

Abstract
GB virus type C (GBV-C) is an apparently nonpathogenic virus that replicates in T and B lymphocytes and is a common cause of persistent human infection. Among HIV-1-infected individuals, persistent coinfection with GBV-C is associated with prolonged survival, and infection of blood mononuclear cells or CD4+ T cells with GBV-C and HIV in vitro results in significantly reduced HIV-1 replication. To date, the viral protein(s) that lead to HIV inhibition have not been identified. The GBV-C nonstructural phosphoprotein (NS5A) is predicted to have pleotropic effects on cells, including interactions with the IFN-induced dsRNA-activated protein kinase (PKR). We studied GBV-C NS5A to determine whether it is involved in inhibition of HIV replication. GBV-C NS5A protein from an isolate that was cleared by IFN therapy did not inhibit PKR, whereas NS5A from an isolate that was not cleared by IFN-inhibited PKR function in a yeast genetic system. Both of these GBV-C NS5A proteins were expressed in a CD4+ T cell line (Jurkat), and both induced a potent, dose-dependent inhibition of HIV-1 replication, thus the effect was independent of PKR inhibition. NS5A induced the release of the chemokine SDF-1 and decreased surface expression of the HIV coreceptor CXCR4, potentially explaining the HIV inhibition. Deletion mapping of the NS5A protein found that an 85-aa region between amino acids 152 and 237 inhibits HIV-1 replication. Thus, GBV-C NS5A protein alters the cellular milieu necessary for HIV-1 replication and may provide a previously undescribed therapeutic approach for anti-HIV therapy.
Keywords: chemokine receptor, chemokines, nonstructural proteins
GB virus C (GBV-C) is a human flavivirus that is present in 1–3% of healthy U.S. blood donors (reviewed in ref. 1). Because of shared modes of transmission, up to 86% of HIV-positive individuals have evidence of active (39.6%) or prior (46%) GBV-C infection (1, 2). Although viremia may persist for decades in some individuals, most immune-competent hosts clear GBV-C infection concurrently with the development of antibodies to the envelope glycoprotein E2 (3, 4), which appear to confer some, although not complete, protection against reinfection (4). Although no disease entity has yet been associated with GBV-C infection (reviewed in refs. 1 and 5), numerous studies found a significant association between persistent GBV-C viremia and prolonged survival among HIV-infected individuals (6; reviewed in ref. 1).
The GBV-C genome organization is similar to that of hepatitis C virus (HCV) (7–9). The single-strand, positive-sense RNA genome encodes a long ORF that is translated into a polyprotein of ≈3,000 aa (10). Based on comparisons with HCV, the structural proteins (E1 and E2) are thought to be cleaved from the polyprotein by cellular signal peptidases, whereas the nonstructural (NS) proteins are predicted to be cleaved by two viral proteases (NS2 and NS3; Fig. 1A) (11). A major difference between GBV-C and HCV is the apparent tissue tropism, because GBV-C appears to replicate in bone marrow cells and both T and B lymphocytes rather than in hepatocytes (10, 12–15).
Fig. 1.

