


Abstract
Complementary DNA encoding a protein, designated Cc RNase, was isolated from the insect Ceratitis capitata. Deduced amino acid sequence analysis demonstrates that the Cc RNase has strong sequence homology with other uncharacterized proteins predicted from EST sequences belonging to different animal species, therefore defining a new protein family, which is conserved from Caenorhabditis elegans to humans. Phylogenetic analysis data in addition to extensive homolog searches in all available complete genomes suggested that all family members are true orthologs. Proteins belonging to this family are composed of 95–101 amino acids. The C.capitata orthologous protein was expressed in Escherichia coli. Despite the fact that the amino acid sequence of Cc RNase does not share any significant similarities with other known ribonucleases, our data give strong evidence in support of the assignment of enzymatic activity to the recombinant protein. The expressed molecule exhibits ribonucleolytic activity against poly(C) and poly(U) synthetic substrates, as well as rRNA. It is also demonstrated that expression of Cc RNase in E.coli inhibits growth of the host cells.
INTRODUCTION
One of the major, but poorly understood, aspects of gene regulation is the control of RNA turnover. In eukaryotes, our knowledge of the way different species of RNA are degraded is based on studies defining cis-acting sequences that function as either regulatory or cleavage targets (1,2). Along these lines, little is known about the identity of RNA-degrading enzymes.
Ribonucleases (RNases) constitute a large superfamily of enzymes demostrating different molecular and enzymatic properties. A number of them, such as RNase A, have been widely studied and the results obtained have successfully contributed to the understanding of their stability, structure and function (3). In addition to their catalytic activity, other members of this superfamily have been shown to possess important biological actions, including neurotoxicity and immunosupressivity, as well as angiogenic and antitumor activity (4–6). RNases also serve as cytotoxic agents, mediating host defense in higher plants and mammals, and their potential use as therapeutic agents has recently been suggested (7). The number of proteins shown to exhibit RNase activity has greatly expanded in recent years in light of the discovery of new classes of RNases involved in cell metabolism and gene regulation (8). RNase protein sequences from many species are known and their molecular evolution has been investigated. RNases are thought to belong to the oldest enzymes, so it is not surprising that they are involved in such a wide spectrum of biological functions (9).
Although RNases have been excessively studied at the biochemical and molecular levels in mammals, amphibians, fungi and bacteria, less is known about insect derived RNases. Drosophila RNase H1 is the best-studied insect RNase (10) and genetic experiments have shown that it is essential for metamorphosis in Drosophila (11). Three different RNA-degrading enzymes have been isolated from the insect Ceratitis capitata. Two of these, purified by the Gavilanes group, have been characterized as alkaline (12) and acidic RNases (13), respectively. A third poly(U) and poly(C)-specific RNase has been isolated and characterized in our laboratory (14,15). Using a polyclonal antibody against this enzyme, a cDNA clone encoding for a new RNase (named Cc RNase) was isolated by immunoscreening a cDNA library prepared from 6-day-old larvae of the insect C.capitata (16). In this report, we present the successful expression of active Cc RNase in Escherichia coli, despite a cytotoxic effect, as well as a phylogenetic study of this protein. Our data strongly support the idea that Cc RNase belongs to a novel highly conserved protein family, which includes members from many metazoan phyla.
MATERIALS AND METHODS
Animals
The insect C.capitata was grown under controlled conditions of temperature and humidity as described previously by Sideris and Fragoulis (14). Six-day-old larvae were collected and stored in liquid nitrogen prior to use.
Materials
Oligonucleotides were custom synthesized by MWG Biotec (Ebersberg, Germany). Restriction enzymes and T4 DNA ligase were purchased from New England Biolabs. The DyNAZyme™ II DNA polymerase was obtained from Finnzymes Oy (Espoo, Finland). Plasmid preparation kits were from Qiagen. All expression vectors, bacterial strains and His-Bind resin were from Novagen. HEPES, poly(A), poly(U), poly(C), poly(G) and poly(I) were purchased from Serva (Heidelberg, Germany). Ultrapure urea was obtained from ICN Biomedicals and dithiothreitol (DTT) was purchased from Sigma Chemical Co. Marker proteins for molecular weight estimations in SDS–PAGE were from Pharmacia (Uppsala, Sweden). All other reagents used were of analytical grade and obtained from Merck (Darmstadt, Germany).
Isolation of a full-length cDNA clone encoding Cc RNase
Total RNA was prepared from 6-day-old larvae of the insect C.capitata as described by Bouhin et al. (17) and poly(A)+ RNA was isolated using Dynabeads® oligo(dT)25 (Dynal). One microgram of poly(A)+ RNA was reverse transcribed using the SMART PCR cDNA synthesis kit (Clontech Laboratories) as specified in the manufacturer’s manual. In order to isolate a unique transcript encoding for Cc RNase from totally synthesized single-stranded cDNAs, a modified version of the hybrid selection technique (18) was employed. A 5′-biotinylated cDNA was synthesized by PCR using the incomplete cDNA clone (R1) isolated previously in our laboratory (16) as template. The biotinylated strand of PCR product was attached to Dynabeads® M-280 Streptavidin (Dynal) as recommended by the manufacturer. Hybridization of the biotinylated probe to the single-stranded cDNA was performed at 63°C, overnight, in 3× SSC, 0.1% SDS, 1.25× Denhardt’s. Following hybridization, the beads were washed three times in 2× SSC, 0.1% SDS at 60°C for 10 min, twice in TE buffer at room temperature and finally resuspended in distilled H2O. The selected hybrids were amplified by PCR using the LD primer (5′-AAG CAG TGG TAA CAA CGC AGA GT-3′), which anneals at the 5′ and 3′ ends of the cDNA hybrids. The PCR products were cloned into the pCR™ 2.1 cloning vector (Invitrogen) and sequenced in both directions.
