

Abstract
MENX is a recessive multiple endocrine neoplasia-like syndrome in the rat. The tumor spectrum in MENX overlaps those of human multiple endocrine neoplasia (MEN) types 1 and 2. We mapped the MenX locus to the distal part of rat chromosome 4, excluding the homologs of the genes responsible for the MEN syndromes (RET and MEN1) and syndromes with an endocrine tumor component (VHL and NF1). We report the fine mapping of the disease locus and the identification of a homozygous frameshift mutation in Cdkn1b, encoding the cyclin-dependent kinase inhibitor p27Kip1. As a consequence of the mutation, MENX-affected rats show dramatic reduction in p27Kip1 protein. We have identified a germ-line nonsense mutation in the human CDKN1B gene in a MEN1 mutation-negative patient presenting with pituitary and parathyroid tumors. Expanded pedigree analysis shows that the mutation is associated with the development of an MEN1-like phenotype in multiple generations. Our findings demonstrate that germ-line mutations in p27Kip1 can predispose to the development of multiple endocrine tumors in both rats and humans.
Keywords: cancer, tumor suppression, cyclin-dependent kinase inhibitor
In humans, the multiple endocrine neoplasia (MEN) syndromes are inherited as autosomal-dominant traits, and they are caused by mutations of either the RET protooncogene (MEN2A, MEN2B) or the MEN1 tumor-suppressor gene (MEN1). We have identified an MEN-like syndrome (MENX) in the rat (1), with a phenotypic overlap of both MEN1 and MEN2. Affected animals develop bilateral pheochromocytomas and parathyroid adenomas, multifocal thyroid C cell hyperplasia, paragangliomas (1), and endocrine pancreas hyperplasia (N.S.P. and M.J.A., unpublished observation). In addition, affected animals develop bilateral cataracts in the first few weeks of life. In contrast to the human syndromes, MENX is inherited recessively (1). We have mapped the locus to a 20-megabase region of distal rat chromosome 4 (2). This linkage analysis has excluded the rat homologs of the RET and MEN1 genes as well as other genes implicated in cancer syndromes with an endocrine tissue component (namely, VHL, NF1, and the succinate dehydrogenase subunits B, C, and D genes). Therefore, the MENX syndrome is caused by an alteration in a gene not previously associated with predisposition to syndromic endocrine cancer.
The identification of the genes responsible for MEN1 and MEN2 has enabled the genetic diagnosis, and hence early detection, for patients with suspected endocrine tumor syndromes. Noteworthy, however, is the number of patients with MEN1 who are lacking a detectable germ-line MEN1 mutation (3, 4). These patients represent a difficult group to counsel and manage clinically. It has been generally assumed that mutation-negative MEN1 patients still have mutations in the MEN1 gene but that the mutations are outside the gene regions that are typically sequenced. Genetic heterogeneity for the MEN1 phenotype in humans is not expected, and it has not been described. Theoretically speaking, mutation-negative, suspected MEN1 cases might also be caused by mutations in still-unidentified predisposing genes.
We describe the fine mapping of the MenX locus and identification of the Cdkn1b gene, which encodes the cyclin-dependent kinase inhibitor (CKI) p27Kip1, as the gene responsible for the MENX syndrome. An 8-bp tandem duplication in Cdkn1b exon 2 causes a frameshift mutation leading to extreme reduction of p27Kip1 in vivo. Supporting our conclusion that mutation of p27Kip1 is responsible for the phenotype is the presence of a germ-line nonsense CDKN1B mutation in a suspected MEN1 patient without mutations in MEN1, thus establishing an association between germ-line mutation of the putative tumor-suppressor p27 and a heritable cancer syndrome.
Results
Fine Mapping of the MenX Locus and Identification of Candidate Genes.
We performed linkage analysis of 151 animals obtained from the [Sprague–Dawley white eye (SDwe) × Wistar–Kyoto] F1 × SDwe/SDwe backcross (for nomenclature, see Materials and Methods). This analysis placed the MenX locus in a genomic segment of ≈3 megabases on rat chromosome 4 (Fig. 1A). After determining the expression profile in silico and the putative function of the genes located in this region, candidate genes were screened for mutations. Among them was Cdkn1b, encoding the CKI p27Kip1. p27Kip1 (hereafter referred to as p27) is expressed in all tissues that develop tumors in affected rats. Moreover, p27-knockout mice develop tumors in the pituitary intermediate lobe (5–7), a tissue that is also affected in the MENX syndrome. The putative tumor-suppressor function of p27 is consistent with the recessive mode of inheritance observed in MENX rats. We sequenced the Cdkn1b gene in affected and unaffected littermates as well as in seven commercially available inbred rat strains. Affected rats (hereafter indicated as mut/mut) are homozygous for a tandem duplication of 8 nt in exon 2 of Cdkn1b: c. 520–528dupTTTCAGAC (NM_1020;013762.2) (Fig. 1B). This mutation was not found in any inbred rat strain. The 8-nt insertion results in a frameshift after codon 177 (p. G177fs), predicting a different C-terminal domain containing 42 p27-unrelated amino acid residues (Fig. 1C). The C terminus of the mutant p27 protein (hereafter referred to as p27_1020;G177fs) has no homology to proteins in the Swiss-Prot database.