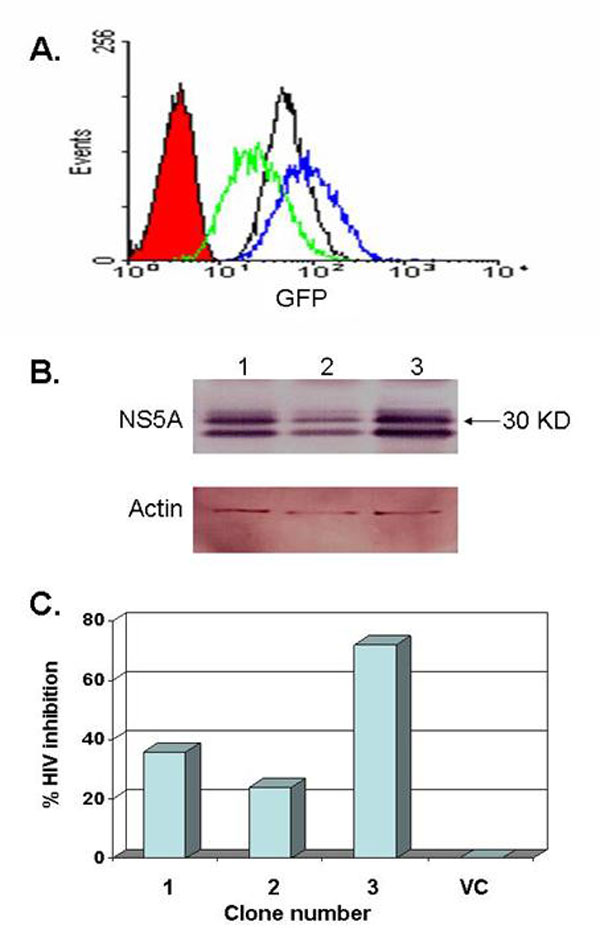
GB virus C NS5A expression. (A) The predicted genome organization of GBV-C is shown, including the 5′ nontranslated region [containing an internal ribosomal entry site (IRES)], envelope glycoproteins (E1 and E2) nonstructural proteins (NS), and 3′ nontranslated region. The predicted 414-aa-long NS5A protein is encoded by nucleotides 6150–7392 (GenBank accession no. AF121950) with the putative N-terminal membrane anchoring domain shown in yellow. (B) GFP expression was detected by flow cytometry in Jurkat cells alone (black) or stably transfected with VC (red), VC-GFP (blue), or expressing the NS5A from an IFN-S isolate (GenBank accession no. DQ177420 in green) and an IFN-R isolate (DQ177421) in purple. (C) Immunoreactive GBV-C NS5A proteins from both the IFN-S and IFN-R isolates were observed in GFP-positive clones, and NS5A expression was reduced when cells were maintained in doxycycline for 48 h before preparing cell lysates.
HIV-1 attachment to cells involves interaction between HIV gp120 and the CD4 receptor, whereas entry into target cells requires interaction with a coreceptor, almost always the chemokine receptor CCR5 or CXCR4 (16, 17). HIV-1 isolates transmitted in vivo generally use CCR5 as their coreceptor and replicate in monocytes, macrophages, and primary CD4+ T cells (R5 viruses) (17, 18). In contrast, HIV-1 isolates that use CXCR4 as their entry coreceptor (X4 viruses) frequently emerge in HIV-infected people later in the course of infection and replicate in T cell lines (17, 18). The natural ligands for CCR5 are the chemokines RANTES, MIP-1α, and MIP-1β, whereas SDF-1 is the ligand for CXCR4 (19, 20). These chemokines inhibit HIV-1 by competing for binding to the chemokine receptors and, thus, inhibiting HIV-1 entry into the cell (19), and by inducing postentry inhibition of HIV reverse transcription (21–23).
Coinfection of peripheral blood mononuclear cells (PBMCs) with GBV-C and HIV-1 results in inhibition of HIV-1 replication (2, 24–27). PBMCs infected with GBV-C release significantly more RANTES, MIP-1α, MIP-1β, and SDF-1 into culture supernatants and have significantly less CCR5 on their surface than do control cells (24, 25), suggesting that modulation of chemokines and chemokine receptors may be a mechanism by which GBV-C influences HIV-1 disease progression. No specific GBV-C viral protein has been implicated in the inhibitory effect of GBV-C on HIV-1 isolates that use the CXCR4 coreceptor.
Based on comparisons with HCV, the GBV-C NS5A protein is thought to be anchored at the N terminus in the endoplasmic reticulum and necessary for RNA replication (10, 28–30). The HCV NS5A appears to have three structured domains, one of which contains a zinc-binding motif that is conserved in GBV-C (31). Like HCV NS5A (10), there are two phosphorylated forms of GBV-C NS5A, which represent basal and hyperphosphorylated forms of the protein (32). HCV NS5A appears to modulate host antiviral responses at least, in part, by inhibiting dsRNA-activated protein kinase (PKR)-mediated phosphorylation of the eukaryotic initiation factor eIF-2α (33, 34). HCV NS5A also has been shown to inhibit apoptosis and induce oxidative stress (35–38). GBV-C NS5A is also phosphorylated and inhibits PKR-mediated phosphorylation of eIF-2 α in a yeast genetic system (32). To characterize the effect of GBV-C NS5A protein on lymphocytes, stable CD4+ Jurkat T cell lines were selected that expressed NS5A in a tetracycline repressible system, and the effect of GBV-C NS5A protein on HIV-1 replication was examined in these cells.