Identification of protein sequences and phylogenetic analysis
The dbEST (Expessed Sequence Tags) (19) database on the National Center for Biotechnology Information World Wide Web server was searched by the TBLASTN software (20) using the deduced amino acid sequence of Cc RNase (accession no. AJ441124) as query. Accession numbers of ESTs retrieved as above are presented in Figure 1. Multiple sequence alignment and production of a phylogenetic tree was performed by the Clustal X 1.8 program (21) using default parameters. The phylogenetic tree of the encoded amino acid sequences was reconstructed by the neighbor joining method (22). One thousand bootstrapping replications (23) were conducted to assess the reliability of the reconstructed tree. The njplot program (24) was used to draw the reconstructed tree.
Figure 1.

(A) Nucleotide and deduced amino acid sequence of Cc RNase cDNA. Numbers refer to nucleotide and amino acid positions. The putative polyadenylation signal is underlined. (B) Alignment of Cc RNase amino acid sequence with the predicted homologous proteins of other animal species. Alignment was performed by the Clustal X 1.8 program using default parameters. Black background, identical; gray, conserved (upper case) or similar (lower case) amino acids in at least 60% of the sequence (MacBoxShade, default parameters).
Construction of the expression plasmids
Amplification of the open reading frame (ORF) of the Cc RNase gene was carried out using high fidelity DNA polymerase (Applied Biosystems) and primers specific to the 5′ (starting at the ATG initiator codon) and 3′ ends of the Cc RNase, based on the nucleotide sequence (accession no. AJ441124). The primers used were designed so that the amplified DNA would contain NcoI and XhoI restriction endonuclease sites at its 5′ and 3′ ends, respectively, in order to facilitate subsequent subcloning. The primer sequences were as follows: sense, 5′-CCA TGG AAA TTT GTG GTC CAA AAT TGT CG-3′; antisense, 5′-CTC GAG TAC GTC AAC GAA CCA ACG AAC-3′. The amplified cDNA fragment was first cloned into pCR™ 2.1 vector and the NcoI–XhoI DNA fragment was purified and subcloned into expression vector pSCREEN-1b(+), creating the PF construct. Using slightly modified sense primers containing different restriction sites, the amplified DNA was cloned into the pET 20b (NdeI–XhoI insert) or pET 37b (ScaI–XhoI insert) expression vectors, creating the Psa and Pcb constructs, respectively. All constructs were confirmed by DNA sequencing.
Bacterial growth assay
The effect of the expressed recombinant protein on E.coli growth was investigated, following the procedure described by Rosenberg (25). Overnight cultures of bacteria transformed with different expression plasmids harboring the Cc RNase gene (PF, Psa or Pcb) were diluted 1:50 in M9 medium supplemented with ampicillin (100 µg/ml) and chloramphenicol (34 µg/ml). Optical densities (600 nm) were recorded at t = 0 and hourly thereafter. When the exponential phase of growth was reached, cultures were split into two halves and isopropyl β-d-thiogalactopyranoside (IPTG) (final concentration 1 mM) was added to one half of the culture to induce the production of recombinant protein. Optical densities were recorded hourly thereafter. Each experiment was repeated four times.
Purification of expressed Cc RNase
Overnight culture of BL21(DE3)pLysS E.coli cells transformed with expression plasmid PF were diluted 1:50 in M9 medium supplemented with ampicillin (100 µg/ml) and chloramphenicol (34 µg/ml). Soon after the OD600 of the culture had reached 0.6, IPTG was added to 1 mM final concentration, and bacteria were harvested after a 4 h induction period.
For preparative purposes, cells from 0.5 l of culture were suspended in 10 ml of buffer A (50 mM Tris–acetate pH 8.4, containing 10 mM EDTA) and sonicated (6 × 1 min cycle). The suspension was then centrifuged at 17 000 g for 30 min at 4°C. The cell pellet was freed from membrane proteins by two washes in 0.1 M Tris–acetate pH 8.4, containing 10 mM EDTA, 2% Triton X-100 and 2 M urea, followed by repeated washes in 0.1 M Tris pH 8.4, containing 10 mM EDTA to eliminates traces of Triton and urea. Inclusion bodies were then dissolved in binding buffer (20 mM Tris–HCl pH 7.9, 0.5 M NaCl, 5 mM imidazole) and any insoluble material was removed by centrifugation. The expressed fusion protein was purified from the supernatant with His binding resin (Novagen) in the presence of 8 M urea, according to the manufacturer’s specifications. The fractions containing the purified protein were pooled and DTT was added to a final concentration of 0.1 M. After incubation at 37°C for 2 h, the fusion protein solution was dialyzed overnight against 20 mM Tris–HCl pH 8.0 and then digested with thrombin. The pure fragment of Cc RNase was isolated by chromatography on His binding resin. Renaturation of the recombinant RNase was achieved by dialyzing the pure fraction against 20 mM Tris–HCl pH 8.0, for at least 24 h at 4°C with three changes. Dialysis conditions for maximal renaturation of the recombinant protein was estimated by RNase activity assay.
Protein purity was assessed by SDS–PAGE according to Laemmli (26). Western blot analysis was performed using a polyclonal antibody, raised in rabbit, against the poly(U), poly(C)-specific RNase isolated from the insect C.capitata (15). Protein concentration was determined using a Bio-Rad (Richmond, CA) protein assay kit based on the Bradford dye binding assay (27). Calibration curves were constructed using bovine serum albumin as standard.
Assay for RNase activity
RNase activity of the recombinant protein against rRNA or various single-stranded polyribonucleotides was determined by the production of perchloric acid-soluble material according to the method of Slifman et al. (28), with slight modifications. The reaction was performed at 37°C in a total volume of 50 µl, containing 50 µg of the various substrates, in 20 mM Tris–HCl pH 8.0, and the appropriate amount of recombinant protein. The reaction was stopped by addition of 0.2 ml ice-cold 7% perchloric acid containing 0.1% uranyl acetate, and acid-soluble ribonucleotides remaining in the supernatant fraction after centrifugation were quantitated spectrophotometrically at 260 nm. The amount of enzyme used in the assays was selected such that the rate of hydrolysis would be linear in the time range examined.