Fig. 1.

Mapping and cloning of the MENX mutation. (A) Haplotype analysis of affected and unaffected animals produced from a (SDwe × Wistar–Kyoto) F1 × SDwe/SDwe backcross for nine microsatellite markers located in the linkage-defined MENX candidate region. Filled areas indicate homozygosity for a given microsatellite marker; empty areas indicate heterozygosity. The number of animals with a specific haplotype and their disease status are listed on the right. Mb, megabases. (B) Pedigree of a breeding group segregating the MENX phenotype. Genomic DNA of the animals was amplified with primers spanning the Cdkn1b mutation, and the alleles (WT and mutated) were resolved on polyacrylamide gels. For each Cdkn1b genotype, the corresponding sequence chromatogram is shown. The insertion in the mut/mut rat sequence is indicated by a rectangle. Open square/circle, unaffected male/female; filled square/circle, affected male/female. M, molecular size marker. (C) Alignment of annotated rat wild-type p27 protein sequence (wt) with the predicted mutant p27 protein (mut). The mutant protein is predicted to be 21 amino acids longer than the WT.
The Mutant Cdkn1bAllele Encodes a Protein That Is Absent or Present at an Extremely Reduced Level in Vivo.
We studied expression of the mutant p27 mRNA in tissues of sex-matched 2-month-old littermates of the three p27 genotypes (+/+, +/mut, mut/mut). Total RNA was extracted from frozen spleen, thymus, and adrenal glands. RT-PCR and sequencing using several primer sets established that the mutant allele is transcribed and spliced at sites used in wild-type (WT) p27 mRNA (Fig. 2A).
Fig. 2.

Analyses of the Cdkn1b mRNA and p27 protein in tissues of MENX-affected and control rats. (A Upper) Schematic representation of the Cdkn1b gene and location of the primers used for RT-PCR. Filled areas represent the coding sequence. The position of the MENX mutation (insertion) in exon 2 is indicated. (A Lower) RT-PCR results obtained by amplifying RNA from three tissues [thymus (T), spleen (S), and adrenals (A)] of normal (+/+), heterozygous (+/m), and homozygous mutant (m/m) rats. Fragments were resolved on polyacrylamide gels and stained with ethidium bromide. The primers used are indicated. (B) Real-time RT-PCR to determine the level of expression of Cdkn1b in rat tissues. The level of Cdkn1b mRNA in tissues of WT animals (+/+) is arbitrarily set at 1. The level of Cdkn1b mRNA in heterozygous (+/m) or mutant (m/m) rat tissues is normalized against the values of the WT tissues. Values are the mean of three experiments performed on three animals per genotype ± standard deviation. In adrenal glands derived from mut/mut rats, there is significantly more Cdkn1b mRNA than in WT animals (∗, P < 0.05; double-sided t test). (C) Expression of p27 protein in rat tissues. The Western blot analysis of p27 in total protein lysates obtained from tissues of 2-month-old rats is shown. The membrane was sequentially probed with antibodies against p27, Cdk2, Cdk4, cyclin E, cyclin D1, and with an anti-β-tubulin antibody to control for equal loading. KMS12 and Granta human lymphoma cell lines were loaded in parallel as positive controls.
To determine the level of expression of the mutant mRNA, we performed quantitative real-time RT-PCR. The primers and probe for this assay allow for the simultaneous amplification and detection of both WT and mutant Cdkn1b transcripts. In thymus and spleen (unaffected tissues) of +/+ and mut/mut rats, there were comparable levels of Cdkn1b mRNA (Fig. 2B). Adrenal glands (affected tissue) of mut/mut animals show a higher level of the mutant p27 transcript than the WT p27 in +/+ animals. The analysis of additional endocrine and nonendocrine tissues showed that the mutant Cdkn1b transcript is expressed at levels comparable with the normal one in pituitary gland, thyroid, liver, and testis and at a lower level than the WT allele in lung and kidney (Fig. 6A, which is published as supporting information on the PNAS web site). In conclusion, mut/mut rats show a tissue-specific relative amount of the mutant Cdkn1b mRNA.
Expression of the p27_1020;G177fs protein was analyzed by Western blotting in the same tissues as in Fig. 2A. p27_1020;G177fs was undetectable in spleen and adrenals of mut/mut rats (Fig. 2C). A faint band was visible in thymus of affected rats, and the size of this band is in agreement with that predicted based on the mutant p27 mRNA sequence (see also Fig. 6B). Heterozygous animals showed a reduced amount of native p27 protein, and the mutant band was not detectable. Blots were stripped and reprobed with antibodies against p27-binding partners and other members of the WAF/KIP CKI protein family. Cyclin D1, cyclin E, and cyclin-dependent kinases 2 and 4 (Cdk2 and Cdk4, respectively) expression is depicted in Fig. 2C, which shows no differences in expression between p27mut/mut and p27+/+ rats. SKP2, p21, and p57 were undetectable in the tissues examined (not shown).