Results
Effect of GBV-C NS5A Protein on HIV Replication.
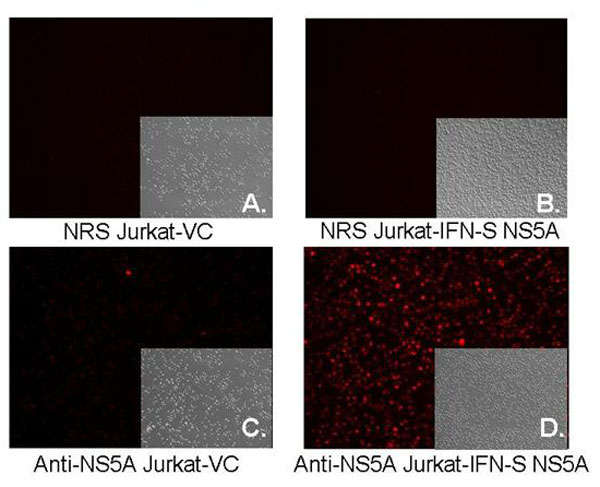
The predicted full-length GBV-C NS5A coding sequences from two isolates were expressed in Jurkat cells under the control of tetracycline. One of the isolates was obtained from a patient whose GBV-C viremia cleared during IFN therapy administered for coexisting HCV infection (IFN-sensitive, IFN-S; GenBank accession no. DQ177420), and the other NS5A coding sequence was obtained from a patient who did not clear GBV-C viremia (IFN-resistant, IFN-R; GenBank accession no. DQ177421) (32). NS5A was expressed in a bicistronic vector that also expressed GFP contained on the same transcript, with translation of NS5A by using capped mRNA, whereas translation of the GFP was directed by the EMC internal ribosomal entry site (IRES). GFP expression was monitored by flow cytometry (Fig. 1B). Cell lines were cloned, and those containing the NS5A sequences demonstrated two immunoreactive proteins with estimated molecular masses of 56 kDa and 58 kDa (Fig. 1C) as described in ref. 31. Expression of IFN-S NS5A was greater than that of IFN-R NS5A; however, expression of both proteins was decreased to undetectable levels when cells were incubated in doxycycline (Fig. 1C). NS5A expression was confirmed by immunofluorescence microscopy (Fig. 7, which is published as supporting information on the PNAS web site).
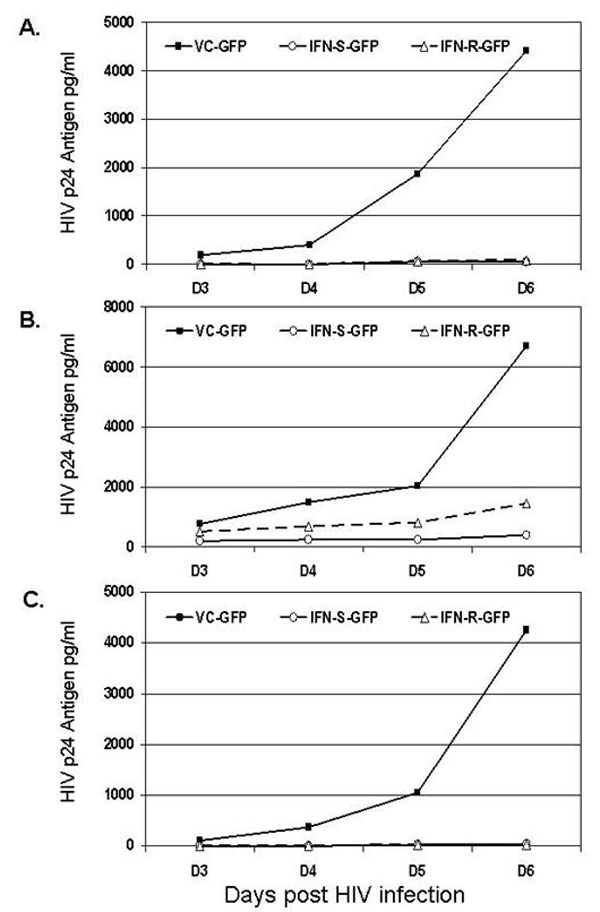
To determine whether GBV-C NS5A influenced HIV-1 replication, cells were maintained in doxycycline to suppress NS5A expression. Doxycycline was removed 4 days before infection with an X4-tropic HIV-1 isolate (NIH repository no. 1073). HIV-1 replication (p24 antigen release into culture supernatants) was reduced by >99% in cells expressing either the IFN-R or the IFN-S NS5A protein compared with the vector control expressing GFP (VC-GFP) or the vector control without GFP (VC) (Fig. 2A). Previous studies demonstrated that the IFN-R NS5A protein inhibited PKR-mediated phosphorylation of eIF-2α, whereas IFN-S NS5A did not (32). Both IFN-R and IFN-S NS5A expression inhibited the replication of all HIV-1 isolates tested, including clade B and clade D isolates (NIH repository nos. 317, 2969, and 2521; Fig. 8, which is published as supporting information on the PNAS web site). To ensure that NS5A was not affecting the HIV p24 antigen assay, HIV-1 infectivity in culture supernatants was measured, confirming the HIV-1 replication inhibition (Fig. 2B).