Activity staining gels
Recombinant Cc RNase activity towards poly(C) was also assayed by activity staining gel (29). Purified RNase was analyzed at 4°C in an 11–15% SDS–polyacrylamide gel, containing 0.25 mg/ml poly(C) as substrate. Following electrophoresis, SDS was removed and the gel was incubated in a buffer of 0.1 M Tris–HCl pH 7.6 at 37°C for 3 h, to allow RNase-catalyzed hydrolysis of poly(C), and stained for 10 min with 0.2% toluidine blue. RNase activity was visualized by means of color disappearance in the substrate degradation positions.
Isolation of rRNA from rat liver
RNA was extracted from rat liver polysomes using the phenol chloroform method described by Perry et al. (30). The polysomal fraction was prepared according to Sideris and Fragoulis (14). The aqueous phase was made 0.1 M in sodium acetate and the RNA was precipitated with 2 vol of cold ethanol. The precipitated RNA was collected by centrifugation, dried with N2, resuspended in a small volume of 20 mM Tris–HCl, pH 7.6 containing 0.1 M NaCl and 1 mM EDTA and stored in aliquots at –80°C.
RESULTS
Isolation and characterization of a Cc RNase cDNA clone
The cDNA sequence encoding Cc RNase reported previously under accession no. Y15384 (16) was determined by a combination of two overlapping cDNA clones, namely R1 and R4. Clone R1 (nucleotides 81–873) was isolated by immunoscreening an expression cDNA library, while clone R4 (nucleotides 1–80) was obtained by means of 5′ RACE–PCR. In the work presented here a hybrid selection approach was employed in order to isolate the full-length cDNA clone encoding the Cc RNase. Poly(A)+ RNA from 6-day-old larvae was reverse transcribed and the single strand cDNA molecules were hybridized with a 5′-biotinylated probe synthesized by PCR using the incomplete cDNA clone R1 as template. The selected hybrids were amplified by PCR and cloned into the pCR™ 2.1 vector. Three cDNA clones were selected, and the authenticity of the clones was verified initially by restriction endonuclease mapping and finally by DNA sequencing. Sequencing results revealed no differences among these clones, although they appeared to differ slightly from the sequence data obtained previously (16). In fact, the observed differences concern, mainly, a number of nucleotide deletions, resulting in the appearance of two putative different open reading frames. The differences between these sequences could be attributed to errors in the nucleotide sequence determination of the R1 clone or to in vivo recombination events, since this clone resulted from a recombinant λ phage that expressed a toxic protein. The cDNA sequence data presented in this work were also confirmed by the nucleotide sequence obtained from a genomic DNA clone, which was synthesized by PCR, using specific primers corresponding to the 5′ and 3′ ends of this cDNA (data not shown). For the above reasons, we corrected the cDNA sequence of Cc RNase and resubmitted it to the EMBL Nucleotide Sequence Database, under accession no. AJ441124.
The isolated Cc RNase cDNA clone has a length of 864 bp and contains an ORF of 288 nt encoding a 95 amino acid polypeptide with a calculated molecular mass of 10 627 Da and pI of 4.02 (Fig. 1A). The first methionine codon is situated at position 149 of the cDNA clone and is the assigned initiation codon. The nucleotide sequence AAAATGA, surrounding the predicted initiator methionine, conforms perfectly to the consensus AAAATGN, which is the translation initiation sequence of many proteins in Drosophila melanogaster (31). Additionally, this ATG was identified as the initiation codon in all EST sequences examined in our phylogenetic analysis. The coding region is followed by a 3′-UTR of 392 nt and a prominent poly(A) tail. A putative polyadenylation signal AAUAUA was found within the 3′-UTR, located 19 nt upstream of the poly(A) tract.
Phylogenetic analysis of the Cc RNase gene
We used the deduced amino acid sequence of the Cc RNase cDNA as an initial probe in an interactive TBLASTN search of the available EST sequence databases using the default settings of the National Center for Biotechnology Information. This search resulted in the retrieval of a large number of EST sequences possessing a high level of similarity to Cc RNase. These tags derived from 19 different animal species, belonging to mammals, amphibians, fish, birds, insects and nematodes. All the retrieved ESTs belonged to genes encoding hypothetical proteins of 95–101 amino acid residues, whose function is still unknown. The human homolog of Cc RNase protein was represented by a large number of EST clones (approximately 200) prepared from various organs and tissues, indicating a wide distribution. It is important to note that a high proportion of these ESTs were derived from tumor and fetal tissues.
Multiple alignment of the deduced amino acid sequences revealed that the Cc RNase shows great similarity to Drosophila, with only 11 substituted amino acids (87.4% identity, 94.7% similarity), and is most divergent from the human counterpart (34.7% identity, 50.5% similarity). It also revealed that the proteins of this family are most conserved in their N- and C-terminus regions (amino acids 4–45 and 61–91, numbering according to C.capitata) (Fig. 1B). Hydropathy plots (32) indicated that these two conserved parts of the sequences contain a highly hydrophobic stretch. Searches in the PROSITE, interProscan and Pfam databases using the conserved regions of Cc RNase did not reveal any matches with known functional domains. All the members of the protein family contain between three and five cysteine residues, among which Cys4, Cys11 and Cys68 (numbering according to C.capitata) are conserved in all species compared.