Immunohistochemistry (IHC) using a monoclonal anti-p27 antibody was performed on a broad panel of endocrine and nonendocrine tissues from 2-month-old rats of the three p27 genotypes. Heterozygous rats showed a pattern of p27 staining similar to that of the +/+ rats, and therefore these results are not shown. The results of the IHC confirm that mut/mut rats have extremely reduced (thyroid, thymus, parathyroid, and brain) or absent (adrenals, pituitary, lung, kidney, liver, and testis) p27 protein expression (Figs. 6C–P and 7, which are published as supporting information on the PNAS web site). In contrast, +/+ animals show strong nuclear staining for p27 in all tissues. The reduced staining of p27 in mut/mut rats could be caused by degradation of the protein during the G1/S phase transit (8, 9) if more cells are proliferating in mut/mut versus +/+ rats. However, staining with the proliferation marker Ki-67 showed no difference in the percentage of proliferating cells between +/+ and mut/mut tissues (data not shown). In conclusion, regardless of the abundance of the mutant Cdkn1b transcript, p27 protein is dramatically reduced/absent in the tissues of mut/mut rats.
Comparison of Phenotype of p27mut/mut Rats and p27−/− Mice.
The protein encoded by the mutant p27 allele (p27_1020;G177fs) is mostly not present in the tissues of affected rats, thereby making the MENX animal model similar to the p27-knockout (−/−) mice. p27−/− mice develop normally, but they display a 20–30% increase in body weight compared with WT littermates (5–7). Although several tissues show hyperplasia (ovaries, testis), because of increased cellularity, p27−/− mice develop pituitary intermediate lobe adenomas as the sole tumor phenotype. Cataracts have not been reported. In agreement with the p27-deficient mice, the p27mut/mut rats also show increased body weight compared with the +/+ littermates (Fig. 8A, which is published as supporting information on the PNAS web site). The size of the thymus of mut/mut rats is ≈3–5 times that of normal littermates (Fig. 8B) because of increased cellularity as in p27−/− mice and not because of malignant growth. Heterozygous p27+/mut rats have an intermediate body weight but no increase in thymic size. The life span of the mutant animals averaged 243 ± 60 days, whereas that of their nonaffected littermates averaged 519 ± 60 days. The overall survival of +/mut rats is the same as that of WT animals, confirming the recessive phenotype of MENX (Fig. 9, which is published as supporting information on the PNAS web site).
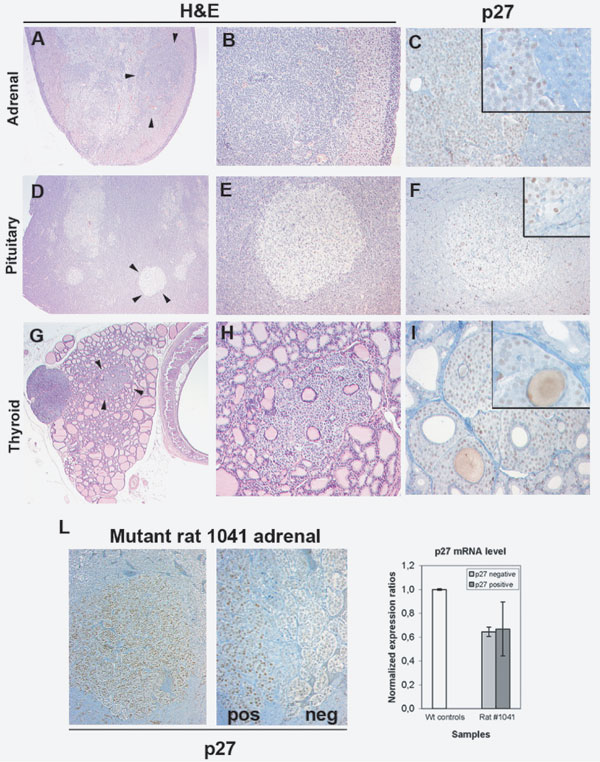
p27_1020;G177fs Expression in Hyperplastic/Tumor Tissues of mut/mut Rats.
To correlate tumor progression with p27 expression, we killed littermates at 2, 4, 6, and 8 months of age. The first lesions occur in the adrenal glands, where hyperplastic areas are visible in affected rats at 2 months of age. At 4 months of age, mut/mut rats also show multifocal pituitary hyperplasia. At 6 months or older, they have developed infiltrating adrenal medullary tumors (pheochromocytomas), multifocal pituitary adenomas, and thyroid medullary cell hyperplasia (Fig. 10 A–I, which is published as supporting information on the PNAS web site). Hyperplastic/tumor cells showed no staining for p27 protein by IHC. Moreover, all tissues, regardless of the stage of the lesions, showed <1% positivity for the proliferation marker antigen Ki-67 (data not shown). Occasionally, tumors in older rats (8 months or older) showed small foci of cells with weak nuclear p27 positivity. To verify whether the expression of p27_1020;G177fs in these tumor areas was caused by an increased transcription rate of the mutant allele, we microdissected p27-positive nodules and adjacent p27-negative areas from the adrenal glands of two independent mutant rats and performed real-time RT-PCR. The transcriptional level of mutant Cdkn1b mRNA in both areas is not significantly different, and it is similar to the level observed in normal adrenal medullary cells (Fig. 10L and data not shown). Thus, the presence of the mutant p27 protein in isolated tumor areas is the result of posttranscriptional regulatory mechanisms associated with tumor progression.