Fig. 2.

Expression of GBV-C NS5A in Jurkat cells inhibits HIV-1 replication. (A) Cells containing IFN-R or IFN-S GBV-C NS5A or VC or VC-GFP were infected with HIV-1 (isolate no. 1073) after doxycyline was removed for 4 days. HIV-1 p24 antigen released into culture supernatants was statistically greater in control cells on days 4–6 after HIV-1 infection. ∗, P < 0.03; ∗∗, P < 0.001. Infections were performed in triplicate, and SE for IFN-R NS5A and vector control cells are shown. (B) The HIV-1 infectious titer (TCID50 in PBMC culture) of culture supernatants was determined for cells expressing the VC-GFP or the IFN-S NS5A.
Control and NS5A expressing cells were maintained in various concentrations of doxycycline for 96 h to decrease NS5A and GFP expression before infection with HIV. Fig. 3A demonstrates that the doxycycline did not alter HIV replication in VC-GFP Jurkat cells; however, dose-dependent inhibition of HIV-1 replication was observed in the NS5A expressing Jurkat cells maintained in doxycycline (Fig. 3B). Similar results were observed in the IFN-R NS5A-expressing cells (data not shown). When IFN-S NS5A expressing cells were maintained in doxycycline (5 μg/ml) for several passages, NS5A was not detectable by immunoblot; however, HIV replication still was inhibited, although to a lesser extent (73% inhibition on day 4; data not shown).
Fig. 3.

Dose relationship between GB virus C NS5A expression and HIV replication in Jurkat cells as well as NS5A expression in infected PBMC cultures. (A and B) Jurkat cells expressing VC-GFP (A) or IFN-S NS5A (B) were maintained in 0, 1, or 5 μg/ml doxycycline. Doxycycline had no significant effect on HIV-1 replication in VC-GFP cells (A), yet in Jurkat cells expressing NS5A, HIV-1 replication increased with increases in doxycycline concentration (decreased NS5A expression; B). HIV-1 replication was significantly lower (>90%) than that observed in control cells when maintained in 5 μg/ml doxycycline. (C) NS5A demonstrates immunoreactive protein in PBMC lysates (200,000 cells) 3 days after GBV-C (G) or mock infection (M) and Jurkat cells with NS5A (5A) or the VC-GFP.
To determine whether the amount of NS5A protein expression in Jurkat cells is comparable with that found in GBV-C-infected PBMCs, 2 × 106 PBMCs were infected with either a clinical GBV-C isolate (39) or a mock-control preparation, and cell lysates were examined for NS5A expression by immunoblot analysis. A 58-kDa immunoreactive protein was observed in the GBV-C-infected PBMCs (Fig. 3C), demonstrating that NS5A protein expression in infected PBMCs is detected at levels comparable with that in Jurkat cells in which HIV inhibition is observed. GBV-C and HIV coinfection in these PMBCs demonstrated significant inhibition in HIV-1 replication as described (data not shown; ref. 24).
GBV-C NS5A Modulates SDF-1 and CXCR4 Expression.
