Scholar]
- 12.Chuang L.S., Ian,H.I., Koh,T.W., Ng,H.H., Xu,G. and Li,B.F. (1997) Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science, 277, 1996–2000. [DOI] [PubMed] [Google Scholar]
- 13.Kimura H. and Shiota,K. (2003) Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J. Biol. Chem., 278, 4806–4812. [DOI] [PubMed] [Google Scholar]
- 14.Baylin S.B. and Herman,J.G. (2000) DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet., 16, 168–174. [DOI] [PubMed] [Google Scholar]
- 15.Baylin S.B., Herman,J.G., Graff,J.R., Vertino,P.M. and Issa,J.P. (1998) Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv. Cancer Res., 72, 141–196. [PubMed] [Google Scholar]
- 16.Baylin S.B., Esteller,M., Rountree,M.R., Bachman,K.E., Schuebel,K. and Herman,J.G. (2001) Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum. Mol. Genet., 10, 687–692. [DOI] [PubMed] [Google Scholar]
- 17.Wu J., Issa,J.P., Herman,J., Bassett,D.,Jr, Nelkin,B.D. and Baylin,S.B. (1993) Expression of an exogenous eukaryotic DNA methyltransferase gene induces transformation of NIH 3T3 cells. Proc. Natl Acad. Sci. USA, 90, 8891–8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bakin A.V. and Curran,T. (1999) Role of DNA 5-methylcytosine transferase in cell transformation by fos. Science, 283, 387–390. [DOI] [PubMed] [Google Scholar]
- 19.Laird P.W., Jackson-Grusby,L., Fazeli,A., Dickinson,S.L., Jung,W.E., Li,E., Weinberg,R.A. and Jaenisch,R. (1995) Suppression of intestinal neoplasia by DNA hypomethylation. Cell, 81, 197–205. [DOI] [PubMed] [Google Scholar]
- 20.Kruijer W., Cooper,J.A., Hunter,T. and Verma,I.M. (1984) Platelet-derived growth factor induces rapid but transient expression of the c-fos gene and protein. Nature, 312, 711–716. [DOI] [PubMed] [Google Scholar]
- 21.Muller R., Bravo,R., Burckhardt,J. and Curran,T. (1984) Induction of c-fos gene and protein by growth factors precedes activation of c-myc. Nature, 312, 716–720. [DOI] [PubMed] [Google Scholar]
- 22.Bigey P., Ramchandani,S., Theberge,J., Araujo,F.D. and Szyf,M. (2000) Transcriptional regulation of the human DNA Methyltransferase (Dnmt1) gene. Gene, 242, 407–418. [DOI] [PubMed] [Google Scholar]
- 23.Lin S.Y., Black,A.R., Kostic,D., Pajovic,S., Hoover,C.N. and Azizkhan,J.C. (1996) Cell cycle-regulated association of E2F1 and Sp1 is related to their functional interaction. Mol. Cell. Biol., 16, 1668–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dyson N. (1998) The regulation of E2F by pRB-family proteins. Genes Dev., 12, 2245–2262. [DOI] [PubMed] [Google Scholar]
- 25.Brehm A. and Kouzarides,T. (1999) Retinoblastoma protein meets chromatin. Trends Biochem. Sci., 24, 142–145. [DOI] [PubMed] [Google Scholar]
- 26.Ohtsubo M., Theodoras,A.M., Schumacher,J., Roberts,J.M. and Pagano,M. (1995) Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol. Cell. Biol., 15, 2612–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson D.G., Cress,W.D., Jakoi,L. and Nevins,J.R. (1994) Oncogenic capacity of the E2F1 gene. Proc. Natl Acad. Sci. USA, 91, 12823–12827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo R.X., Postigo,A.A. and Dean,D.C. (1998) Rb interacts with histone deacetylase to repress transcription. Cell, 92, 463–473. [DOI] [PubMed] [Google Scholar]
- 29.Kimura H., Takeda,T., Tanaka,S., Ogawa,T. and Shiota,K. (1998) Expression of rat DNA (cytosine-5) methyltransferase (DNA MTase) in rodent trophoblast giant cells: molecular cloning and characterization of rat DNA MTase. Biochem. Biophys. Res. Commun., 253, 495–501. [DOI] [PubMed] [Google Scholar]