A Germ Line CDKN1B Mutation Is Found in a Suspected MEN1 Patient with No Mutations in Menin.
Not all patients with classical MEN syndromes show mutations in RET or MEN1. Specifically, up to 30% of suspected MEN1 cases show no mutations in Menin (the protein encoded by the MEN1 gene) (3, 4). Although some of this gap is expected to be caused by inactivating mutations in the noncoding regions of MEN1, some MEN1 mutation-negative cases are candidates for an MENX-like mutation in CDKN1B. Patient N, a 48-year-old Caucasian female, developed acromegaly and had a 3-cm pituitary tumor removed when she was 30. Histologically, the tumor was an invasive pituitary adenoma with growth hormone hyperproduction, high mitotic activity, and cell atypia. Sixteen years later, she was diagnosed with primary hyperparathyroidism, likely caused by parathyroid hyperplasia or adenoma, but she has not yet been operated. Sequencing of MEN1 revealed no mutations despite an MEN1 phenotype. We sequenced the complete coding region and 140 bp of the 5′ untranslated region of the CDKN1B gene in this patient, and we identified a germ-line heterozygous TGG → TAG nonsense mutation at codon 76 (c. G692A, NM_1020;004064) (Fig. 3A). This mutation predicts premature truncation of the p27 protein at codon 76 (p. W76X) (Fig. 3A). This mutation was not observed in 380 unrelated healthy controls, confirming that it is neither a polymorphism nor a rare variant but indeed a unique pathogenic mutation. Additional family members agreed to take part in the study, and the partial pedigree of the proband's family is shown in Fig. 3B. The proband's mother (I-2) and one sister (II-2) are mutation-negative. The proband's older sister II-4 is a mutation carrier. She was diagnosed at age 55 with renal angiomyolipoma, an MEN1-associated nonendocrine tumor (10). The proband's youngest sister (II-3; age 44) and her teenage daughter carry the germ-line mutation. They reported no symptoms, but they did not undergo a thorough medical examination; therefore, the presence of early endocrine lesions cannot be excluded. Interestingly, in MEN1, up to 40% of patients manifest a first phenotypic feature after 40 years of age (11). No mutations in the MEN1 gene were found in the CDKN1B mutation-positive individuals II-3 and II-4. The proband's father had acromegaly, as the proband, and her brother (II-5) died at age 39 from hypertension (pheochromocytoma cannot be excluded). The son of the mutation-positive sister, individual III-3, developed testicular cancer at age 28. No samples from the father (I-1) and from individuals II-5 and III-3 were available for further analysis. A series of microsatellite markers in close proximity to CDKN1B on chromosome 12 was used to trace the inheritance of CDKN1B alleles over generations (Fig. 3C). We infer that the affected haplotype is inherited from the proband's father. The renal angiomyolipoma of sister II-4 was available for molecular analysis; direct sequencing revealed the presence of both CDKN1B alleles in tumor DNA (Fig. 3D). The absence of loss of heterozygosity of the WT allele was confirmed by amplifying the markers shown in Fig. 3C in both normal and tumor tissue DNA (data not shown). We performed RT-PCR on normal and tumor tissue RNA, and then we sequenced the product obtained; both alleles are transcribed in both tissue areas (Fig. 3D). Although the tumor tissue retains the CDKN1B WT allele and expresses it at the RNA level, it shows no p27 protein staining (Fig. 3D).
Fig. 3.

Identification of a germ-line mutation in CDKN1B in a suspected MEN1 patient and segregation analyses. (A Upper) A PCR fragment corresponding to part of CDKN1B exon 1 was obtained from the proband, cloned, and both alleles were separately sequenced. The germ line of the proband showed a heterozygous nonsense mutation at codon 76 (c. G692A). (A Lower) The position of the mutation in the CDKN1B gene and p27 protein is shown. (B) Proband family pedigree. Generation numbers are represented by Roman numerals, and individual numbers are in Arabic numerals. The proband is II-1, indicated by the arrow. Mut+ indicates mutation-positive individuals. AM, acromegaly; PT, pituitary adenoma; PC, parathyroid cancer; RA, renal angiomyolipoma; HT, hypertension; TC, testicular cancer. The age of onset is indicated in parentheses. (C) Haplotype analysis. The microsatellite markers on chromosome 12 and their positions are indicated on the right. The affected haplotype is shown in black. (D) Analysis of the tumor tissue of individual II-4. Sequencing chromatograms show the G692GA mutation in both normal and tumor tissue DNA and RNA. Immunohistochemical staining with an anti-p27 antibody (Right) shows a lack of p27 protein in the tumor tissue. The arrows indicate infiltrating lymphocytes used as a positive control for p27 staining. (Immunoperoxidase original magnification: ×650.)
