


Abstract
The nuclear receptor CAR (NR1I3) regulates transcription of genes encoding xenobiotic- and steroid-metabolizing enzymes. Regulatory processes that are mediated by CAR are modulated by a structurally diverse array of chemicals including common pharmaceutical and environmental agents. Here we describe four in-frame splice variants of the human CAR receptor gene. The variant mRNA splice transcripts were expressed in all human livers evaluated. Molecular modeling of the splice variant proteins predicts that the structural effects are localized within the receptor’s ligand-binding domain. Assays to assess function indicate that the variant proteins, when compared with the reference protein isoform, exhibit compromised activities with respect to DNA binding, transcriptional activation and coactivator recruitment.
INTRODUCTION
CAR (constitutive androstane receptor, NR1I3) is a nuclear receptor that mediates the transcriptional activation of genes encoding phase 1 (e.g. CYP2B6 and CYP3A4) and phase 2 (e.g. UDP-glucoronysyltransferase 1A1) xenobiotic- and steroid-metabolizing enzymes, as well as a number of xenobiotic transporter proteins (e.g. multidrug resistance-associated proteins 2 and 3) (1–5). Nuclear receptors, such as CAR, the pregnane X receptor (PXR), peroxisome proliferator-activated receptor α (PPARα) and the aryl-hydrocarbon receptor, transcriptionally and coordinately regulate the expression of a battery of enzymes that function to metabolize a vast array of lipophilic molecules in mammalian tissues (6–9). These receptors are themselves activated or regulated by a similarly overlapping network of exogenous and endogenous substances. CAR is an essential mediator of the transcriptional response to a subclass of these structurally diverse chemicals, represented by the prototypical inducing agent, phenobarbital (PB) (10).
CAR is expressed primarily in the liver and to a lesser extent in the intestinal epithelium and the stomach (11). It is a member of the NR1I subfamily that also includes the vitamin D receptor (VDR) and PXR (12). Although most nuclear receptors have five domains, the human CAR (hCAR) protein contains three: a highly conserved DNA-binding domain (C domain); a hinge domain (D domain); and a divergent ligand-binding/dimerization/transcriptional activation domain (E domain). hCAR lacks an A/B domain that typically confers a ligand-independent transactivation response, and the more poorly characterized hypervariable F domain (13). CAR interacts with DNA as a heterodimer with retinoid X receptor (RXR) (14). The heterodimer can bind an array of prototypical nuclear receptor-binding sites, including DR-5 (direct repeat-5), DR-4, DR-3, ER-6 (everted repeat-6) and IR-8 (inverted repeat-8) motifs (2,15). A number of these response elements have been identified in the promoters of PB-responsive human genes (1,4,16). As with many nuclear receptors, CAR interacts with transcriptional coregulators, including p160 family members—steroid receptor coactivators (SRC-1, -2 and -3), small heterodimerization protein and nuclear receptor corepressors (11,17–21).
hCAR activity may be positively, or negatively, regulated by ligand interactions. For example, clotrimazole is an hCAR ligand than functions as an inverse agonist, an activity typical of most identified hCAR ligands (22), whereas 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime has been identified recently as an hCAR agonist (23). However, PB and other PB-like inducers do not function as direct CAR ligands, but rather activate hCAR by stimulating its nuclear translocation from the cytosol (24). The latter process appears ultimately important in governing receptor activity and remains relatively poorly understood (3).
In mice, the hepatocyte transcriptional response to PB is dependent on the CAR receptor and independent of PXR (10,25). However, due to significant species differences in the PB induction response, results from mouse studies do not necessarily represent the processes occurring in human hepatocytes. For example, studies suggest that PB is an activating ligand of human PXR (22,26,27) and that hPXR can control the transcriptional expression of a number of PB-inducible genes (7). Other results indicate that the human VDR also plays a role in regulation of the PB-inducible genes, CYP2B6, CYP3A4 and CYP2C9 (28). These findings suggest that an array of nuclear receptors are involved in regulating responses to PB-like inducing agents in humans, consistent with the idea that a collection of receptors probably function as a xenobiotic ‘safety net’ (29).
Considering the structural diversity of PB-like inducing agents, it appears unlikely that CAR, PXR and VDR exclusively mediate the biological responses to these agents. Alternatively spliced isoforms of such nuclear receptors may possess properties distinct from their reference counterparts, enabling expansion of receptor functional diversity and perhaps differential responsiveness to given subsets of PB-like inducing substances. Specifically, we hypothesized that splice variants of hCAR exist, possessing unique structures with altered receptor activities. In this investigation, we report the cloning and characterization of four distinct splice forms of the hCAR derived from human liver cDNA samples. All of the variants encode insertions or deletions of the ligand-binding domain. Examination of cDNA transcript levels from a human liver bank revealed that CAR transcript isoforms are commonly expressed in individuals. Functional characterization indicated that the variants possess altered capacities of DNA interaction, transcriptional activation and coactivator recruitment.
MATERIALS AND METHODS
PCR-based cloning of hCAR variants
Human hepatocyte cDNA samples (30) were amplified for 30 cycles at an annealing temperature of 60°C using VENT DNA polymerase (New England Biolabs, Beverly, MA). DNA primers used for the PCR amplification were: hCAR FP, with an incorporated EcoRI site (5′-GATCGAATTCGTCATGGCCAGTAGGGAAGATGAG-3′), and hCAR RP, with an incorporated EcoRV + stop site (5′-GATCGATATCTCAGCTGCAGATCTCCTGGAGCCAG-3′). PCR cycling was performed at 95°C for 30 s, 60°C for 30 s and 72°C for 90 s. Purified products were separated by agarose gel electrophoresis, and fragments in the range of 800–1200 bp were purified using a Qiagen gel purification kit (Qiagen, Chatsworth, CA). Purified fragments were blunt-end ligated into the SmaI site of pBluescript and transformed into DH5α ultracompetent cells (Invitrogen, Carlsbad, CA). Colonies were screened by PCR to determine the presence of an insert within the vector. Positive clones were grown overnight in 2 ml cultures containing ampicillin, purified, and sequenced using an ABI 310 automated sequencer (Applera Corp., Foster City, CA).
Plasmids constructs
The full-length coding sequence for each hCAR isoform was subcloned into the mammalian expression vector pTracer CMV.2 (Invitrogen) using EcoRI–EcoRV restriction sites. To generate GST–hCAR fusions, the hCAR-coding sequence was subcloned in-frame with GST in the bacterial pGEX4T-1 vector using EcoRI–EcoRV sites. For bacterial expression, T7-hCAR fusion constructs were prepared by cutting the hCAR-coding sequence from the pTracer CMV.2 vector with EcoRI and NotI and subcloning the released fragment into the pET21a bacterial expression vector (Novagen, Madison, WI). Human RXRα (a gift from P. Chambon, IGBMC, Universite Louis Pasteur, Illkirch, France) was also subcloned into the EcoRI site in pTracer CMV.2. The GST–hRXRα bacterial expression plasmid was a gift from Dr Karen Swisshelm (University of Washington).
pGL-3 Basic PBREM-TK-Luciferase was generated by insertion of one copy of the CYP2B6 PB-responsive enhancer module (PBREM). The DNA primers CYP2B6 BS-FP (5′-TTGGTTCAGGAAAGTCCATGCTGCCACCTCTTCAGGGTCAGGAAAGTACAG-3‘) and CYP2B6 BS-RP (5′-ACTGTACTTTCCTGACCCTGAAGAG-3′) were annealed and extended with Klenow DNA polymerase. The fragment was then blunt-end ligated into the SmaI restriction site in pGL3-Basic Vector (Promega, Madison, WI), containing a thymidine kinase promoter (TK-luc3). TK-luc3 was prepared as described previously (16). Sequence analysis identified clones that were positive for a single full-length copy of the PBREM element.