Because GBV-C induces chemokines in primary lymphocyte cultures (24), and because SDF-1 inhibits X4 HIV-1 replication (40), chemokine release from Jurkat cells with and without NS5A was examined. After removal of doxycycline from Jurkat cells, significantly more SDF-1 was released into culture supernatants in the cells expressing GBV-C NS5A compared with VC cells (Fig. 4A). There were no differences in release of RANTES, MIP-1α, or MIP-1β between the NS5A expressing and control Jurkat cells (data not shown). The detection of CXCR4 was significantly reduced on IFN-R NS5A cells compared with vector control cells (49.6% lower mean fluorescence; Fig. 4B; P = 0.003). Similar reductions were seen on IFN-S NS5A cells (data not shown).
Fig. 4.

Effect of GBV-C on SDF-1 release and CXCR4 surface density. (A) SDF-1 in culture supernatants was measured by ELISA in triplicate on sequential days after removal of doxycycline. There were no differences between VC or parental Jurkat cells; however, SDF-1 increased in Jurkat cells expressing NS5A (t test, P < 0.001 for days 5 and 6). (B) Surface CXCR4 on VC cells or cells stably expressing NS5A was determined by flow cytometry by using isotype control (IC) or anti-CXCR4 antibodies. (C) The effect of adding neutralizing antibody to SDF-1 to cell cultures or isotype control antibody (IC) only partially decreased the HIV-1 inhibitory effect (HIV p24 antigen in culture supernatants 4 days after infection).
To determine whether the HIV-1 inhibition was related solely to SDF-1 release, cells were maintained in neutralizing anti-SDF-1 antibodies (concentration 10 μg/ml) before and after HIV-1 infection. This concentration was shown to block SDF-1 anti-HIV effects in PBMCs (24). As expected, HIV-1 replication increased in VC cells incubated in anti-SDF-1 antibody after HIV infection (Fig. 4C). Anti-SDF-1 also led to increased HIV-1 replication in NS5A-expressing cells; however, these cells still had significantly less HIV-1 replication than control cells (Fig. 4C; P < 0.001 for IFN-S NS5A and P = 0.02 for IFN-R NS5A). Thus, NS5A-induced release of SDF-1 does not appear to completely explain the HIV-1 inhibitory effect observed in these cells. Inclusion of isotype control and anti-SDF-1 antibodies in the media was associated with a reduction in HIV inhibition (Fig. 4C), although the difference between NS5A-expressing cells and VC cells still was statistically significant. HIV replication was greater in IFN-R NS5A-expressing Jurkat cells, possibly due to the fact that lower levels of NS5A are expressed in these cells (Fig. 1C). The decreased surface density of CXCR4 was not altered when cells were incubated in neutralizing anti-SDF-1 antibodies (10 μg/ml; data not shown), suggesting that the effect of NS5A on CXCR4 expression is not due simply to increased SDF-1 release into culture supernatants. CD4 or CCR5 expression on Jurkat cells was not altered in GBV-C NS5A-expressing and VC cells (data not shown).
GBV-C NS5A Deletion Mapping.
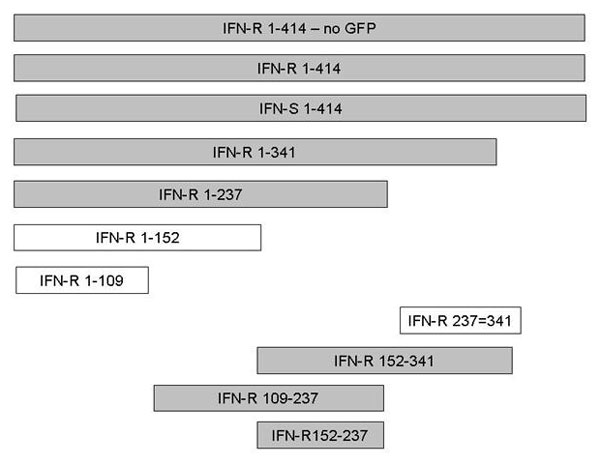
To characterize which portion of the GBV-C NS5A protein is required for HIV inhibition, a series of Jurkat cell lines expressing deletion mutants were constructed. HIV replication in Jurkat cells expressing full-length and key NS5A deletion mutants is shown in Fig. 5. HIV replication also was examined in additional NS5A deletion expressing cell lines (Fig. 9, which is published as supporting information on the PNAS web site). The cell line expressing an NS5A deletion mutant containing amino acids 152–237 demonstrated HIV-1 inhibition (Fig. 5), as did all other mutants that include this region. In contrast, NS5A deletions 1–152 and 237–341 did not inhibit HIV-1. Thus, the 85 aa between 152 and 237 are sufficient to inhibit HIV replication when expressed in Jurkat cells. Further evidence that this region inhibits HIV is shown in Fig. 10, which is published as supporting information on the PNAS web site, because HIV-1 inhibition correlated with the level of this NS5A deletion protein expressed in individual cloned Jurkat cell lines. It is of note that there are two forms of this immunoreactive 85-aa protein in Jurkat cells (Fig. 10), indicating that this peptide fragment serves as a substrate for intracellular phosphorylation.