- 30.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 31.Mertineit C., Yoder,J.A., Taketo,T., Laird,D.W., Trasler,J.M. and Bestor,T.H. (1998) Sex-specific exons control DNA methyltransferase in mammalian germ cells. Development, 125, 889–897. [DOI] [PubMed] [Google Scholar]
- 32.Yoshida M., Kijima,M., Akita,M. and Beppu,T. (1990) Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem., 265, 17174–17179. [PubMed] [Google Scholar]
- 33.Sherr C.J. (1996) Cancer cell cycles. Science, 274, 1672–1677. [DOI] [PubMed] [Google Scholar]
- 34.Smale S.T. and Baltimore,D. (1989) The “initiator” as a transcription control element. Cell, 57, 103–113. [DOI] [PubMed] [Google Scholar]
- 35.Javahery R., Khachi,A., Lo,K., Zenzie-Gregory,B. and Smale,S.T. (1994) DNA sequence requirements for transcriptional initiator activity in mammalian cells. Mol. Cell. Biol., 14, 116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ikeda M.A. and Nevins,J.R. (1993) Identification of distinct roles for separate E1A domains in disruption of E2F complexes. Mol. Cell. Biol., 13, 7029–7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kishikawa S., Murata,T., Kimura,H., Shiota,K. and Yokoyama,K.K. (2002) Regulation of transcription of the Dnmt1 gene by Sp1 and Sp3 zinc finger proteins. Eur. J. Biochem., 269, 2961–2970. [DOI] [PubMed] [Google Scholar]
- 38.Takahashi Y., Rayman,J.B. and Dynlacht,B.D. (2000) Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev., 14, 804–816. [PMC free article] [PubMed] [Google Scholar]
- 39.Yamasaki L., Jacks,T., Bronson,R., Goillot,E., Harlow,E. and Dyson,N.J. (1996) Tumor induction and tissue atrophy in mice lacking E2F-1. Cell, 85, 537–548. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y., Sun,L. and Jost,J.P. (1996) In differentiating mouse myoblasts DNA methyltransferase is posttranscriptionally and posttranslationally regulated. Nucleic Acids Res., 24, 2718–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiao H., Hasegawa,T. and Isobe,K. (1999) Both Sp1 and Sp3 are responsible for p21waf1 promoter activity induced by histone deacetylase inhibitor in NIH3T3 cells. J. Cell. Biochem., 73, 291–302. [PubMed] [Google Scholar]
- 42.Harper J.W., Adami,G.R., Wei,N., Keyomarsi,K. and Elledge,S.J. (1993) The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell, 75, 805–816. [DOI] [PubMed] [Google Scholar]
- 43.Reeves R., Gorman,C.M. and Howard,B. (1985) Minichromosome assembly of non-integrated plasmid DNA transfected into mammalian cells. Nucleic Acids Res., 13, 3599–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kass S.U., Landsberger,N. and Wolffe,A.P. (1997) DNA methylation directs a time-dependent repression of transcription initiation. Curr. Biol., 7, 157–165. [DOI] [PubMed] [Google Scholar]
- 45.Horton L.E., Qian,Y. and Templeton,D.J. (1995) G1 cyclins control the retinoblastoma gene product growth regulation activity via upstream mechanisms. Cell Growth Differ., 6, 395–407. [PubMed] [Google Scholar]