The coding sequence of the SRC-1 receptor interaction domain (RID; amino acids 632–754) was amplified with Platinum Taq (Invitrogen), using a human SRC-1 FP that incorporated an EcoRI site (5′-GATCGAATTCAAACTAGTGCAGCTTTTGAC-3′), and human SRC-1 RP that included a translation stop signal (5′-GAATTCTCATCAATCATCCAGGCTCAGGTTTGGAG-3′). The PCR fragment was cloned in pGEM-T (Promega). Clones were characterized by PCR and by DNA sequencing. To generate a GST-fused construct, the SRC-1 RID fragment was subcloned into the EcoRI restriction site of pGEX4T-1 (Amersham Pharmacia Biotech AB, Piscataway, NJ).
hCAR antibody
A rabbit polyclonal antibody was generated against an epitope of the hCAR ligand-binding domain, N-KGQQRRPRDRFLYAKL-C (Quality Control Biochemicals, Hopkinton, MA). Antibody was affinity purified and dialyzed against phosphate-buffered saline (PBS) as described in the supplier’s protocol. The specificity of the antibody was determined by western blot of crude bacterial lysates containing recombinantly expressed hCAR. The antibody reacted with all isoforms of hCAR, but not with any components present in crude lysates.
Molecular modeling
CAR was modeled based on the reporter structure of the VDR, 1DB1(31). Models were generated with the HOMOLOGY module of INSIGHTII software (Accelrys, San Diego, CA) using established protocols. Protein backbone coordinates were taken from VDR for all helices, strands and loops with identical lengths.
Detection of hCAR transcript variants in human liver
Access to a human liver tissue bank was generously provided by Dr Kenneth Thummel, University of Washington. cDNA fractions were prepared from sample livers and subjected to PCR amplification with primer pairs designed to generate amplicons that spanned the exon 6–7 splice junction, the exon 7–8 junction, or both splice junctions combined. The amplicons were separated by agarose gel electrophoresis and stained with ethidium bromide. Primers used for detection of the reference (wt) and 12 bp insertion at the exon 6–7 junction were Exon 6 FP (5′-GACCAGATCTCCCTTCTCAAG-3′) and Exon 7 RP (5′-CTCAGGCTCTTGGAGCTGCAG-3′). Primers used for detection of the wt and 15 bp insertion at the exon 7–8 junction were Exon 7 FP (5′-GGAGTTGCTCTTTCACTTCCA-3′) and Exon 8 RP (5′-CCTTCGCTGCTGGCCCTTGATG-3′). Primers used for detection of the exon 7 deletion were Exon 6 FP and Exon 8 RP. PCR amplifications were performed in a 50 µl reaction volume containing 3.0 mM MgCl2, 25 pmol of each primer and 0.5 µl of cDNA. Cycling was conducted at 95°C for 30 s, 60°C for 30 s and 72°C for 30 s. For detection of the 12 bp (exon 6–7 junction) and 15 bp (exon 7–8 junction) insertions, the amplification was carried out for 32 cycles, whereas detection of the exon 7 deletion variant was completed in 35 cycles.
Protein expression
COS-1. Approximately 400 000 COS-1 cells were plated in each well of a 6-well plate and the cells were allowed to attach overnight. Cells were transfected using Lipofectamine (Invitrogen) with either 2 µg of hCAR expression plasmid or empty vector control, in accordance with the manufacturer’s protocol. Whole-cell lysates were generated 24 h post-transfection by application of RIPA buffer (50 mM Tris–HCl pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA) containing a protease inhibitor cocktail (Calbiochem, La Jolla, CA).
BL21. The GST or GST-fused proteins (GST–hCAR, GST– SRC-1-RID) were expressed using the pGEX4T-1 bacterial expression plasmid (Amersham Pharmacia Biotech AB) in the Escherichia coli strain BL21(DE3). In brief, 3 ml of overnight bacterial growth was added to 27 ml of 2× YT broth containing 100 µg/ml ampicillin and allowed to reach an optical density of 0.6–1.0 (1–2 h) at 37°C. Bacteria were then induced by addition of isopropyl-β-d-thiogalactopyranoside (IPTG) to 0.5 mM and incubated for an additional 3 h at room temperature. Cells were collected by centrifugation and lysed by sonication in PBS containing protease inhibitors (Calbiochem). Cell debris was removed by centrifugation, and the supernatant was added to 300 µl of glutathione– Sepharose 4B (Amersham Pharmacia Biotech AB). GST fusion proteins were bound to the resin during rotation at room temperature and then washed four times with PBS, in a total wash volume of 4 ml. Depending on the procedure, proteins were either eluted in glutathione elution buffer (10 mM reduced glutathione in 50 mM Tris–HCl pH 8.0) or were equilibrated with GST interaction buffer (2 mg/ml bovine serum albumin, 25 mM HEPES pH 7.6, 100 mM NaCl, 20% glycerol and 1 mM EDTA) (32). Eluted proteins were visualized with PAGE and subsequent Coomassie staining. The hCAR isoforms were also expressed in bacteria as T7-tagged proteins using the pET21a vector. T7-tagged proteins were expressed using the same procedure; however, no affinity purification followed the sonication step.
In vitro transcription and translation
PCR products were generated that contained an open reading frame encoding hCAR isoforms or RXR, in addition to a 5′ T7 promoter and a region directing 3′ polyadenylation. The PCR products were then used as templates for transcription in the TNT-coupled reticulocyte lysate (Promega Corp.). Protein expression was carried out in the presence of [35S]methionine as described in the manufacturer’s protocol.
Western immunoblotting
Soluble fraction proteins from 30 µg of total transfected COS-1 cell lysate, 20 µg of crude bacterial lysate (soluble fraction) or 200 µg of whole human liver lysate were separated by denaturing PAGE and transferred to a PDVF membrane. The membrane was incubated with an hCAR antibody (1:5000) generated in our laboratory, as described above. The primary antigen reaction was followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit antibody (Biorad, Hercules, CA). Proteins were visualized using the enhanced chemiluminescence reagent kit (Amersham Pharmacia Biotech AB).
Electrophoretic mobility shift assay (EMSA)
EMSAs were performed in a similar manner to the procedures already described (16) with the exception that bacterially expressed and purified GST-fused proteins were used. Briefly, GST, GST–hCAR isoforms and GST–hRXRα were expressed as described above and proteins were incubated in combination with double-stranded 32P-labeled oligonucleotides encoding either the CYP2B6 NR1 or NR2 element (100 000 c.p.m. per reaction). DNA–protein complex formation was conducted at room temperature in the presence of the previously described binding buffer and 2 µg of poly(dI/dC). Complexes were resolved in 5% non-denaturing polyacrylamide gels. The gels were dried and mobility shifts were visualized by autoradiography.
Culture and transfection of COS-1 cells
Cells were maintained in high glucose Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS). One day prior to transfection, cells were trypsinized and plated onto 24-well plates (100 000 cells/well). For determination of transcriptional activity of the variant hCAR forms, cells were transfected using Lipofectamine with 50 ng of pRL (Renilla luciferase; Promega) for normalization, 50 ng of pTracer CMV.2-hRXRα, 200 ng of pGL-3-PBREM-TK-Luciferase and 200 ng of either pTracer CMV.2-hCAR or variant hCAR constructs. Transfections were performed according to the manufacturer’s recommendations.
Transcriptional activation assays
Dual-luciferase assays were performed as per the manufacturer’s protocol (Promega, Dual-Luciferase Reporter Assay System).
In vitro interaction assay
GST ‘pull-down’ assays were performed as previously described (32). After the last wash, complexes were suspended in 2 ml of interaction buffer (described above), and 150 µl of the mixture was used for each reaction. A total of 350 µl of addition interaction buffer was then aliquoted into each tube. An additional 150 µl of the GST and GST–SRC-1 RID suspension was eluted from the beads in 10 mM reduced glutathione and separated in a pre-cast 10% Tris–HCl gel (Biorad). The gel was Coomassie stained to visualize expression of the proteins. To each reaction, 5 µl of 35S-labeled in vitro translated hCAR or hCAR variant was added and the complexes were incubated overnight at 4°C with rotation. The complexes were washed four times with a total of 4 ml of interaction buffer and were eluted in 50 µl of 10 mM glutathione. Eluted protein was separated in a pre-cast 10% Tris–HCl gel (Biorad), and the gel was dried and exposed to film.