In order to identify the human, Drosophila, mouse, Anopheles and Caenorhabditis elegans genes encoding the retrieved ESTs, a search in genome sequence databases was conducted. In all cases, the family is represented by a single gene, which contains three exons interrupted by two introns. Genomic DNA and cDNA sequence comparisons in parallel with consensus splice site identification determined exon– intron boundaries. In humans the gene is localized at chromosome 17, map position 17p13.2, corresponding to the LOC201233 gene locus. The finding that only one functional gene exists in all of the genomes analyzed, in combination with the high level of similarity that extends along the entire length of homologs, suggested that these proteins belong to a unique orthologous family (33). Extensive homolog searches in other complete genomes (plants, archaea, prokarya and fungi) did not result in the finding of any significant similarities.
Phylogenetic distances among amino acid sequences from multiple species were calculated and represented in a phylogenetic tree (Fig. 2). Based on sequence homology levels, this phylogenetic tree can be subdivided into four subfamilies, which include insects, mammals, nematodes and the chordates from other taxa (amphibians, fish, birds and urochordates). The phylogenetic tree presented here includes representatives of the two major subdivisions of the animal kingdom, protostomes and deuterostomes. The common ancestor of human, C.capitata and C.elegans predates the division of protostomes–deuterostomes (34). Consequently, the novel RNase described here was probably present in the earliest metazoan. We have not been able to identify prokaryotic or yeast genes that match our family of sequences. Therefore, it appears that although this protein was absent in primitive eukaryotes, it became established early in eukaryote evolution.
Figure 2.

A phylogenetic tree showing the relationships among the proteins of the Cc RNase family The phylogenetic tree was reconstructed by the neighbor joining method (28). The accession number following each species name refers to the EST sequence used in this study. Bootstrap values (>50%) are shown on interior branches. The scale bar beneath the tree represents 10 substitutions per 100 residues.
Expression of the Cc RNase in E.coli
The coding region of the Cc RNase cDNA was cloned into expression vectors pET-20b and pET-37b, creating plasmids Psa and Pcb, respectively. The target protein in the Psa construct was expressed bearing a C-terminus His tag, whereas in the Pcb construct it was expressed as a fusion protein containing the cellulose-binding domain. Both plasmids produced transformants in Nova blue host cells. On the other hand, no colonies were obtained in BL21(DE3) host cells. In order to suppress the basal expression of T7 RNA polymerase prior to induction, BL21(DE3) host cells carrying the pLysS or pLysE plasmid were used (35). In these strains both expression plasmids produced transformants, although a reduction in transformation efficiency was observed when compared to control vectors pET-20b and pET-37b. These observations led us to the conclusion that the expression plasmids Psa and Pcb produced toxic effects in E.coli, with a wide spectrum of severity. The inhibitory effect was also studied in IPTG-induced liquid cultures. As shown in Figure 3A and B, the uninduced control cultures of E.coli harboring the Pcb or Psa plasmids were seen to have normal growth, while cells in the IPTG-induced cultures stopped dividing. The use of BL21(DH3)pLysE host cells, which provide a higher level of lysozyme as an additional means of reducing the recombinant protein expression level, did not eliminate the toxicity of Cc RNase (Fig. 3C). It is therefore possible that the production of even a very small amount of the recombinant product is sufficient to cause a severe toxic effect in E.coli.
Figure 3.

Growth of E.coli expressing Cc RNase BL21(DE3)pLysS E.coli host cells harboring the plasmid Pcb (A), Psa (B) or PF (D) and BL21(DE3)pLysS host cells containing the plasmid Psa (C) were grown in M9 medium supplemented with ampicillin and the optical densities (600 nm) were recorded hourly. When exponential phase growth was achieved, 1 mM IPTG was added to half of each culture. Each experiment was repeated four times and the mean values are reported. Solid diamonds, after IPTG induction; open squares, without induction.
In order to overcome the toxic effect caused by the Psa and Pcb constructs, an attempt to express the Cc RNase gene in the pSCREEN vector was undertaken, creating the plasmid PF. In this construct the target protein was expressed as a fusion containing the first 260 amino acids of the bacteriophage T7 gene 10 major capsid protein at its N-terminus. Transform ation of the PF plasmid into BL21(DE3), BL21(DE3)pLysS or BL21(DE3)pLysE E.coli host cells did not produce any toxic effects. In addition, liquid cultures of cells harboring this plasmid showed a normal growth rate despite IPTG induction (Fig. 3D). Analysis of induced and non-induced cell extracts by SDS–PAGE also revealed that the viability of E.coli is accompanied by overproduction of the Cc RNase fusion protein (Fig. 4). An explanation for the above observation could be that the Cc RNase expressed from the PF plasmid forms insoluble and non-toxic aggregates, which are deposited into inclusion bodies and thus do not inhibit cell growth. Alternatively, the addition of 260 amino acids residues at the N-terminus of Cc RNase may be the cause of a conformational change in the expressed protein molecule that is responsible for suppressing the toxic effect.
Figure 4.