- 46.el-Deiry W.S., Nelkin,B.D., Celano,P., Yen,R.W., Falco,J.P., Hamilton,S.R. and Baylin,S.B. (1991) High expression of the DNA methyltransferase gene characterizes human neoplastic cells and progression stages of colon cancer. Proc. Natl Acad. Sci. USA, 88, 3470–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robertson K.D., Uzvolgyi,E., Liang,G., Talmadge,C., Sumegi,J., Gonzales,F.A. and Jones,P.A. (1999) The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res., 27, 2291–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu J.W., Field,S.J., Gore,L., Thompson,M., Yang,H., Fujiwara,Y., Cardiff,R.D., Greenberg,M., Orkin,S.H. and DeGregori,J. (2001) E2F1 and E2F2 determine thresholds for antigen-induced T-cell proliferation and suppress tumorigenesis. Mol. Cell. Biol., 21, 8547–8564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Humbert P.O., Verona,R., Trimarchi,J.M., Rogers,C., Dandapani,S. and Lees,J.A. (2000) E2f3 is critical for normal cellular proliferation. Genes Dev., 14, 690–703. [PMC free article] [PubMed] [Google Scholar]
- 50.Humbert P.O., Rogers,C., Ganiatsas,S., Landsberg,R.L., Trimarchi,J.M., Dandapani,S., Brugnara,C., Erdman,S., Schrenzel,M., Bronson,R.T. and Lees,J.A. (2000) E2F4 is essential for normal erythrocyte maturation and neonatal viability. Mol. Cell, 6, 281–291. [DOI] [PubMed] [Google Scholar]
- 51.Lindeman G.J., Dagnino,L., Gaubatz,S., Xu,Y., Bronson,R.T., Warren,H.B. and Livingston,D.M. (1998) A specific, nonproliferative role for E2F-5 in choroid plexus function revealed by gene targeting. Genes Dev., 12, 1092–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sage J., Mulligan,G.J., Attardi,L.D., Miller,A., Chen,S., Williams,B., Theodorou,E. and Jacks,T. (2000) Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev., 14, 3037–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dannenberg J.H., van Rossum,A., Schuijff,L. and te Riele,H. (2000) Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev., 14, 3051–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robertson K.D., Ait-Si-Ali,S., Yokochi,T., Wade,P.A., Jones,P.L. and Wolffe,A.P. (2000) DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nature Genet., 25, 338–342. [DOI] [PubMed] [Google Scholar]
- 55.Fuks F., Burgers,W.A., Brehm,A., Hughes-Davies,L. and Kouzarides,T. (2000) DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nature Genet., 24, 88–91. [DOI] [PubMed] [Google Scholar]
- 56.Fournel M., Sapieha,P., Beaulieu,N., Besterman,J.M. and MacLeod,A.R. (1999) Down-regulation of human DNA-(cytosine-5) methyltransferase induces cell cycle regulators p16ink4A and p21WAF/Cip1 by distinct mechanisms. J. Biol. Chem., 274, 24250–24256. [DOI] [PubMed] [Google Scholar]
- 57.Milutinovic S., Knox,J.D. and Szyf,M. (2000) DNA methyltransferase inhibition induces the transcription of the tumor suppressor p21WAF1/CIP1/sdi1. J. Biol. Chem., 275, 6353–6359. [DOI] [PubMed] [Google Scholar]
- 58.Young J.I. and Smith,J.R. (2001) DNA methyltransferase inhibition in normal human fibroblasts induces a p21-dependent cell cycle withdrawal. J. Biol. Chem., 276, 19610–19616. [DOI] [PubMed] [Google Scholar]
- 59.Cormier R.T. and Dove,W.F. (2000) Dnmt1N/+ reduces the net growth rate and multiplicity of intestinal adenomas in C57BL/6-multiple intestinal neoplasia (Min)/+ mice independently of p53 but demonstrates strong synergy with the modifier of Min 1AKR resistance allele. Cancer Res., 60, 3965–3970. [PubMed] [Google Scholar]
- 60.Lei H., Oh,S.P., Okano,M., Juttermann,R., Goss,K.A., Jaenisch,R. and Li,E. (1996) De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development, 122, 3195–3205. [DOI] [PubMed] [Google Scholar]