Fig. 5.

Deletion mutants of GB virus C NS5A inhibit HIV replication in Jurkat cells. Schematic representation of the full-length NS5A from the IFN-S GBV-C isolate, and key deletion mutants from IFN-R isolate NS5A are shown. Numbering represents the NS5A amino acids included in the expression clone. HIV replication (measured by p24 antigen release into culture supernatant) in each cell line on days 0, 4, 5, 6, and 7 is shown. NS5A proteins that inhibited HIV replication compared with the VC expressing GFP are shaded.
To ensure that the observed HIV inhibition was due to expression of NS5A protein and not due to effects of the transcribed NS5A RNA in the Jurkat cells, we created a frameshift mutation, placing an AUG in front of the BstE II site at nucleotide 6509 (AF121950). The AUG starts out of frame at NS5A amino acid 111 and encodes 27 missense amino acids before a stop codon. NS5A RNA continues to nucleotide 6856 (NS5A amino acid 237) and is followed by the EMC internal ribosomal entry site and GFP coding region. Stably transfected Jurkat cells were selected (Fig. 6A), and no NS5A was expressed (Fig. 6B), although the NS5A DNA sequence was present in the cells when assessed by DNA PCR (data not shown). HIV replication was not significantly inhibited in these cells compared with vector control cells (P > 0.05 on all days after HIV infection; Fig. 6C), demonstrating that NS5A protein, and not RNA, is responsible for the HIV inhibition.
Fig. 6.

HIV inhibition requires GBV-C NS5A protein expression. Jurkat cells containing a plasmid transcribing NS5A RNA containing a frameshift mutation were selected (FS). An AUG initiates translation at nucleotide 6509 of GBV-C (NS5A amino acid 111) and RNA extends to nucleotide 6856 (to amino acid 237). The frameshift construct encodes 27 missense amino acids. (A and B) GFP expression was examined by flow cytometry (A), and NS5A expression was assessed by immunoblot analysis for the vector control (VC-GFP), FS, and IFN-S NS5A expressing cells (B). (C) HIV replication (NIH reagent program isolate 1073) was not inhibited significantly in cells expressing the frameshift RNA compared with the VC-GFP cells.
Discussion
GBV-C viremia is associated with prolonged survival among HIV-infected people in most, although not all, studies (41). A metaanalysis combining studies of 1,294 HIV-infected individuals found that the relative risk of death was 0.41 (95% confidence intervals 0.23–0.69) for those with persistent GBV-C infection compared with GBV-C uninfected (6). Additional beneficial findings associated with GBV-C viremia include slower CD4 decline, lower plasma HIV RNA concentration, and improved response to antiretroviral therapy, further supporting a beneficial role for GBV-C infection (reviewed in refs. 1 and 6). Epidemiological studies cannot determine whether this association is caused by GBV-C infection or whether GBV-C viremia serves as either a marker of HIV-1 replication or a beneficial host factor in HIV-infected individuals. Studies to identify potential biological mechanisms by which GBV-C might influence HIV-1 disease progression are required to either support or refute a causal role of GBV-C for this epidemiological observation.
Because GBV-C replicates in lymphocytes, including CD4+ T cells (10, 13, 14), it has been postulated that GBV-C might alter HIV-1 replication through direct or indirect mechanisms (1, 24, 42). Previous studies found that in vitro coinfection of PBMCs with GBV-C and HIV-1 resulted in decreased HIV-1 replication for both R5 and X4 viruses (2, 24–26). However, no specific GBV-C protein mediating this HIV inhibitory effect has been identified. In this study, GBV-C NS5A protein expressed in Jurkat cells resulted in potent inhibition of HIV-1 replication (Figs. 2, 3, and 5). The effect was associated, in part, with increased release of SDF-1 into culture supernatants and by decreased expression of CXCR4 on the cell surface (Fig. 4). Although SDF-1 contributes to HIV-1 replication inhibition, neutralizing antibodies to SDF-1 only partially blocked this inhibitory effect, suggesting that NS5A inhibits HIV-1 replication through additional mechanisms.