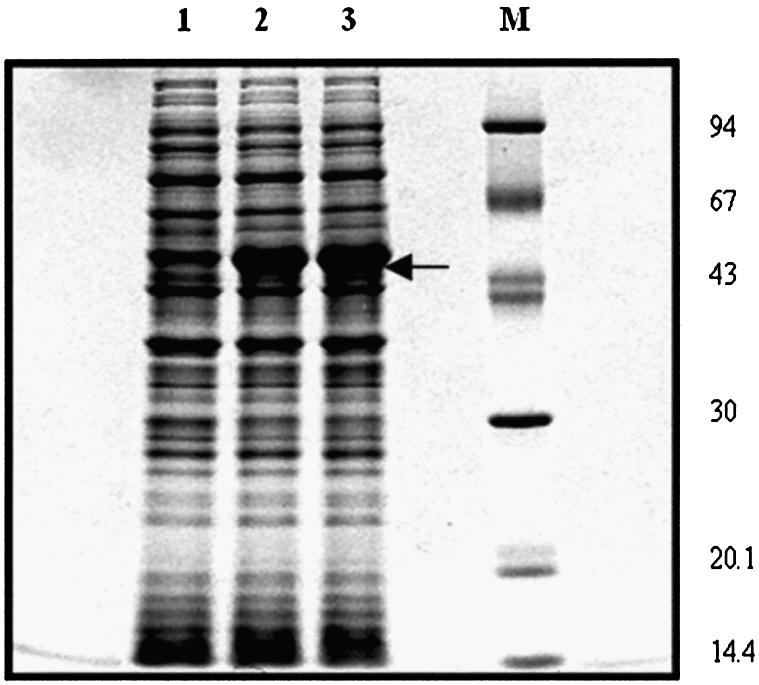
Expression of Cc RNase in E.coli. Total extracts from equivalent amounts of E.coli harboring the expression plasmid PF were analyzed by SDS–PAGE and the gel was stained with Coomassie brilliant blue R250. Lane 1 shows the total cell extract before IPTG induction, whereas lanes 2 and 3 demostrate proteins obtained after IPTG induction at 2 and 4 h, respectively. Cc RNase is expressed as a fusion protein exhibiting a molecular mass of 45 kDa. Marker proteins of known molecular weight (94, 67, 43, 30, 20.1 and 14.4 kDa top to bottom) are presented in lane M.
Purification and characterization of the recombinant Cc RNase
The recombinant fusion protein expressed in BL21(DE3) pLysS E.coli cells harboring the PF plasmid was purified using a Sepharose–Ni column in the presence of 8 M urea. The purified fusion protein was reduced using 0.1 M DTT and then digested with thrombin. The fragment containing Cc RNase was isolated by chromatography using the same column and exhibited a molecular mass of 16.0 kDa as estimated by SDS–PAGE (Fig. 5, lane 4). When the ribonucleolytic activity of the purified fraction was checked by SDS–PAGE in the presence of poly(C), a single band corresponding to the molecular mass of the purified protein was detected in the gel, followed by zymography (Fig. 5, lane 2). On the other hand, no band in the activity gel was observed when the recombinant protein was incubated at 99°C for 5 min (Fig. 5, lane 1). The relationship between the recombinant Cc RNase and the native poly(U), poly(C)-specific RNase isolated from the insect C.capitata (15) was investigated by western blot analysis. As shown in Figure 5 (lane 3), the polyclonal antibody raised against the native RNase also recognizes the recombinant protein.
Figure 5.

SDS–PAGE, activity staining and immunoblot analyses of the purified Cc RNase Aliquots of 2 µg of the purified recombinant protein (lane 2) and an equivalent amount of this protein preincubated at 99°C for 5 min (lane 1) were analyzed on an 11–15% polyacrylamide gel containing 0.25 mg/ml poly(C). After electrophoresis the gel was stained for RNase activity as described in Materials and Methods. The same protein amount (lane 3) was analyzed by SDS–PAGE, electro-transferred onto a nitrocellulose membrane and detected using a polyclonal antibody raised against the native poly(U), poly(C)-specific RNase isolated from the insect C.capitata. Purified recombinant Cc RNase (lane 4) and a set of marker proteins of known molecular weight (94, 67, 43, 30, 20.1 and 14.4 kDa top to bottom, lane M) were run in parallel and the proteins were stained with Coomassie blue.
The substrate specificity of the purified recombinant Cc RNase was assayed under the conditions described in Materials and Methods, using different homopolyribonucleotides and rRNA as substrates. As shown in Figure 6A, Cc RNase preferentially degrades poly(C), while it degrades poly(U) at a lower rate. The purified enzyme also displayed ribonucleolytic activity towards native rRNA. In all cases, the rate of degradation is linear with increasing enzyme concentration. Under the same assay conditions no significant degradation of poly(A), poly(G) or poly(I) was observed. Control experiments performed under identical conditions using denatured enzyme or buffer further confirmed the specificity of the ribonucleolytic activity of the expressed protein. Recombinant Cc RNase enzymatic activity towards rRNA was also tested using an agarose gel assay. As shown in Figure 6B, incubation of rRNA with the recombinant protein resulted in an extensive degradation of 28S and 18S RNA in a time-dependent manner.
Figure 6.

Ribonucleolytic activity of the recombinant protein. (A) Standard amounts (50 µg) of poly(C) (solid diamonds), poly(U) (solid triangles) and rRNA (open squares) were incubated at 37°C for 3 h with different amounts of purified recombinant Cc RNase as described in Materials and Methods. The reaction was stopped by addition of perchloric acid containing uranyl acetate and acid-soluble ribonucleotides remaining in the supernatant fraction after centrifugation were quantitated spectrophotometrically at 260 nm. Error bars represent means ± SE (n = 3). (B) An aliquot of 2 µg of rRNA from rat liver was incubated with 0.4 µg of purified recombinant Cc RNase at 37°C for 5 (lane 2), 10 (lane 3) and 15 min (lane 4). An equal amount of rRNA without Cc RNase was also incubated at 37°C for 15 min (lane 1). The reaction products were analyzed by 1% agarose gel electrophoresis and visualized with ethidium bromide staining. Arrows show 28S and 18S rat rRNA.
The enzymatic properties of the purified recombinant Cc RNase were tested using poly(C) as substrate. The enzyme exhibits maximum activity at pH 8.0. It is a thermolabile molecule, which loses most of its activity immediately following preincubation at 60°C. The purified enzyme seems to remain almost unaffected in the presence of Ca2+, while 1 mM Zn2+ or Hg2+ causes total inhibition of its ribonucleolytic activity.