Results
Cloning of hCAR splice variants
To identify novel isoforms of hCAR, we designed oligonucleotide primers targeted to the amino acid-coding sequence of the reference mRNA (14). PCR was performed on an individual human liver cDNA sample and the products were subcloned into pBluescript. Twenty clones were selected at random and subjected to DNA sequencing. A distribution of five reference, four 12 bp insertion, eight 15 bp insertion and one 12/15 bp double insertion clones was obtained (Fig. 1B and C). Two additional clones contained complete intron 8 insertions, probably a product of incompletely processed mRNA. The exon 7 deletion plasmid was detected in a similar manner from a separate set of cDNA clones. The nomenclature of the hCAR variants that is used throughout this manuscript is presented in Table 1.
Figure 1.




Description of hCAR isoforms. (A) Diagram detailing the hCAR protein structure aligned to the exonic regions of the corresponding mRNA, the DNA-binding domain (DBD), the hinge domain (H) and the ligand-binding domain (LBD), respectively. (B) A schematic representation (top) of hCAR exons 1–9 in the context of the gene (our unpublished observations and Locuslink ID no. 9970/genomic contig: NT_026945). The region of mRNA that demonstrates the alternative splicing events identified in this study, exons 7, 8 and 9, is depicted in greater detail. The sizes of the introns and exons noted above and below the amplified schematic are derived from the reference isoforms, obtained from the Locuslink data (as above). The mRNA isoform containing the 12 bp insertion is generated by an alternative splice acceptor site within intron 6 leading to a 5′ extension of exon 7. Incorporation of these nucleotides leads to an in-frame 4 amino acid insertion. The mRNA isoform containing the 15 bp insertion is generated by an alternative splice acceptor site within intron 7 leading to a 5′ extension of exon 8. Incorporation of these 15 nucleotides leads to an in-frame 5 amino acid insertion. Individual clones that contained both the 12 and 15 bp insertion were also identified. An additional mRNA isoform is generated by the complete removal of exon 7, leading to an in-frame deletion of 39 amino acids. (C) Detail of the nucleotide sequence surrounding the splice junctions between exons 6 and 7 (top) and exons 7 and 8 (bottom). Reference sequences are represented in upper case letters, and incorporated nucleotides due to alternative splicing are represented by lower case letters. Canonical splice donor and splice acceptor sites present in the genomic DNA are shown in bold lower case. The reading frame of each sequence is shown below each sequence. Splice acceptor sites are indicated by arrows. (D) ClustalW alignment of the predicted hCAR isoform amino acid sequences, Reference, 4aaINS, 5aaINS and 39aaDEL (53).
Table 1. hCAR isoform nomenclature.
| mRNA/DNA clone | Protein |
|---|---|
| Reference, wild type | hCAR-REF |
| 12 bp insertion, exon 6–7 splice variant | hCAR-4aaINS |
| 15 bp insertion, exon 7–8 splice variant | hCAR-5aaINS |
| Double insertion, combined 6–7 and 7–8 splice variant | hCAR-dblINS |
| Exon 7 deletion | hCAR-39aaDEL |
The reference isoform of hCAR is a 348 amino acid protein with a theoretical molecular weight of 39.558 kDa. The genomic sequence of human CAR is located in contig NT_026945.9. As shown in Figure 1A and B, the reference mRNA isoform is composed of nine exons (NCBI LocusLink Report number 9970) (Fig. 1A and B) and the DNA-binding domain is encoded by exons 2, 3 and 4 (Fig. 1A). A small hinge domain is localized to a portion of exon 4, while the ligand-binding domain is encoded by exons 4–8 together with the 5′ portion of exon 9 (Fig. 1A).
The 12 bp insertion hCAR is localized between exon 6 and exon 7 and is produced by an alternative 3′ acceptor site that incorporates 12 nucleotides of intron 6 (Fig. 1B). The transcript encodes an in-frame insertion of 4 amino acids (SPTV) in the ligand-binding domain (Fig. 1C). The theoretical molecular weight of this isoform is 39.942 kDa. It is noteworthy that an isoform of CAR was cloned recently from rhesus monkey (GenBank accession: AY116212) that contains a 12 bp insertion identical to that identified for hCAR, perhaps indicating that the respective 4 amino acid CAR isoform is primate specific. A similar 4 amino acid insertion (SLSR), produced by apparently identical splicing events, occurs in both human and mouse RXRβ3 (33–35).
The hCAR 15 bp insertion occurs between exon 7 and exon 8 and is produced by an alternative 3′ acceptor site that incorporates 15 bp of intron 7 (Fig. 1B). The transcript encodes an in-frame insertion of 5 amino acids (APYLT) (Fig. 1C). The theoretical molecular weight of this isoform is 40.103 kDa. Independent confirmation of the existence of the transcript is found in the AceView database (LocusID 9970, mRNA variant c and d). Following sequence alignment and structural analysis, a similar insertion point of 8 amino acids (GVYTFLSS) was identified in estrogen receptor α, and shown to be involved in receptor dimerization (36).
CAR transcripts that encode a deletion of exon 7 are predicted to result in a protein containing 39 fewer amino acids. The theoretical molecular weight of this CAR isoform is 35.042 kDa. Independent confirmation of the exon 7 deletion transcript is indicated by examination of the Aceview database (LocusID 9970, mRNA variant c).
mRNAs encoding the variant isoforms of hCAR exist in all human livers
Clones of the variant hCAR isoforms were observed in cDNA derived from four human livers. However, the question still existed as to whether the mRNAs encoding the isoforms occur at large in the human population. To evaluate this issue, PCR amplifications using primer sets designed to detect each of the identified different isoforms of hCAR were performed on cDNA samples derived from a tissue bank of 25 human livers. Each of the hCAR isoforms was detected readily by PCR analyses in all of the 25 liver samples (data not shown). Data from representative groups of liver samples are presented in Figure 2. Different livers are represented in the respective panels due to limitations in the available tissue resource. The amplification of the exon 6–7 junction produced two bands that were sequence verified to correspond to cDNA encoding the hCAR-REF isoform and the cDNA encoding the hCAR-4aaINS isoforms, respectively (Fig. 2A). Amplification of the exon 7–8 junction produced three bands, two of which corresponded to the anticipated hCAR-REF and hCAR-5aaINS clones (Fig. 2B). When the third lower mobility band was subjected to DNA sequence analysis, the data demonstrated that the fragment encoded only the 15 bp CAR insertion. This same banding pattern was reproduced by amplification with different primer pairs and different cDNA preparations and, therefore, its migration is anomalous. The exon 7 deletion isoform of hCAR was detected by use of an exon 6 forward primer and exon 8 reverse primer. In Figure 2C, a doublet band is present in the position of the exon 7 deletion amplicon, probably consisting of an mRNA isoform containing the exon 7 deletion plus 15 nucleotides of intron 7 (as found in the mRNA encoding the hCAR-5aaINS). The existence of the latter isoform was supported by data contained within Aceview database (LocusID 9970, mRNA variant c).
Figure 2.

Alternatively spliced mRNAs encoding the reference, 12 bp insertion, 15 bp insertion and the exon 7 deletion of hCAR are present in human liver. cDNAs derived from different individuals are noted above each the gel. (A) Amplicons from PCR of human liver cDNA using the primers Exon 6 FP and Exon 7 RP. The amplicon from the mRNA isoform containing the 12 bp insertion runs slightly higher than the reference isoform. Identical amplifications were performed on clones of these two isoforms and used as markers (not shown). (B) Amplicons from PCR of human liver cDNA using the primers Exon 7 FP and Exon 8 RP. The amplicon from the mRNA isoform containing the 15 bp insertion runs slightly higher than the reference isoform. Identical amplifications were performed on clones of these two isoforms in order to run as markers; these lanes are noted as cREF (reference clone) and c15 (15 bp insertion clone). (C) Amplicons from PCR of human liver cDNA using the primers Exon 6 FP and Exon 8 RP. The amplicon from the mRNA isoform containing the deletion of exon 7 runs lower than the reference isoform. Identical amplifications were performed on clones of two isoforms to use as markers, noted as cREF (reference clone) and cDEL (exon 7 deletion).