Intracellular Localization of the Mutant p27 Proteins.
In many cellular systems, p27 is regulated posttranscriptionally at the levels of protein translation, stability, and subcellular localization (8, 9). Cytoplasmic relocation, the result of phosphorylation or sequestration by partner proteins, plays an important role in regulating p27 function. To determine the effect of the rat and human p27 mutations on protein localization, we generated the following Myc-tagged proteins: WT p27, p27_1020;G177fs (MENX), p27_1020;G177X (artificial truncation), and p27_1020;W76X (human mutation) (Fig. 4 A). The p27_1020;G177X construct was generated to determine whether the presence of the p27-unrelated C-terminal region of the p27_1020;G177fs protein affects the localization of the protein.
Fig. 4.

Expression and subcellular localization of exogenous proteins during transfection. (A) Schematic representation of Myc-tagged p27 proteins used for transfections. The shaded area at the C terminus of p27_1020;G177fs represents the p27-unrelated amino acids present in the MENX mutant protein. (B and C) Immunoblots showing the expression of the transfected proteins. (B) MCF-7 cells were transfected with 1 μg of the indicated constructs. Cell lysates were prepared at the indicated times after transfection; 50 μg of total protein was separated by electrophoresis, blotted, and probed with antibodies to the Myc tag. To control for equal loading, the membrane was probed with the anti-tubulin monoclonal antibody. (C) MCF-7 cells were transfected as in B. Twenty-four hours later, cells were harvested, and proteins were extracted. Different amounts of total protein lysates (indicated) were loaded onto the gel. p27_1020;G177fs and of p27_1020;W76X proteins seem to be ≈6-fold less abundant than the WT p27 protein. (D) Subcellular localization of Myc-tagged p27 proteins. Rat2 cells were transfected with the Myc-WTp27, Myc-p27_1020;G177fs, Myc-p27_1020;G177X, and p27_1020;W76X (indicated on the right) constructs. Forty-eight hours later, the transfected cells were fixed and permeabilized, and then they were stained with an antibody to the Myc tag visualized with Cy3 (red). Nuclei were counterstained with Hoechst (blue). (Left) Cy3. (Center) Hoechst. (Right) Merge. (Scale bars: 10 μm.)
Transient transfections were performed in Rat2 immortalized rat fibroblasts and in human MCF-7 and embryonic kidney (HEK) 293T cell lines. Western blotting showed that the fusion proteins are all expressed (Fig. 4B). Notably, the p27_1020;G177fs and p27_1020;W76X proteins are expressed at a lower level compared with the WT p27 or the p27_1020;G177X fusion proteins, despite comparable transfection efficiencies (assessed by fluorescence-activated cell sorting). Dilution experiments showed that p27_1020;G177fs and p27_1020;W76X are reduced ≈6-fold compared with the WT p27 protein (Fig. 4C). This phenomenon was reproducibly observed in all cell lines and for independent DNA clones of the same constructs. We determined the cellular localization of the fusion proteins by using an anti-Myc antibody and indirect immunofluorescence (Fig. 4D). During transfection, both WT p27 and p27_1020;G177fs localized mainly in the nucleus (Fig. 4D). Both the p27_1020;G177X and the p27_1020;W76X proteins are retained in the cytoplasm (Fig. 4D). Similar results were obtained in all cell lines. These experiments show that p27_1020;G177fs retains the ability to transfer into the nucleus, but it is expressed at a lower level than WT p27 (in agreement with the in vivo data of the IHC), whereas the W76X mutation prevents the truncated p27 protein from entering the nucleus, the cell compartment where p27 exerts its function as a cell cycle inhibitor.
Discussion
We report here a previously unknown gene mutation mechanism responsible for MEN, detecting germ-line mutations in both the rat Cdkn1b gene and the human CDKN1B gene. The mutations we identified behave as loss-of-function mutations: the rat Cdkn1b insertion is responsible for the loss of protein in vivo, whereas the truncated human p27 protein is not transferred to the nucleus, where p27 is required to bind to cyclin–Cdk complexes and inhibit cell cycle progression.