Based on our observation that reduction, prior to refolding, is necessary in order to obtain an enzymatically active recombinant protein, we investigated the role of disulfide bonding in Cc RNase. When 50 mM DTT was added to the assay reaction mixture, the ribonucleolytic activity was decreased by 75%. It has been reported that reduced proteins can be alkylated with iodoacetamide to modify free thiol groups so that disulfides cannot be re-acquired after removal of the reducing agent (36). When the reduced recombinant Cc RNase was alkylated with iodoacetamide, the activity of the enzyme was decreased by 92% as compared to that of the unreduced, unalkylated enzyme. Alkylation alone of the refolded Cc RNase caused only a 5% decrease in the activity. Therefore, Cc RNase is nearly inactive in the reduced state. These data strongly suggested that Cc RNase acquires at least one intramolecular disulfide bond which is important for protein folding into a functional structure.
DISCUSSION
In our previous work we presented a cDNA sequence obtained from two overlapping clones, encoding a poly(U), poly(C)-specific RNase from the insect C.capitata (Cc RNase) (16). The putative ORF of this sequence, showing a relatively weak homology to the RNase A superfamily, was used for the construction of a recombinant protein. The expressed protein was recognized by the anti-Cc RNase polyclonal antibody but did not possess ribonucleolytic activity.
In this work we report the isolation, characterization and expression of the full-length cDNA clone coding for Cc RNase. Nucleotide alignment between the previous and the recently obtained clones showed a number of minor nucleotide deletions, resulting in the appearance of a different putative ORF. This ORF encodes a protein consisting of 95 amino acids, which has strong sequence homology with other uncharacterized proteins predicted from ESTs, isolated from 20 different animal species. In addition, the recombinant protein expressed from this ORF, apart from demonstrating immunological reaction with the anti-Cc RNase polyclonal antibody, also degrades poly(U) and poly(C) synthetic substrates, as well as rRNA, in a linear manner in terms of incubation time and enzyme concentration. For the above reasons, we strongly believe that the Cc RNase cDNA sequence presented in this work is the correct one.
The significant protein sequence conservation among Cc RNase and apparent homologs in evolutionarily distant species defines a new family of proteins conserved from C.elegans to humans. Data obtained from phylogenetic analysis suggested that all family members are true orthologs. This finding was also supported by the fact that no paralogous sequences were identified in any species analyzed. A phylogenetic tree was constructed in order to study the evolutionary relationships between family members. Vertebrate sequences cluster together in a separate branch. It is noted that the human member has an extremely conserved counterpart in all vertebrates analyzed. Similarly, insects, nematodes and lower chordates group in well differentiated lineages. It was also observed that all insect family members share more similarity with nematode counterparts than they do with mammals. This finding is in agreement with the current view of invertebrate molecular phylogeny, according to which nematodes and arthropods reside in a common evolutionary clade, the Ecdysozoa (37). The phylogenetic distribution of Cc RNase indicated that this enzyme originated from a common ancestor of many metazoan phyla early in eukaryote evolution. Since we have not yet been able to identify yeast genes that would match our family sequences, this protein may be absent in primitive eukaryotes. The wide distribution of Cc RNase in the animal kingdom suggests strong and continuous selection pressure to maintain this enzyme, suggesting that it performs a very basic biological function. This suggestion is also supported by the fact that a large number of human ESTs derived from multiple adult tissues, as well as from fetal tissues and carcinomas, were identified as encoding the human counterpart of this protein family. It is well established that wide expression is a characteristic shared by genes with a basic housekeeping cellular function (38).
Cc RNase is a novel RNase. Searches for nuclear localization signals and conserved protein domains did not yield any significant results. Protein alignment of this novel family showed that the conserved residues were mainly clustered in two hydrophobic domains at the N- and C-termini of the protein sequences. The lack of histidines in the Cc RNase sequence raises a significant point, as histidines participate in the general acid–base catalysis of most RNases (39). For example, two defined histidine residues are known to be involved in the catalytic mechanism of all members of the RNase A superfamily in humans (40). On the other hand, one histidine and one glutamic acid are considered to be critical for the acid–base catalysis in some microbial RNases belonging to the T1 family (41). There are also RNases, such as RNase III and RNase H type II, whose ribonucleolytic activity has no dependence on histidine residues. Structural biochemistry studies in combination with mutation and subsequent activity analyses revealed that RNA catalysis by these RNases is carried out by acidic anino acid residues (42,43). Although experimental data concerning the catalytic mechanism of Cc RNase have not been obtained so far, residues Lys7, Glu40 and Asp46 (numbering according to C.capitata) were found to be conserved in all family members studied, therefore indicating a possible participation in catalysis.
The newly defined protein sequences of this family contain between three and five cysteine residues, three of which, Cys4, Cys11 and Cys68 (numbering according to C.capitata), are conserved in all species examined. The finding that recombinant Cc RNase expressed in E.coli was not enzymatically active until a reduction step was performed prior to renaturation suggested that correct folding of Cc RNase requires the formation of a disulfide bond(s). This observation is also supported by the fact that the alkylated recombinant protein was inactive. In contrast, when an alkylating agent was added after the renaturation step no effect on the activity of Cc RNase was observed, suggesting that a sulfydryl group(s) is not involved in the catalytic mechanism.
This study also demonstrates that the expression of Cc RNase is toxic to E.coli. Similar data were obtained when other RNases, like RNase L (44), ECP (25) and human RNase 7 (45), were expressed in E.coli. Cytotoxicity is closely associated with a group of RNases bearing specific biological actions in addition to their catalytic activity. This group includes a variety of RNases, such as BS-RNase (46), onconase (47) and modified RNase A (48). It has been proposed that these proteins enter cells by absorptive endocytosis (49), degrade cellular RNA and consequently inhibit protein synthesis and cause cell death (7). This RNase-based mechanism is employed in different biological processes such as prevention of self-pollination in plants, defense against parasites, viral infections and tumors in animals (50).
In conclusion, this study defines a novel highly conserved eukaryotic protein family whose biological role is not yet clear. The fact that the C.capitata counterpart of this family possesses ribonucleolytic and antibacterial activities indicates a multiplicity of biological functions for this molecule. In light of these findings, aspects of the biological significance of these novel protein family members, such as gene expression, physiological functions and their role in host defense mechanisms, in addition to the investigation of their cellular topology and biochemical characterization, remain to be explored.