Western blot of human liver extracts using hCAR antibody reveals several immunoreactive species
We assessed whether hCAR variants were detectable as stable protein isoforms in human liver samples. Western immunoblot analyses were conducted using the soluble fractions of human liver whole-cell lysates (Fig. 3). The antibody used in these experiments targets an epitope external to the regions of polymorphism and is therefore capable of detecting all hCAR isoforms described in this report (Fig. 1D). In addition, the antibody produces only minimal background immunoreactivity within the 30–50 kDa size range (Fig. 4B and C, empty vector lanes). However, the antibody reacted with four distinctive immunoreactive bands, apparent in Figure 3. The strongest band (no. 2) migrates at ∼40 kDa, corresponding to the estimated size of the CAR REF isoform, as well as the 4aaINS, 5aaINS and dblINS isoforms. Because these isoforms differ from one another by only a few amino acids, their unambiguous resolution is difficult using 10% PAGE. However, additional immunoreactive protein bands were detected in several liver samples, including band no. 3, migrating at ∼35 kDa, a size anticipated to correspond to the hCAR-39aaDEL isoform. Immunoreactive bands were also apparent just below 50 kDa (no. 1) and just below 35 kDa (no. 4). Despite the difficulty inherent in resolving proteins differing by <1 kDa, the western immunoblot data provided suggestive evidence for an array of immunoreactive CAR protein species in human liver.
Figure 3.
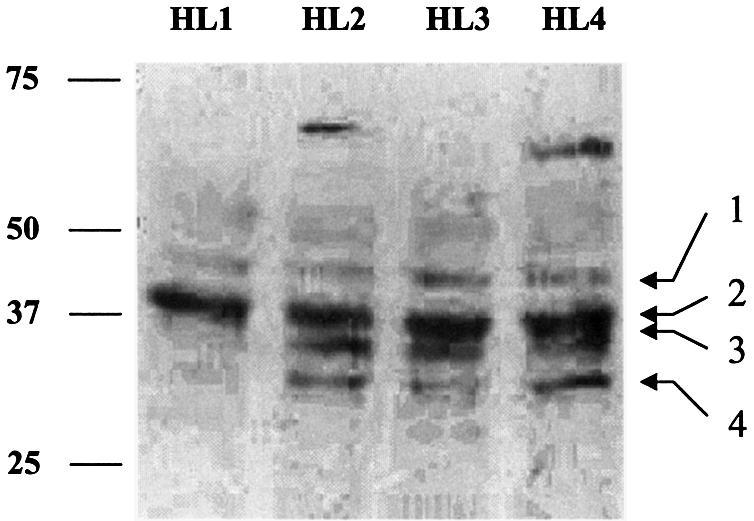
Multiple hCAR immunoreactive species are detected in whole-cell liver extracts. A western immunoblot of 200 µg/lane, whole-cell lysate from four different human livers is presented. Proteins were separated by electrophoresis on a 7.5% polyacrylamide gel and transferred to a PDVF membrane. The blot was probed with an antibody preparation generated in our laboratory (described in Materials and Methods). A number of immunoreactive bands (labeled 1–4) are apparent within the size range predicted for hCAR isoforms.
Figure 4.

hCAR protein isoforms are stable when transiently expressed. (A) hCAR isoforms were expressed using a rabbit reticulocyte lysate system as described in Materials and Methods. Product proteins were visualized by autoradiography of 35S-labeled proteins. (B) hCAR isoforms expressed in bacteria [BL21 (DE3)]. A total of 20 µg/lane of crude bacterial lysate was applied. (C) COS-1 cells were transfected with 2 µg of empty expression plasmid or expression plasmid containing different hCAR isoforms. Whole-cell lysates were prepared from the transfected cells and 30 µg/lane of total lysate was applied to the gel. hCAR antibody developed in our laboratory was used to detect immunoreactive species.
hCAR isoforms can be expressed to full length in a number of recombinant protein expression systems
We questioned whether the cloned variant hCAR cDNAs were capable of producing stable full-length proteins. To this end, hCAR protein isoforms were expressed in three separate systems, in E.coli, in a rabbit reticulocyte lysate system and in mammalian cells transfected with the respective expression constructs. Figure 4A presents the results of the TNT- coupled transcription and reticulocyte translation assays, conducted in the presence of [35S]methionine with subsequent visualization of the proteins separated by SDS–PAGE. The data demonstrate that all of the identified hCAR isoforms are expressed and resolved in this system. The full-length and 4, 5 or 9 (dbl) amino acid variant hCARs migrated at ∼40 kDa, with slight, yet discernible separation of the bands as predicted by theoretical molecular weight analysis. The hCAR-39aaDEL isoform was also clearly apparent at its correspondingly lower molecular weight. In addition, Figure 4B illustrates that each of the respective hCAR protein isoforms is expressed stably in E.coli. Similarly, Figure 4C further demonstrates the expression of each hCAR variant in mammalian cells, using transfection approaches in COS-1 cells.
Variant hCAR transcripts encode modifications of the ligand-binding domain
We conducted computer homology modeling studies to better assess the potential structural impact of the various amino acid insertions and to predict their possible functional effects. Currently, a crystal structure does not exist for any mammalian CAR protein. Therefore, our modeling approach required the use of homologous structures in the crystal database, and several criteria were taken into consideration. First, since all structural variation identified in our study mapped to the hCAR ligand-binding domain (amino acids 102–348), it was required that the homologous template structure must align to that region. Secondly, in order to generate a model of significant accuracy, the template needed to exhibit a sequence identity of >30% (37). Thirdly, since CAR is constitutively active, it was desirable to identify a template that was crystallized in the presence of an agonist and therefore likely to exhibit the structural state of an active receptor. A Psi-blast of the protein sequences database derived from the Brookhaven Protein Data Bank was performed with the hCAR amino acid sequence as the query. The iterative search revealed two possible model structures that fit the above criteria. hPXR (1ILH) (38) exhibited 57% sequence identity to hCAR, while hVDR (1DB1) (31) exhibited 37% sequence identity. Relative to the CAR structure, hPXR and hVDR contain large insertions between helix 1 and helix 3 of their respective ligand-binding domains that do not align with hCAR. Although the amino acid sequence of the crystallized hPXR protein retains this insertion, the insertion was largely removed from the sequence of the crystallized hVDR protein (31) (Fig. 5C). Therefore, despite the higher overall sequence identity with hPXR, hVDR (1DB1) was selected as the most appropriate template structure to model the hCAR variant receptors.
Figure 5.


hCAR structure model suggests that hCAR isoforms possess altered ligand-binding domain structure. (A) hCAR-REF was modeled using the hVDR X-ray crystallographic structure [PDB accession code 1DB1(31)] as a template. α-Helices are labeled 1–12. β-Strands are labeled S1, 2 or 3, according to the VDR structure. (B) hCAR-dblINS was generated using the same procedure as for the hCAR-REF model. Secondary structure is labeled as in the reference model. Inserted amino acid sequences are shown in green and are labeled with their corresponding amino acid sequences. The 4 amino acid insertion (SPTV) is predicted to extend α-helix 6 in the C-terminal direction. The 5 amino acid insertion (APYLT) is predicted to extend the loop between α-helices 8 and 9. (C) hCAR (upper sequence) was aligned with the sequence of hVDR (lower sequence) used in the generation of the 1DB1 structure. α-Helices are indicated by asterisks, and β-strands are indicated by dashes. The locations of amino acid insertions are indicated by arrows and shown as text in bold font above the sequences. The location of the 51 amino acid deletion in the VDR model is also indicated.
Figure 5A illustrates a model of hCAR-REF indicating features of a fold that are typical of a nuclear receptor ligand-binding domain, including 10 α-helices and a single β-sheet composed of three β-strands. The root mean square deviation between our model and hVDR over 245 superimposed Cα positions was 0.61 Å. Visual evaluation of the superimposed structures of hCAR-REF and 1DB1 indicated little deviation within the core of the structure (helix 3–helix 10/11) with the exception of the loop between helix 9 and 10, the loop between helix 3 and 4, and the β-turn between S2 and S3. In regions corresponding to the loop between helix 1 and helix 3, and the loop between H10/11 and H12, there was substantial deviation of the model from the template due to additional amino acids present in 1DB1 that are not present in hCAR. In addition to this deviation predicted for the hCAR-REF structure, the 4 or 5 amino acid insertions embedded in the hCAR isoforms produced marked deviation from the local structural alignment.