HIV replication inhibition required NS5A amino acids 152–237, a region that includes differing sequence polymorphisms in IFN-S and IFN-R GBV-C isolates (32). IFN-R and IFN-S GBV-C NS5A proteins were shown to have different inhibitory effects on PKR function, and only the IFN-R NS5A inhibited PKR-mediated phosphorylation of eIF-2α (32). The fact that both IFN-R and IFN-S NS5A proteins inhibited HIV-1 to a similar extent indicates that the effect of NS5A on PKR function is distinct from the HIV-1 inhibitory effect. It has been proposed that GBV-C genotypes present in diverse geographic regions may have differential effects on HIV-1 (43). The IFN-S NS5A protein comes from a genotype 3 isolate (32), whereas the IFN-R NS5A is genotype 2, suggesting that genotypic differences in NS5A do not appear to influence HIV-1 inhibition. The full-length GBV-C NS5A protein and the 152–237 NS5A deletion protein both appear to be good phosphorylation substrates in Jurkat cells (Figs. 1 and 10). Because of the large number of potential phosphorylation sites in the 85 NS5A amino acids that inhibit HIV (15 for IFN-S and 18 for IFN-R), it is tempting to speculate that phosphorylation of this peptide interferes with chemokine or chemokine receptor pathways or other intracellular pathways involved in HIV replication. Thus, identification of cellular proteins that interact with NS5A, and determination of the effects of the protein on cellular gene expression, appears warranted.
In previous studies, we and others were not able to demonstrate reduced CXCR4 levels in GBV-C-infected PBMCs (24, 25). During GBV-C infection of PBMCs, NS5A is expressed only in cells supporting active GBV-C replication, and the protein is not released from cells, nor is it part of the virus particle. Hence, the effect of NS5A may be limited to actively infected cells. Because <5% of PBMCs are actively infected with GBV-C and in vitro GBV-C replication in PBMCs is not robust (39), the effect of GBV-C on CXCR4 may be insufficient to be detected by flow cytometry. However, PBMCs infected with GBV-C ex vivo demonstrated similar levels of NS5A expression, as did the Jurkat cells in which HIV replication was inhibited (Fig. 3C). By controlled expression of NS5A in a stably transformed population of Jurkat cells, we were able to demonstrate that NS5A leads to decreased surface density of CXCR4 (Fig. 4). This decreased surface expression was not related to blockade of CXCR4 detection by SDF-1, because maintaining the cells in neutralizing anti-SDF-1 antibodies did not increase the detection of CXCR4 on the cell surface.
HIV-positive people with acute dengue or measles viruses infection have marked reductions in plasma HIV RNA concentration, suggesting viral interference (44, 45). Although measles viruses has been shown to inhibit HIV replication independent of chemokines in vitro (46), several members of the Flavivirus family (HCV, Japanese Encephalitis Virus and Dengue Virus) alter the cellular release of cytokines and chemokines (47–49). Thus, chemokine and cytokine modulation may be a common feature of Flavivirus replication, and the effects observed with GBV-C NS5A protein may be found in related viruses as well. Nevertheless, a more prominent HIV inhibitory effect would be expected for GBV-C infection compared with other members of the Flaviviridae because of its propensity to result in persistent viremia (present in up to 39% of HIV-positive people; ref. 2) and because it replicates in CD4+ and CD8+ T lymphocytes and B lymphocytes (13). Thus, modulation of cellular chemokines occurs in the local milieu of HIV replication. In addition, GBV-C appears to be nonpathogenic (5), and, therefore, there does not appear to be a risk of comorbidity. These data provide support for the hypothesis that GBV-C replication is related causally to the observed epidemiological association between persistent GBV-C viremia and prolonged survival in HIV-infected people (6).
GBV-C inhibits multiple HIV-1 clades (Fig. 8; ref. 25) and a zidovudine-resistant HIV-1 isolate (isolate no. 1073). Because the effect of the GBV-C NS5A protein appears to occur at the cellular level and not by targeting an HIV-1 replication enzyme or receptor binding domain, the barrier for HIV-1 to develop resistance to this effect would be expected to be greater than that seen with current treatment strategies. Thus, further characterization of this inhibitory effect may lead to previously undescribed approaches for antiretroviral therapy.