Acknowledgments
ACKNOWLEDGEMENTS
This work is part of the PhD thesis of Mr Th. Rampias and was supported by the Hellenic State Scholarship Foundation.
DDBJ/EMBL/GenBank accession no. AJ441124
REFERENCES
- 1.Wellington C.L., Greenberg,M.E. and Belasco,J.G. (1993) The destabilizing elements in the coding region of c-fos mRNA are recognized as RNA. Mol. Cell. Biol., 13, 5034–5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coutts M., Krowczynska,A. and Brawerman,G. (1993) Protection of mRNA against nucleases in cytoplasmic extracts of mouse sarcoma ascites cells. Biochim. Biophys. Acta, 1173, 49–56. [DOI] [PubMed] [Google Scholar]
- 3.Beintema J.J. and Kleineidam,R.G. (1998) The ribonuclease A superfamily: general discussion. Cell Mol. Life Sci., 54, 825–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benner S.A. and Allemann,R.K. (1989) The return of pancreatic ribonucleases. Trends Biochem. Sci., 14, 396–397. [DOI] [PubMed] [Google Scholar]
- 5.D’Alessio G., Di Donato,A., Parente,A. and Piccoli,R. (1991) Seminal RNase: a unique member of the ribonuclease superfamily. Trends Biochem. Sci., 16, 104–106. [DOI] [PubMed] [Google Scholar]
- 6.Vallee B.L. and Riordan,J.F. (1997) Organogenesis and angiogenin. Cell Mol. Life Sci., 53, 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matousek J. (2001) Ribonucleases and their antitumor activity. Comp. Biochem. Physiol., 129C, 175–191. [DOI] [PubMed] [Google Scholar]
- 8.Zuo Y. and Deutscher,M.P. (2001) Exoribonuclease superfamilies: structural analysis and phylogenetic distribution. Nucleic Acids Res., 29, 1017–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beintema J.J., Schuller,C., Irie,M. and Carsana,A. (1988) Molecular evolution of the ribonuclease superfamily. Prog. Biophys. Mol. Biol., 51, 165–192. [DOI] [PubMed] [Google Scholar]
- 10.Filippov V., Filippova,M. and Gill,S.S. (1997) Functional characterization of RNase H1 from Drosophila melanogaster. Biochem. Biophys. Res. Commun., 240, 844–849. [DOI] [PubMed] [Google Scholar]
- 11.Filippov V., Filippova,M. and Gill,S.S. (2001) Drosophila RNase H1 is essential for development but not for proliferation. Mol. Genet. Genomics, 265, 771–777. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Segura J.M., Fominaya,J.M., Orozco,M.M. and Gavilanes,J.G. (1985) Alkaline ribonuclease from the insect Ceratitis capitata. Biochim. Biophys. Acta, 826, 129–136. [Google Scholar]
- 13.Garcia-Segura J.M., Orozco,M.M., Fominaya,J.M. and Gavilanes,J.G. (1986) Purification, molecular and enzymic characterization of an acid RNase from the insect Ceratitis capitata. Eur. J. Biochem., 15, 367–372. [DOI] [PubMed] [Google Scholar]
- 14.Sideris D.C. and Fragoulis,E.G. (1987) Purification and characterization of a ribonuclease specific for poly(U) and poly(C) from the larvae of Ceratitis capitata. Eur. J. Biochem., 164, 309–315. [DOI] [PubMed] [Google Scholar]
- 15.Lalioti V.S., Sideris,D.C. and Fragoulis,E.G. (1992) Further characterization of a poly(U), poly(C) ribonuclease from the insect Ceratitis capitata. Insect Biochem. Mol. Biol., 22, 125–135. [DOI] [PubMed] [Google Scholar]
- 16.Sideris D.C., Rampias,T.N. and Fragoulis,E.G. (2000) Isolation and sequencing of a cDNA encoding for a ribonuclease from the insect Ceratitis capitata. Insect Biochem. Mol. Biol., 30, 153–161. [DOI] [PubMed] [Google Scholar]
- 17.Bouhin H., Charles,J.P., Quennedey,B. and Delachambre,J. (1992) Developmental profiles of epidermal mRNAs during the pupal-adult molt of Tenebrio molitor and isolation of a cDNA clone encoding an adult cuticular protein: effects of a juvenile hormone analogue. Dev. Biol., 149, 112–122. [DOI] [PubMed] [Google Scholar]
- 18.Tagle D.A., Swaroop,M., Lovett,M. and Collins,F.S. (1993) Magnetic bead capture of expressed sequences encoded within large genomic segments. Nature, 361, 751–753. [DOI] [PubMed] [Google Scholar]
- 19.Boguski M.S., Lowe,T.M. and Tolstoshev,C.M. (1993) dbEST—database for ‘expressed sequence tags’. Nature Genet., 4, 332–333. [DOI] [PubMed] [Google Scholar]
- 20.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhahg,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson J.D., Gibson,T.J., Plewniak,F., Jeanmougin,F. and Higgins,D.G. (1997) The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res., 25, 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saitou N. and Nei,M. (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol., 4, 406–425. [DOI] [PubMed] [Google Scholar]
- 23.Felsenstein J. (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution, 39, 783–791. [DOI] [PubMed] [Google Scholar]
- 24.Perriere G. and Gouy,M. (1996) WWW-query: an on-line retrieval system for biological sequence banks. Biochimie, 78, 364–369. [DOI] [PubMed] [Google Scholar]
- 25.Rosenberg H.F. (1995) Recombinant human eosinophil cationic protein. Ribonuclease activity is not essential for cytotoxicity. J. Biol. Chem., 270, 7876–7881. [DOI] [PubMed] [Google Scholar]