Figure 5B depicts the predicted effects resulting from the incorporation of the 4 amino acid insertion, SPTV CAR variant. The predicted structural impact of this insertion includes a C-terminal extension of helix 6 producing an alteration in the structure of the ligand-binding cavity (Fig. 5C). This prediction is supported by modeling studies of hCAR performed by others that noted the integral role of helix 6 in the formation of the ligand-binding pocket (5).
Insertion of the five amino acids (APYLT), corresponding to the 15 bp insertion hCAR variant, expands the loop between helix 8 and helix 9 (Fig. 5B and C). Superimposing our hCAR model containing the 5 amino acid insertion over that of PPARγ, within the PPARγ–RXRα heterodimer structure, 1FM9 (39), enables the prediction that the 5 amino acid insertion in hCAR would produce a bulged loop within this domain, probably resulting in steric hindrance of the dimerization potential of CAR with RXR.
Removal of the amino acids encoded by the exon 7 deletion variant characterized in this report is consistent with the removal of α-helices 7 and 8 of the CAR reference protein structure, and therefore is likely to result in the collapse of the hCAR ligand-binding domain.
Variant hCAR proteins exhibit compromised DNA binding
A well-characterized property of the reference form of hCAR is its ability to bind a subset of DNA response elements as a heterodimeric complex with RXRα (14,16). The predicted structural variation inherent in the identified hCAR isoforms predominantly impacts the protein domain participating in heterodimerization, a prerequisite for DNA binding. Therefore, it is possible that the variant isoforms of hCAR exhibit altered DNA-binding capacities.
To examine this latter issue, we performed EMSAs using NR1 and NR2 elements derived from the CYP2B6 PBREM (16) using expressed, affinity-purified, GST fusion proteins of hRXRα and the hCAR isoforms (Fig. 6A). GST alone did not bind to either element (Fig. 6B, lane 1). We were surprised to find that all of hCAR isoforms interacted with DNA in the absence of RXR (Fig. 6B, lanes 2–5), in particular the NR2 element. These results indicate that hCAR may possess a limited ability to homodimerize. Similarly, hRXRα alone interacted with the NR1 element, although not with NR2 (Fig. 6, lane 6). hCAR-REF–RXR bound robustly to both the NR1 and NR2 elements (Fig. 6, lane 7). The hCAR-4aaINS–RXR complex interacted with both the NR1 and NR2 elements, albeit at much weaker levels than the hCAR-REF–RXR complex (Fig. 6, lane 8 versus lane 7). The two remaining hCAR isoforms also appeared to bind weakly to NR1 elements in the presence of RXR. Since GST itself may dimerize and therefore mask apparent dimerization of GST–CAR with GST–RXR, we conducted additional studies in the presence of an excess (50×) of free GST. Inclusion of excess free GST did result in a reduced NR1 DNA interaction for the hCAR-5aaINS–RXR and hCAR-dlbINS–RXR complexes, but excess GST did not affect any of the remaining protein–DNA interactions (data not shown). As might be predicted from the extent of the deletion, results from additional experiments employing the hCAR-39aaDEL construct demonstrated that it lacked the ability to bind either the NR1 or NR2 element as a heterodimeric complex with RXR (data not shown).
Figure 6.


hCAR-REF and hCAR-4aaINS bind to the CYP2B6 PBREM NR1 and NR2 elements. EMSA studies were performed using recombinantly expressed proteins (A). (B) GST alone did not bind the respective DNA elements (lane 1, top and bottom gel). All hCAR isoforms appeared to interact with the tested DNA elements in the absence of RXRα, forming a more rapidly migrating complex (lanes 2, 3, 4 and 5). This effect was more pronounced with the NR2 element. RXRα bound weakly to the NR1, but not the NR2 element (lane 6, top gel). The hCAR-REF–RXRα complex bound to both of the previously characterized hCAR response elements (lane 7, top and bottom gels). The hCAR-4aaINS–RXRα complex also bound to both elements, although this interaction appeared to be weaker when compared with the hCAR-REF complex. In the top gel, it appears that the hCAR-dblINS–RXRα complex bound DNA (lane 10, top gel); however, subsequent attempts to reproduce this result were unsuccessful.
Variant hCAR proteins exhibit compromised transactivation
The reference form of hCAR transactivates gene expression through a number of response elements observed in PB-responsive genes, with the human PBREM response element of CYP2B6 representing a well-characterized region (16). We generated a PBREM-TK-luciferase reporter and evaluated the transactivation capacity of the different hCAR isoforms in co-transfection studies. Reporter expression was assessed following co-transfection with an RXRα expression plasmid, the Renilla luciferase reporter (for transfection normalization), and either hCAR reference, hCAR variants or empty vector expression plasmids. The reference isoform induced the reporter expression ∼3-fold over the empty vector control (Fig. 7A). The only other isoform noted to exhibit activity was hCAR-4aaINS, which activated the reporter ∼2-fold (Fig. 7A). A TK-luciferase reporter without the PBREM sequence was not activated by any of the hCAR isoforms.
Figure 7.

hCAR-REF and hCAR-4aaINS transactivate a reporter containing the PBREM sequence. Transfections were performed in COS-1 cells. All data are presented as values normalized to Renilla luciferase. (A) Transfection of hCAR-REF in combination with hRXRα resulted in an ∼3-fold activation of the PBREM reporter. Identical transfections with hCAR-4aaINS led to 2-fold activation of the same reporter. Transfection of hRXRα with empty expression vector or any of the other variants isoforms of hCAR did not result in detectable transactivation. (B) A 100 ng aliquot of hCAR-REF and 50 ng of hRXRα (1× REF/RXR) were co-transfected with either 200 ng (2×) of empty vector (V), 4aaINS, 5aaINS, dblINS or 39aaDEL hCAR expression constructs. None of the hCAR variant isoforms exhibited dominant-negative activity. However, the 4aaINS isoform appears to cooperate with hCAR-REF in transactivating the reporter. (C) Cells were transfected as before and treated with either vehicle (acetone) or 10 µM clotrimazole. As expected, hCAR-REF transactivation activity was diminished by addition of clotrimazole. The transactivation by the 4aaINS isoform was also suppressed by clotrimazole. Each bar represents the mean ± SD of four separate measurements.
We reasoned that hCAR variants may function to transactivate reporter expression in a secondary manner, e.g. acting in concert with hCAR-REF, or perhaps through dominant-negative mechanisms. Remarkably, the hCAR-4aaINS isoform appeared to act in concert with the reference isoform when transactivating the reporter (Fig. 7B). However, when the reference isoform of hCAR was co-transfected sequentially with a 2-fold molar excess of each of the other hCAR variants, no further alterations of hCAR-REF activity were observed (Fig. 7B).
Another property of reference hCAR that can be evaluated using the transfection approach is repression of its transactivation potential by the ligand clotrimazole (22). Clotrimazole additions repressed hCAR-REF transactivation, and also repressed transactivation stimulated by the hCAR-4aaINS isoform. Clotrimazole additions produced no observable effect on any of the other hCAR isoforms (Fig. 7C).
Variant hCAR proteins exhibit weaker interactions with the SRC-1 receptor interaction domain
It has been reported that CAR interacts with SRC-1 in the absence of ligand (15). Analysis of our model, whereby hCAR was superimposed upon 1FM9 (the 1FM9 structure was derived by co-crystallization with an SRC-1 coactivator fragment), led us to predict that the hCAR variant insertions would not disrupt the interaction with SRC-1. To evaluate the SRC-1 interactions more directly, we performed protein interaction assays with in vitro translated 35S-labeled hCAR protein (Fig. 8A) and the human SRC-1 RID fused to GST (Fig. 8B). The interaction of GST–SRC-1 with hCAR-REF was robust (Fig. 8C, lane 2), with no detectable interaction apparent for hCAR-REF and GST alone (Fig. 8C, lane 1). Interactions of the hCAR variant isoforms with GST–SRC-1 were weaker, but specific (Fig, 8C; lanes 4, 6, 8 and 10).
Figure 8.