After the identification of p27 as a key regulator of cell cycle progression, somatic mutations of the CDKN1B gene have been sought by various groups in the major tumor types. Only a handful of somatic mutations (and no germ-line mutations) have been reported in the hundreds of human tumor samples so far analyzed (12–14). Because mutations in p27 are extremely rare in human cancers, there has been some debate as to whether p27 is a bona fide tumor-suppressor gene. The presence of hemizygous loss of p27 in hematopoietic malignancies (15, 16) and the reduced expression in many tumor types (17) support the hypothesis of a tumor-suppressor role of p27 in tumorigenesis. The finding that a germ-line heterozygous truncating CDKN1B mutation predisposes to an MEN1-like phenotype provides definitive proof that in endocrine cells, p27 plays an important role as a tumor suppressor, and its inactivation leads to multitissue tumor formation in humans. It also establishes a causal role between p27 alterations and tumor formation in humans.
p27-deficient mice are relatively free of malignancies, with the exception of hyperplasia of the pituitary intermediate lobe, which develops into adenoma with age (5–7). When they are challenged with mutagenic agents (18), heterozygous p27+/− mice are predisposed to increased tumor multiplicity in diverse sites, suggesting that p27 acts as a dose-dependent tumor suppressor in mice. A similar dosage effect could be at work in human tumors because hemizygous deletions of the p27 locus 12p13 have been detected in leukemias and lymphomas without mutation of the wild-type allele (15, 16). The only tumor sample from a CDKN1B mutation-positive individual available for molecular analysis (i.e., renal angiomyolipoma of individual II-4) showed expression of both alleles at the mRNA level but no p27 protein expression. Therefore, in this tumor, p27 behaves as a classical tumor suppressor.
Although p27-null mice show hyperplasia in multiple organs, they develop pituitary adenomas as the sole tumor phenotype. p27mut/mut rats develop a broader spectrum of neuroendocrine tumors. The difference in the sensitivity of various tissues to p27 loss could be the result of defined species-specific temporal and spatial expression patterns of p27. The eye phenotype is an example of species-specific effects in mouse versus rat (see below).
Although the eye phenotype is not the focus of the present study, it is worth mentioning that p27mut/mut rats also develop bilateral, juvenile cataracts (1). The presence of both tumors and cataract lesions in affected rats suggests that failure to commit to terminal differentiation may be responsible for the MENX phenotype. In mice, loss of p27 does not affect lens development because the main pathway regulating this process involves p57 (a member of the same CKI family; ref. 19). p27 expression and consequent cell cycle arrest become important in the absence of p57 because mice mutant for both of these CKIs have more severe developmental defects in the lens, and they show nuclei in the lens fiber compartment, signs of ectopic cell proliferation and failure of terminal differentiation during lens fiber formation (19). In agreement with those observations, also in p27mut/mut rats numerous epithelial cells remain within the lens tissue (Fig. 11, which is published as supporting information on the PNAS web site).
Recent studies suggest a link between p27 and the genes causing the human MEN syndromes. Menin, the product of the MEN1 gene, interacts with mixed-lineage leukemia family proteins in a histone methyltransferase complex that activates transcription of p27 and p18 (20, 21). It has also been reported that inhibition of oncogenic RET activity results in down-regulation of p27 in thyroid medullary carcinoma cells (22), likely through the Akt-mediated phosphorylation of the transcription factor AFX/FKHR (23). These studies indicate that p27 is an important readout of the Menin signaling pathway and a putative downstream target of oncogenic RET in endocrine cells. Whether p27 represents a “gatekeeper” in endocrine tumorigenesis warrants further studies.
Down-regulation of p27 has been implicated in a variety of human cancers, and an inverse correlation between p27 protein levels and prognosis has been reported for several tumor types (17). Therefore, restoring p27 expression (function) in tumor cells might be beneficial for the clinical outcome of the patient, making p27 a viable therapeutic target. Developing p27-specific intervention strategies requires understanding its role and regulation in normal and pathologic states. Our MENX model system of endocrine tumorigenesis offers the opportunity to expand the current knowledge about p27 functions and to study the response to therapeutic approaches for neuroendocrine cancers in vivo.
Materials and Methods
Animals and Crosses.
The MENX phenotype was identified in a Sprague–Dawley (SD) rat colony, and it was maintained as reported in ref. 1. Animals expressing the mutant phenotype were indicated as SDwe/SDwe for Sprague–Dawley white eye in reference to the presence of juvenile cataracts (1). To fine map the MenX gene, we crossed SDwe/SDwe homozygous male rats with Wistar–Kyoto female rats (both strains were obtained from Charles River Laboratories, Sulzfeld, Germany). We backcrossed F1 (Wistar–Kyoto × SDwe) females with SDwe/SDwe males to generate 151 F2 animals, of which 57 (38%) were affected. Anatomical and histologic analysis of rats was done as reported in Supporting Methods, which is published as supporting information on the PNAS website. See also ref. 24.
Human Subjects and Analyses.
The participating subjects signed an informed-consent form as approved by the Ethics Committees of the Universities of Göttingen and Munich. Unrelated control DNA samples were obtained from the Institute of Human Genetics, GSF, and comprise 380 healthy Germans (KORA Study Group) (25). DNA was extracted from peripheral venous blood by using the QIAmp kit (Qiagen, Hilden, Germany) and from formalin-fixed, paraffin-embedded tissue as reported in ref. 26. A PCR fragment containing the heterozygous CDKN1B mutation was cloned into the TA vector (Invitrogen, Karlsruhe, Germany), and clones carrying the two alleles were sequenced separately. Sequencing of the coding region and promoter of CDKN1B was performed with the BigDye terminator kit (Applied Biosystems, Darmstadt, Germany), and sequences were run on an ABI377 sequencer (Applied Biosystems). Primer sequences are reported in Supporting Methods.