- 26.Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 27.Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 28.Slifman N.R., Loegering,D.A., McKean,D.J. and Gleich,G.J. (1986) Ribonuclease activity associated with human eosinophil-derived neurotoxin and eosinophil cationic protein. J. Immunol., 137, 2913–2917. [PubMed] [Google Scholar]
- 29.Blank A., Sugiyama,R.H. and Dekker,C.A. (1982) Activity staining of nucleolytic enzymes after sodium dodecyl sulfate-polyacrylamide gel electrophoresis: use of aqueous isopropanol to remove detergent from gels. Anal. Biochem., 120, 267–275. [DOI] [PubMed] [Google Scholar]
- 30.Perry R.P., La Torre,J., Kelley,D.E. and Greenberg,J.R. (1972) On the lability of poly(A) sequences during extraction of messenger RNA from polyribosomes. Biochim. Biophys. Acta, 262, 220–226. [DOI] [PubMed] [Google Scholar]
- 31.Cavener D.R. and Ray,S.C. (1991) Eukaryotic start and stop translation sites. Nucleic Acids Res., 19, 3185–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kyte J. and Doolittle,R.F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol., 157, 105–132. [DOI] [PubMed] [Google Scholar]
- 33.Gogarten J.P. (1994) Which is the most conserved group of proteins? Homology-orthology, paralogy, xenology and the fusion of independent lineages. J. Mol. Evol., 39, 541–543. [DOI] [PubMed] [Google Scholar]
- 34.Giribet G. (2002) Current advances in the phylogenetic reconstruction of metazoan evolution. A new paradigm for the Cambrian explosion? Mol. Phylogenet. Evol., 24, 345–357. [DOI] [PubMed] [Google Scholar]
- 35.Studier F.W. (1991) Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J. Mol. Biol., 219, 37–44. [DOI] [PubMed] [Google Scholar]
- 36.Kishigami S., Akiyama,Y. and Ito,K. (1995) Redox states of DsbA in the periplasm of Escherichia coli. FEBS Lett., 364, 55–58. [DOI] [PubMed] [Google Scholar]
- 37.Adoutte A., Balavoine,G., Lartillot,N., Lespinet,O., Prud’homme,B. and de Rosa,R. (2000) The new animal phylogeny: reliability and implications. Proc. Natl Acad. Sci. USA, 97, 4453–4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gerard C.J., Andrejka,L.M. and Macina,R.A. (2000) Mitochondrial ATP synthase 6 as an endogenous control in the quantitative RT-PCR analysis of clinical cancer samples. Mol. Diagn., 5, 39–46. [DOI] [PubMed] [Google Scholar]
- 39.Walsh C. (1979) Enzymatic Reaction Mechanisms. W.H. Freeman and Co., San Francisco, CA.
- 40.Zhang J., Dyer,K.D. and Rosenberg,H.F. (2002) RNase 8, a novel RNase A superfamily ribonuclease expressed uniquely in placenta. Nucleic Acids Res., 30, 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steyaert J., Hallenga,K., Wyns,L. and Stanssens,P. (1990) Histidine-40 of ribonuclease T1 acts as base catalyst when the true catalytic base, glutamic acid-58, is replaced by alanine. Biochemistry, 29, 9064–9072. [DOI] [PubMed] [Google Scholar]
- 42.Sun W. and Nicholson,A.W. (2001) Mechanism of action of Escherichia coli ribonuclease III. Stringent chemical requirement for the glutamic acid 117 side chain and Mn2+ rescue of the Glu117Asp mutant. Biochemistry, 40, 5102–5110. [DOI] [PubMed] [Google Scholar]
- 43.Chapados B.R., Chai,Q., Hosfield,D.J., Qiu,J., Shen,B. and Tainer,J.A. (2001) Structural biochemistry of a type 2 RNase H: RNA primer recognition and removal during DNA replication. J. Mol. Biol., 307, 541–556. [DOI] [PubMed] [Google Scholar]
- 44.Diaz-Guerra M., Esteban,M. and Martinez,J.L. (1997) Growth of Escherichia coli in acetate as a sole carbon source is inhibited by ankyrin-like repeats present in the 2′,5′-linked oligoadenylate-dependent human RNase L enzyme. FEMS Microbiol Lett., 149, 107–113. [DOI] [PubMed] [Google Scholar]
- 45.Zhang J., Dyer,K.D. and Rosenberg,H.F. (2003) Human RNase 7: a new cationic ribonuclease of the RNase A superfamily. Nucleic Acids Res., 31, 602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mastronicola M.R., Piccoli,R. and D’Alessio,G. (1995) Key extracellular and intracellular steps in the antitumor action of seminal ribonuclease. Eur. J. Biochem., 230, 242–499. [DOI] [PubMed] [Google Scholar]
- 47.Ardelt W., Mikulski,S.M. and Shogen,K. (1991) Amino acid sequence of an anti-tumor protein from Rana pipiens oocytes and early embryos. Homology to pancreatic ribonucleases. J. Biol. Chem., 266, 245–251. [PubMed] [Google Scholar]
- 48.Bretscher L.E., Abel,R.L. and Raines,R.T. (2000) A ribonuclease A variant with low catalytic activity but high cytotoxicity. J. Biol. Chem., 275, 9893–9896. [DOI] [PubMed] [Google Scholar]
- 49.Kim J.S., Soucek,J., Matousek,J. and Raines,R.T. (1995) Mechanism of ribonuclease cytotoxicity. J. Biol. Chem., 270, 31097–31102. [DOI] [PubMed] [Google Scholar]
- 50.Youle R.J., Newton,D., Wu,Y.N., Gadina,M. and Rybak,S.M. (1993) Cytotoxic ribonucleases and chimeras in cancer therapy. Crit. Rev. Ther. Drug Carrier Syst., 10, 1–28. [PubMed] [Google Scholar]