All isoforms of hCAR interact specifically with the SRC-1 RID. (A) 35S-Labeled hCAR protein isoforms expressed in rabbit reticulocyte lysate used for the GST pull-down experiments. Bands representing the [35S]hCAR isoforms are indicated by arrows. (B) Bacterially expressed GST and GST–SRC-1-RID used in the GST pull-down experiments. Arrows indicate bands of the expected size. (C) GST pull-down experiments using immobilized GST–SRC-1-RID (even lanes) or GST (odd lanes) proteins and reticulocyte lysate expressed [35S]hCAR protein isoforms. hCAR-REF interacted robustly and specifically with GST–SRC-1-RID (lane 2 versus lane 1). The other hCAR isoforms interacted specifically with GST–SRC-1-RID, but the interactions were weaker than that obtained with the reference isoform. Equal input c.p.m. levels were applied to each pull-down reaction.
Discussion
Previous reports have documented alternative splicing events in the mouse and human that likely impart structural and functional diversity within the NR1I subfamily (39–44). PXR and VDR splice variants have been described that appear to possess altered receptor activities (40,42–44). An alternatively spliced isoform of mCAR was identified in earlier studies (39). More recently, data obtained from the use of ribonuclease protection assays suggested alternative splicing within the ligand binding domain of hCAR (41). Combined, these observations lend support to the concept that alternative splicing may serve as an important mechanism for expanding the repertoire of xenobiotic receptors available to the mammalian cell.
In this investigation, we have identified and partially characterized activities associated with four novel isoforms of hCAR. These isoforms are encoded by alternatively spliced transcripts of the hCAR gene. One transcript incorporates 12 nucleotides of intron 6 through an alternative splice acceptor site, encoding an in-frame 4 amino acid insertion product, the hCAR-4aaINS isoform. Another transcript incorporates 15 nucleotides of intron 7 through an alternative splice acceptor site, encoding an in-frame 5 amino acid insertion variant, the hCAR-5aaINS isoform. A third transcript incorporates both splicing alterations simultaneously, encoding hCAR-dblINS. The fourth variant transcript, encoding hCAR-39aaΔ, results from a complete deletion of exon 7, also translationally in-frame. Each of the respective hCAR isoforms are predicted to possess altered structure, residing in the receptor’s E domain. The E domain, commonly referred to as the ligand-binding domain, performs integrated functions of dimerization, coregulator recruitment and transactivation (45). In this report, these functions were evaluated for each of the variant hCAR receptors.
An important issue in defining receptor function involves demonstration of its stable expression, both in vivo and in vitro. We have presented evidence supporting the biological relevance of variant hCAR receptors. First, mRNA transcripts encoding each of the hCAR isoforms characterized herein were detected using RT–PCR analysis in 25 human liver tissues sampled to date. Secondly, each of the hCAR protein transcripts was efficiently and stably generated in multiple in vitro expression systems, including transformed bacteria, reticulocyte lysates and transfected mammalian cells. Thirdly, each of the identified hCAR isoforms codes for in-frame translation variants and therefore would not be expected to be targeted by nonsense-mediated mRNA decay mechanisms that degrade premature termination products (46). Fourthly, western blots derived from human liver extracts suggest the presence of multiple CAR protein isoforms.
Amplification of cDNA derived from liver samples indicated that variant isoforms of hCAR are likely to be present in all humans. However, the RT–PCR analysis presented here does not yield an accurate account of the abundance and ratios of these transcripts. These values may exhibit a large interindividual variability. It is already known that hCAR mRNA transcript levels vary significantly between liver samples (47), and that activation of certain signaling cascades alters levels of CAR mRNA (41,48). Consequently, one intriguing question is whether genetic variables play a role in expression of hCAR mRNA and/or the ratio of CAR transcripts. A precedent for signal transduction cascades that directly impinge on pre-mRNA splicing events has been presented (49). Hence, a further intriguing question is whether treatments with dexamethasone or interleukin-6 affect the ratio of CAR transcripts and, in turn, what effect such exposures may have on responsiveness to inducing agents.
EMSA analysis revealed that of the hCAR variants assessed, the hCAR-4aaINS isoform was uniquely capable of interacting with both NR1 and NR2 elements, when heterodimerized with RXR. Based on our results from computer homology modeling, a potential explanation for the latter result is advanced. Insertion of the 5 amino acid sequence, APYLT, between helices 8 and 9 was predicted to stericly hinder the dimerization interface with RXR. Even more dramatically, removal of 39 amino acids, as encoded by the exon 7 deletion variant, would be likely to lead to the collapse of the dimerization interface.
In our studies, the DNA interaction of the hCAR-4aaINS–RXR was weak relative to the reference hCAR isoform complex. The hCAR modeling analysis predicts that the 4 amino acid insertion point for this variant receptor lies immediately adjacent to the dimerization interface and probably results in a C-terminal extension of helix 6, extending the helix in the direction of the dimerization interface. Thus the 4 amino acid insertion is also predicted to impact the receptor’s heterodimerization and DNA-binding functions, supporting the EMSA results presented here. Studies of hRXRβ3 that contains a 4 amino acid insertion in a structurally homologous position also reveal a compromised ability to form heterodimers with the retinoic acid receptor (35). It is of interest to note that Mahajna et al. (35) also noted that the hRXRβ variant possessed an enhanced capacity to homodimerize, suggesting an alternative means by which the hCAR-4aaINS might interact with DNA (35). In addition, it is possible that the rather weak interaction of the hCAR-4aaINS–RXR heterodimer with DNA in vitro may be more robust in vivo. For example, accessory factors may bind the PBREM sequence to form an enhanceosomal architecture, facilitating the interaction of the variant hCAR-4aaINS complex with its cognate response element. It is also possible that the hCAR-4aaINS isoform may be subjected to unique post-translational modifications in COS-1 cells, modifications that may modify its capacity to bind DNA. A number of nuclear receptors undergo post-translational modification by phosphorylation, consequently modifying receptor function (50). Preliminary analysis of the of hCAR-4aaINS amino acid sequence using NetPhos 2.0 (51) indicates that the inserted amino acids possess a high-scoring putative phosphorylation site (0.992) at the serine residue of the peptide GARVSPTVG. Thus, alternative regulation strategies may impact hCAR receptor function and need to be evaluated further.
Studies conducted with the thyroid hormone receptor have suggested that structural alterations of the ligand-binding domain can alter the repertoire of DNA-binding elements involved in receptor interaction (52). It therefore seems reasonable to speculate that variant isoforms of hCAR may bind to a different subset of response elements, distinct from previously defined hCAR–RXR response elements, as tested here. The biological implications of this suggestion may be important, as variant isoforms of hCAR may then be involved in the regulation of different subsets of human genes, possibly distinct from that of the reference form of hCAR. Efforts are underway in the laboratory to evaluate these possibilities.
Since all the hCAR isoforms assessed in the current studies were capable of interacting specifically with nuclear receptor coactivator SRC-1, two possibilities are suggested: (i) that the isoforms of hCAR may be capable of dominant-negative activity by acting to squelch coactivator recruitment; and/or (ii), as suggested above, that the variant hCAR isoforms may be capable of interacting with currently undefined DNA response elements, or might be recruited to promoters through other bridging factors, thereby modulating the transcriptional process through recruitment of coregulatory factors to target promoters. Results of transfection experiments presented in Figure 7B indicated that the former suggestion is not likely to be correct. In order to test the latter suggestion, experiments are currently underway examining the effects of artificial recruitment of hCAR isoforms to DNA using mammalian two-hybrid strategies that allow the evaluation of coactivator recruitment.
The variant isoforms of hCAR are predicted to encode structurally unique ligand-binding domains. Molecular modeling analysis reveals that the ligand-binding pocket structure would probably not be impacted in the hCAR-5aaINS insertion variant. However, the hCAR-4aaINS variant is predicted to impart a direct extension of helix 6, probably resulting in structural alteration of the ligand pocket. Receptors with this amino acid insertion would also be likely to possess an alteration in the pocket’s side chain composition, potentially resulting in altered ligand binding specificity of the isoform, an observation that would be agreement with studies of mouse RXRβ3 (34). This prediction raises an important question: does this hCAR isoform respond to a set of ligands/pharmacophores that are distinct from the reference CAR isoform? One ligand evaluated in the current investigation, clotrimazole, exhibited inverse agonist activity with both the reference and the 4aaINS isoforms of hCAR. It remains curious that, unlike clotrimazole, PB and a number of PB-like inducing agents have been identified that are not ligands for the reference CAR isoforms (15,19,22). This observation leads to the compelling question as to whether certain inducing compounds may interact alternatively with the ligand-binding pocket of hCAR-4aaINS, or other CAR isoforms. Assays are being developed to evaluate this intriguing question.