Rat DNA Extraction and Analyses.
DNA was extracted from rat tail tips with the DNeasy extraction kit (Qiagen). Primers to amplify the microsatellite markers used for genotyping were from public databases (UniSTS), or they were designed based on the rat genome sequence (D4GSF primers, available on request). Sequencing of the Cdkn1b was done as for the human CDKN1B gene by using primers reported in Supporting Methods. Seven commercially available rat strains from Charles River (Sprague–Dawley, Copenhagen, Lewis, Wistar–Kyoto, SHR, F344, Brown Norway) were used for sequence analysis.
Rat RNA Extraction and Analysis.
We extracted total RNA from snap-frozen tissues from affected (p27mut/mut) and unaffected (p27+/mut and p27+/+) animals by using the TRIzol protocol (Invitrogen). We synthesized the first-strand cDNA by using random hexamers and SuperScript II (Invitrogen) (for details, see Supporting Methods). The primers for RT-PCR are reported in Supporting Methods. Quantitative RT-PCR was done with TaqMan assay-on-demand primers and probes for rat Cdkn1b gene and for β2-microglobulin as internal control (Applied Biosystems). The assays were run as reported in ref. 26 (for details, see Supporting Methods). RNA from microdissected formalin-fixed and paraffin-embedded tissues was extracted and processed as reported in ref. 26.
Western Blotting.
For protein extraction from rat snap-frozen tissues, serial 50-μm tissue sections were resuspended in protein lysis buffer and sonicated. For protein extraction from transfected cells, cells were collected, washed twice in PBS, and lysed inlysis buffer for 20 min at 4°C. For details, see Supporting Methods. Antibodies used were: anti-p27 monoclonal antibody (BD Biosciences, Heidelberg, Germany); anti-cyclin D1, anti-cyclin E, anti-Cdk2, and anti-Cdk4 (Santa Cruz Biotechnology, Santa Cruz, CA); anti-Myc tag monoclonal antibody (BD Biosciences). To control for equal protein loading, β-tubulin immunostaining was performed (Santa Cruz Biotechnology).
Mammalian Expression Systems.
We obtained full-length +/+ and mut/mut Cdkn1b cDNA by RT-PCR from rat brain total RNA by using SuperScript II. We also generated by PCR a rat Cdkn1b cDNA containing a stop codon at position 177 (named p27_1020;G177X) and a human CDKN1B cDNA having a nonsense mutation at codon 76 (p27_1020;W76X). We fused the various p27 constructs with the Myc tag (pCMV-Myc; BD Biosciences) to obtain protein tagged at their N termini with the Myc domain. All constructs were confirmed by sequencing.
Cell Lines and Transfections.
Rat2 (normal rat fibroblasts), HEK 293T (normal embryonic human fibroblasts), and MCF-7 (breast cancer) were cultivated in DMEM supplemented with 10% FCS. Cells were grown in six-well plates and transfected when 70–80% confluent by using the FuGENE 6 reagent (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's instructions. The pEGFP.C3 vector (BD Biosciences) was transfected in parallel to monitor transfection efficiency by immunofluorescence. We also cotransfected the Myc fusion proteins with pBB14 overexpressing a membrane-bound EGFP protein (27) to compare the transfection efficiency among our Myc-tagged constructs by fluorescence-activated cell sorting.
Indirect Immunofluorescence.
Cells were grown on coverslips and processed for immunocytochemistry as described in ref. 28 with minor modifications. For details, see Supporting Methods.
Supplementary Material
Acknowledgments
We thank the family members for their collaboration; S. Liyanarachchi (Ohio State University, Columbus, OH) for statistical analyses; L.W. Enquist (Princeton University, Princeton, NJ) for plasmid pBB14; J. Müller, N. Kink, and C. Kloos for help with animal care; P. Lichtner for help with genotyping; M. Rosemann for discussion; and E. Schaeffer, P. Hutzler, A. Voss, F. Cattaneo, S. Schwarz, D. Angermeier, and U. Reich for technical assistance. This work was supported in part by Grant GRK333 from the Deutsche Forschungsgemeinshaft (DFG) (to M.J.A.).