Alternative splicing of mRNA is one of the primary avenues for diversifying the proteome, and this process presents itself as a representation of nature’s efficiency, enabling an array of functions to be encoded in a single gene. The isoforms of hCAR presented in this study may be an example of this efficiency. Diversifying the proteome of the xenosensor system by mechanisms such as alternative splicing would probably facilitate an organism’s ability to adapt to its chemical environment. Further characterization of the variant isoforms of hCAR in animal models will be likely to lead to deciphering specific biological functions in the adaptive xenobiotic induction pathway.
Acknowledgments
Acknowledgements
The authors would like to acknowledge Samir Kalada, Heather Klintworth and Sean Boyle for their technical contributions and fruitful discussions. This paper is dedicated to the memory of Dr Christopher Hassett, a loyal friend, outstanding colleague and meticulous scientist. This study was supported by a grant from the National Institute of General Medical Sciences, GM66411 (C.J.O.), a Center Grant from the National Institute of Environmental Health Sciences (ES07033) and an NIEHS Training Grant, ES07032 (S.S.A.).
REFERENCES
- 1.Goodwin B., Hodgson,E., D’Costa,D.J., Robertson,G.R. and Liddle,C. (2002) Transcriptional regulation of the human CYP3A4 gene by the constitutive androstane receptor. Mol. Pharmacol., 62, 359–365. [DOI] [PubMed] [Google Scholar]
- 2.Kast H.R., Goodwin,B., Tarr,P.T., Jones,S.A., Anisfeld,A.M., Stoltz,C.M., Tontonoz,P., Kliewer,S., Willson,T.M. and Edwards,P.A. (2002) Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor and constitutive androstane receptor. J. Biol. Chem., 277, 2908–2915. [DOI] [PubMed] [Google Scholar]
- 3.Sueyoshi T. and Negishi,M. (2001) Phenobarbital response elements of cytochrome P450 genes and nuclear receptors. Annu. Rev. Pharmacol. Toxicol., 41, 123–143. [DOI] [PubMed] [Google Scholar]
- 4.Sugatani J., Kojima,H., Ueda,A., Kakizaki,S., Yoshinari,K., Gong,Q.H., Owens,I.S., Negishi,M. and Sueyoshi,T. (2001) The phenobarbital response enhancer module in the human bilirubin UDP-glucuronosyltransferase UGT1A1 gene and regulation by the nuclear receptor CAR. Hepatology, 33, 1232–1238. [DOI] [PubMed] [Google Scholar]
- 5.Xiong H., Yoshinari,K., Brouwer,K.L. and Negishi,M. (2002) Role of constitutive androstane receptor in the in vivo induction of Mrp3 and CYP2B1/2 by phenobarbital. Drug Metab. Disposition, 30, 918–923. [DOI] [PubMed] [Google Scholar]
- 6.Cherkaoui-Malki M., Meyer,K., Cao,W.Q., Latruffe,N., Yeldandi,A.V., Rao,M.S., Bradfield,C.A. and Reddy,J.K. (2001) Identification of novel peroxisome proliferator-activated receptor alpha (PPARalpha) target genes in mouse liver using cDNA microarray analysis. Gene Expr., 9, 291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maglich J.M., Stoltz,C.M., Goodwin,B., Hawkins-Brown,D., Moore,J.T. and Kliewer,S.A. (2002) Nuclear pregnane X receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol. Pharmacol., 62, 638–646. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt J.V. and Bradfield,C.A. (1996) Ah receptor signaling pathways. Annu. Rev. Cell. Dev. Biol., 12, 55–89. [DOI] [PubMed] [Google Scholar]
- 9.Ueda A., Hamadeh,H.K., Webb,H.K., Yamamoto,Y., Sueyoshi,T., Afshari,C.A., Lehmann,J.M. and Negishi,M. (2002) Diverse roles of the nuclear orphan receptor CAR in regulating hepatic genes in response to phenobarbital. Mol. Pharmacol., 61, 1–6. [DOI] [PubMed] [Google Scholar]
- 10.Wei P., Zhang,J., Egan-Hafley,M., Liang,S. and Moore,D.D. (2000) The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature, 407, 920–923. [DOI] [PubMed] [Google Scholar]
- 11.Wei P., Zhang,J., Dowhan,D.H., Han,Y. and Moore,D.D. (2002) Specific and overlapping functions of the nuclear hormone receptors CAR and PXR in xenobiotic response. Pharmacogenom. J., 2, 117–126. [DOI] [PubMed] [Google Scholar]
- 12.Nuclear Receptors Nomenclature Committee (1999) A unified nomenclature system for the nuclear receptor superfamily. Cell, 97, 161–163. [DOI] [PubMed] [Google Scholar]
- 13.Robyr D., Wolffe,A.P. and Wahli,W. (2000) Nuclear hormone receptor coregulators in action: diversity for shared tasks. Mol. Endocrinol., 14, 329–347. [DOI] [PubMed] [Google Scholar]
- 14.Baes M., Gulick,T., Choi,H.S., Martinoli,M.G., Simha,D. and Moore,D.D. (1994) A new orphan member of the nuclear hormone receptor superfamily that interacts with a subset of retinoic acid response elements. Mol. Cell. Biol., 14, 1544–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tzameli I., Pissios,P., Schuetz,E.G. and Moore,D.D. (2000) The xenobiotic compound 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene is an agonist ligand for the nuclear receptor CAR. Mol. Cell. Biol., 20, 2951–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sueyoshi T., Kawamoto,T., Zelko,I., Honkakoski,P. and Negishi,M. (1999) The repressed nuclear receptor CAR responds to phenobarbital in activating the human CYP2B6 gene. J. Biol. Chem., 274, 6043–6046. [DOI] [PubMed] [Google Scholar]
- 17.Dussault I., Lin,M., Hollister,K., Fan,M., Termini,J., Sherman,M.A. and Forman,B.M. (2002) A structural model of the constitutive androstane receptor defines novel interactions that mediate ligand-independent activity. Mol. Cell. Biol., 22, 5270–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forman B.M., Tzameli,I., Choi,H.S., Chen,J., Simha,D., Seol,W., Evans,R.M. and Moore,D.D. (1998) Androstane metabolites bind to and deactivate the nuclear receptor CAR-beta. Nature, 395, 612–615. [DOI] [PubMed] [Google Scholar]
- 19.Makinen J., Frank,C., Jyrkkarinne,J., Gynther,J., Carlberg,C. and Honkakoski,P. (2002) Modulation of mouse and human phenobarbital-responsive enhancer module by nuclear receptors. Mol. Pharmacol., 62, 366–378. [DOI] [PubMed] [Google Scholar]
- 20.Min G., Kemper,J.K. and Kemper,B. (2002) Glucocorticoid receptor-interacting protein 1 mediates ligand-independent nuclear translocation and activation of constitutive androstane receptor in vivo. J. Biol. Chem., 277, 26356–26363. [DOI] [PubMed] [Google Scholar]
- 21.Seol W., Choi,H.S. and Moore,D.D. (1996) An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors. Science, 272, 1336–1339. [DOI] [PubMed] [Google Scholar]
- 22.Moore L.B., Parks,D.J., Jones,S.A., Bledsoe,R.K., Consler,T.G., Stimmel,J.B., Goodwin,B., Liddle,C., Blanchard,S.G., Willson,T.M. et al. (2000) Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J. Biol. Chem., 275, 15122–15127. [DOI] [PubMed] [Google Scholar]
- 23.Maglich J.M., Parks,D.J., Moore,L.B., Collins,J.L., Goodwin,B., Billin,A.N., Stoltz,C.A., Kliewer,S.A., Lambert,M.H., Willson,T.M. and Moore,J.T. (2003) Identification of a novel human CAR agonist and its use in the identification of CAR target genes. J. Biol. Chem., 278, 17277–17283. [DOI] [PubMed] [Google Scholar]