Abbreviations
- Cdk
cyclin-dependent kinase
- CKI
cyclin-dependent kinase inhibitor
- IHC
immunohistochemistry
- MEN
multiple endocrine neoplasia
- SD
Sprague–Dawley
- we
white eye
- WT
wild type.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
References
- 1.Fritz A, Walch A, Piotrowska K, Rosemann M, Schaffer E, Weber K, Timper A, Wildner G, Graw J, Hoefler H, et al. Cancer Res. 2002;62:3048–3051. [PubMed] [Google Scholar]
- 2.Piotrowska K, Pellegata NS, Rosemann M, Fritz A, Graw J, Atkinson MJ. Mamm Genome. 2004;15:135–141. doi: 10.1007/s00335-003-3027-8. [DOI] [PubMed] [Google Scholar]
- 3.Namihira H, Sato M, Matsubara S, Ohye H, Bhuiyan M, Murao K, Takahara J. Endocr J. 1999;46:811–816. doi: 10.1507/endocrj.46.811. [DOI] [PubMed] [Google Scholar]
- 4.Sakurai A, Katai M, Yumita W, Minemura K, Hashizume K. Endocrine. 2004;23:45–49. doi: 10.1385/ENDO:23:1:45. [DOI] [PubMed] [Google Scholar]
- 5.Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY, Nakayama K. Cell. 1996;85:707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- 6.Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A. Cell. 1996;85:721–732. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- 7.Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, et al. Cell. 1996;85:733–744. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- 8.Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- 9.Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A, Pagano M. Genes Dev. 1999;13:1181–1189. doi: 10.1101/gad.13.9.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong Q, Debelenko LV, Chandrasekharappa SC, Emmert-Buck MR, Zhuang Z, Guru SC, Manickam P, Skarulis M, Lubensky IA, Liotta LA, et al. J Clin Endocrinol Metab. 1997;82:1416–1420. doi: 10.1210/jcem.82.5.3944. [DOI] [PubMed] [Google Scholar]
- 11.Glascock MJ, Carty SE. Surg Oncol. 2002;11:143–150. doi: 10.1016/s0960-7404(01)00031-7. [DOI] [PubMed] [Google Scholar]
- 12.Kawamata N, Morosetti R, Miller CW, Park D, Spirin KS, Nakamaki T, Takeuchi S, Hatta Y, Simpson J, Wilcyznski S, et al. Cancer Res. 1995;55:2266–2269. [PubMed] [Google Scholar]
- 13.Morosetti R, Kawamata N, Gombart AF, Miller CW, Hatta Y, Hirama T, Said JW, Tomonaga M, Koeffler HP. Blood. 1995;86:1924–1930. [PubMed] [Google Scholar]
- 14.Spirin KS, Simpson JF, Takeuchi S, Kawamata N, Miller CW, Koeffler HP. Cancer Res. 1996;56:2400–2404. [PubMed] [Google Scholar]
- 15.Sato Y, Suto Y, Pietenpol J, Golub TR, Gilliland DG, Davis EM, Le Beau MM, Roberts JM, Vogelstein B, Rowley JD, et al. Blood. 1995;86:1525–1533. [PubMed] [Google Scholar]
- 16.Komuro H, Valentine MB, Rubnitz JE, Saito M, Raimondi SC, Carroll AJ, Yi T, Sherr CJ, Look AT. Neoplasia. 1999;1:253–261. doi: 10.1038/sj.neo.7900033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Philipp-Staheli J, Payne SR, Kemp CJ. Exp Cell Res. 2001;264:148–168. doi: 10.1006/excr.2000.5143. [DOI] [PubMed] [Google Scholar]
- 18.Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. Nature. 1998;396:177–180. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang P, Wong C, DePinho RA, Harper JW, Elledge SJ. Genes Dev. 1998;12:3162–3167. doi: 10.1101/gad.12.20.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milne TA, Hughes CM, Lloyd R, Yang Z, Rozenblatt-Rosen O, Dou Y, Schnepp RW, Krankel C, Livolsi VA, Gibbs D, et al. Proc Natl Acad Sci USA. 2005;102:749–754. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, Meyerson M, Kim SK. Proc Natl Acad Sci USA. 2005;102:14659–14664. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drosten M, Hilken G, Bockmann M, Rodicker F, Mise N, Cranston AN, Dahmen U, Ponder BA, Putzer BM. J Natl Cancer Inst. 2004;96:1231–1239. doi: 10.1093/jnci/djh226. [DOI] [PubMed] [Google Scholar]
- 23.Medema RH, Kops GJ, Bos JL, Burgering BM. Nature. 2000;404:782–787. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- 24.Quintanilla-Martinez L, Kremer M, Specht K, Calzada-Wack J, Nathrath M, Schaich R, Höfler H, Fend F. Am J Pathol. 2003;162:1449–1461. doi: 10.1016/S0002-9440(10)64278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Filipiak B, Heinrich J, Schafer T, Ring J, Wichmann HE. Clin Exp Alergy. 2001;31:1829–1838. doi: 10.1046/j.1365-2222.2001.01246.x. [DOI] [PubMed] [Google Scholar]
- 26.Specht K, Richter T, Müller U, Walch A, Werner M, Höfler H. Am J Pathol. 2001;158:419–429. doi: 10.1016/S0002-9440(10)63985-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalejta RF, Brideau AD, Banfield BW, Beavis AJ. Exp Cell Res. 1999;248:322–328. doi: 10.1006/excr.1999.4427. [DOI] [PubMed] [Google Scholar]
- 28.Pellegata NS, Cajot JF, Stanbridge EJ. Oncogene. 1995;11:337–349. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}