- 24.Kawamoto T., Sueyoshi,T., Zelko,I., Moore,R., Washburn,K. and Negishi,M. (1999) Phenobarbital-responsive nuclear translocation of the receptor CAR in induction of the CYP2B gene. Mol. Cell. Biol., 19, 6318–6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie W., Barwick,J.L., Simon,C.M., Pierce,A.M., Safe,S., Blumberg,B., Guzelian,P.S. and Evans,R.M. (2000) Reciprocal activation of xenobiotic response genes by nuclear receptors SXR/PXR and CAR. Genes Dev., 14, 3014–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann J.M., McKee,D.D., Watson,M.A., Willson,T.M., Moore,J.T. and Kliewer,S.A. (1998) The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Invest., 102, 1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moore L.B., Maglich,J.M., McKee,D.D., Wisely,B., Willson,T.M., Kliewer,S.A., Lambert,M.H. and Moore,J.T. (2002) Pregnane X receptor (PXR), constitutive androstane receptor (CAR) and benzoate X receptor (BXR) define three pharmacologically distinct classes of nuclear receptors. Mol. Endocrinol., 16, 977–986. [DOI] [PubMed] [Google Scholar]
- 28.Drocourt L., Ourlin,J.C., Pascussi,J.M., Maurel,P. and Vilarem,M.J. (2002) Expression of CYP3A4, CYP2B6 and CYP2C9 is regulated by the vitamin D receptor pathway in primary human hepatocytes. J. Biol. Chem., 277, 25125–25132. [DOI] [PubMed] [Google Scholar]
- 29.Xie W. and Evans,R.M. (2001) Orphan nuclear receptors: the exotics of xenobiotics. J. Biol. Chem., 276, 37739–37742. [DOI] [PubMed] [Google Scholar]
- 30.Sandberg M., Hassett,C., Adman,E.T., Meijer,J. and Omiecinski,C.J. (2000) Identification and functional characterization of human soluble epoxide hydrolase genetic polymorphisms. J. Biol. Chem., 275, 28873–28881. [DOI] [PubMed] [Google Scholar]
- 31.Rochel N., Tocchini-Valentini,G., Egea,P.F., Juntunen,K., Garnier,J.M., Vihko,P. and Moras,D. (2001) Functional and structural characterization of the insertion region in the ligand binding domain of the vitamin D nuclear receptor. Eur. J. Biochem., 268, 971–979. [DOI] [PubMed] [Google Scholar]
- 32.Blumberg B., Sabbagh,W.,Jr, Juguilon,H., Bolado,J.,Jr, van Meter,C.M., Ong,E.S. and Evans,R.M. (1998) SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev., 12, 3195–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Epplen C. and Epplen,J.T. (1992) The human cDNA sequence homologous to the mouse MHC class I promoter-binding protein gene contains four additional codons in lymphocytes. Mamm. Genome, 3, 472–475. [DOI] [PubMed] [Google Scholar]
- 34.Fujita A. and Mitsuhashi,T. (1999) Differential regulation of ligand-dependent and ligand-independent functions of the mouse retinoid X receptor beta by alternative splicing. Biochem. Biophys. Res. Commun., 255, 625–630. [DOI] [PubMed] [Google Scholar]
- 35.Mahajna J., Shi,B. and Bruskin,A. (1997) A four-amino-acid insertion in the ligand-binding domain inactivates hRXRbeta and renders dominant negative activity. DNA Cell Biol., 16, 463–476. [DOI] [PubMed] [Google Scholar]
- 36.Gampe R.T. Jr, Montana,V.G., Lambert,M.H., Miller,A.B., Bledsoe,R.K., Milburn,M.V., Kliewer,S.A., Willson,T.M. and Xu,H.E. (2000) Asymmetry in the PPARgamma/RXRalpha crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Mol. Cell, 5, 545–555. [DOI] [PubMed] [Google Scholar]
- 37.Baker D. and Sali,A. (2001) Protein structure prediction and structural genomics. Science, 294, 93–96. [DOI] [PubMed] [Google Scholar]
- 38.Watkins R.E., Wisely,G.B., Moore,L.B., Collins,J.L., Lambert,M.H., Williams,S.P., Willson,T.M., Kliewer,S.A. and Redinbo,M.R. (2001) The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science, 292, 2329–2333. [DOI] [PubMed] [Google Scholar]
- 39.Choi H.S., Chung,M., Tzameli,I., Simha,D., Lee,Y.K., Seol,W. and Moore,D.D. (1997) Differential transactivation by two isoforms of the orphan nuclear hormone receptor CAR. J. Biol. Chem., 272, 23565–23571. [DOI] [PubMed] [Google Scholar]
- 40.Hustert E., Zibat,A., Presecan-Siedel,E., Eiselt,R., Mueller,R., Fuss,C., Brehm,I., Brinkmann,U., Eichelbaum,M., Wojnowski,L. et al. (2001) Natural protein variants of pregnane X receptor with altered transactivation activity toward CYP3A4. Drug Metab. Disposition, 29, 1454–1459. [PubMed] [Google Scholar]
- 41.Pascussi J.M., Gerbal-Chaloin,S., Fabre,J.M., Maurel,P. and Vilarem,M.J. (2000) Dexamethasone enhances constitutive androstane receptor expression in human hepatocytes: consequences on cytochrome P450 gene regulation. Mol. Pharmacol., 58, 1441–1450. [DOI] [PubMed] [Google Scholar]
- 42.Dotzlaw H., Leygue,E., Watson,P. and Murphy,L.C. (1999) The human orphan receptor PXR messenger RNA is expressed in both normal and neoplastic breast tissue. Clin. Cancer Res., 5, 2103–2107. [PubMed] [Google Scholar]
- 43.Ebihara K., Masuhiro,Y., Kitamoto,T., Suzawa,M., Uematsu,Y., Yoshizawa,T., Ono,T., Harada,H., Matsuda,K., Hasegawa,T. et al. (1996) Intron retention generates a novel isoform of the murine vitamin D receptor that acts in a dominant negative way on the vitamin D signaling pathway. Mol. Cell. Biol., 16, 3393–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sunn K.L., Cock,T.A., Crofts,L.A., Eisman,J.A. and Gardiner,E.M. (2001) Novel N-terminal variant of human VDR. Mol. Endocrinol., 15, 1599–1609. [DOI] [PubMed] [Google Scholar]
- 45.Steinmetz A.C., Renaud,J.P. and Moras,D. (2001) Binding of ligands and activation of transcription by nuclear receptors. Annu. Rev. Biophys Biomol. Struct., 30, 329–359. [DOI] [PubMed] [Google Scholar]
- 46.Byers P.H. (2002) Killing the messenger: new insights into nonsense-mediated mRNA decay. J. Clin. Invest., 109, 3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang T.K., Bandiera,S.M. and Chen,J. (2003) Constitutive androstane receptor and pregnane X receptor gene expression in human liver: interindividual variability and correlation with CYP2B6 mRNA levels. Drug Metab. Disposition, 31, 7–10. [DOI] [PubMed] [Google Scholar]
- 48.Pascussi J.M., Gerbal-Chaloin,S., Pichard-Garcia,L., Daujat,M., Fabre,J.M., Maurel,P. and Vilarem,M.J. (2000) Interleukin-6 negatively regulates the expression of pregnane X receptor and constitutively activated receptor in primary human hepatocytes. Biochem. Biophys. Res. Commun., 274, 707–713. [DOI] [PubMed] [Google Scholar]
- 49.Maniatis T. and Tasic,B. (2002) Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature, 418, 236–243. [DOI] [PubMed] [Google Scholar]
- 50.Shao D. and Lazar,M.A. (1999) Modulating nuclear receptor function: may the phos be with you. J. Clin. Invest., 103, 1617–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blom N., Gammeltoft,S. and Brunak,S. (1999) Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol., 294, 1351–1362. [DOI] [PubMed] [Google Scholar]
- 52.Nagaya T., Nomura,Y., Fujieda,M. and Seo,H. (1996) Heterodimerization preferences of thyroid hormone receptor alpha isoforms. Biochem. Biophys. Res. Commun., 226, 426–430. [DOI] [PubMed] [Google Scholar]
- 53.Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]