Materials and Methods
Expression of NS5A Proteins in Jurkat Cells.
GBV-C nucleotide sequences predicted to encode the full-length NS5A protein were amplified from plasma of individuals with GBV-C viremia as described in refs. 10 and 32. NS5A sequences were subcloned into the pTRE2-Hgy plasmid (Clontech, Mountain View, CA) modified to include a stop codon after NS5A, followed by the EMC internal ribosomal entry site element directing the translation of GFP. NS5A deletion mutants were generated by using convenient restriction sites or by PCR mutagenesis and subcloned into the pTRE2-Hgy plasmid. All sequences were confirmed by automated fluorescent dye terminator cycle sequencing (University of Iowa DNA Core Facility; automated DNA sequencer 373A, Applied Biosystems, Foster City, CA). Tet-Off Jurkat cells (Clontech) were transfected with NS5A-containing plasmids or either the VC or with VC-GFP by using the Amaxa (Gaithersburg, MD) nucleofection method. After selection in hygromycin and neomycin (200 μg/ml), cell lines were selected for hygromycin and G418 resistance and cloned at least twice by terminal dilution.
Cells were maintained in RPMI medium 1640 containing 10% FCS with or without doxycycline to regulate NS5A protein expression. To monitor NS5A expression, Jurkat cells were lysed in RIPA buffer containing protease and phosphatase inhibitors, clarified (13,000 × g, 2 min and 4°C) and subjected to SDS/PAGE before transfer to nitrocellulose membranes (Bio-Rad, Hercules, CA) as described in ref. 32. Immunoreactive proteins were identified by using the GE3 anti-GBV-C NS5A rabbit serum kindly provided by Jungsuh Kim (Genelabs Technologies, Redwood City, CA), which was generated against GBV-C nucleotide sequences 6615–6977 expressed in Escherichia coli (50) or by an anti-NS5A peptide sera as described in ref. 32. Alternatively, NS5A was identified in cells by indirect immunofluorescence microscopy by using the GE3 antibody (51).
HIV Infections.
Three HIV-1 isolates (X4) representing clade B (NIH AIDS Research and Reference Reagent Program, catalog nos. 1073, 317, and 2969) and one X4 isolate representing clade D (catalog no. 2521) were studied. HIV-1 was applied to Jurkat cells (200 pg of HIV p24 antigen per 106 cells) containing NS5A sequences (with or without doxycycline to repress expression) or vector control cells with or without doxycycline. After 3 h of attachment, cells were washed and maintained in fresh media. Culture supernatants were obtained at various time points to measure HIV-1 replication and GBV-C NS5A expression. All infections were performed in duplicate or triplicate and repeated at least three times with similar results. HIV-1 replication was determined either by measuring HIV p24 antigen in culture supernatants (Retro-Tek HIV-1 p24 antigen ELISA kits; Zeptometrix, Buffalo, NY) or by measuring infectivity of culture supernatants in PBMC cultures determining the TCID50 as described in refs. 2 and 24.
Characterization of Cell Surface Receptors and Chemokine Release.
CD4, CCR5, and CXCR4 expression on the surface of Jurkat cells (with and without NS5A and doxycycline) were determined by flow cytometry as described in refs. 2 and 24. Polyclonal rabbit anti-CD4 (PE-conjugated), anti-CCR5 (FITC-conjugated), and anti-CXCR4 (PE-conjugated) antibodies (BD Pharmingen, San Jose, CA) were used in these studies. Flow cytometry was performed by using a FACScan (Becton Dickinson, San Jose, CA). SDF-1, RANTES, MIP-1α, and MIP-1β were detected in culture supernatant fluids by ELISA (R & D Systems, Minneapolis, MN) as described in ref. 24. Neutralizing anti-SDF-1 antibody was obtained from R & D Systems.
Statistics.
All statistics were performed by using SigmaStat software V2.03S (Jandel Scientific, Chicago, IL). Comparisons of two samples used t tests, and comparisons of more than three samples used ANOVA.
Supplementary Material
Acknowledgments
We thank Dr. Jungsuh Kim (Genelabs Technologies, Redwood City, CA) for providing GE3 anti-NS5A antisera. The University of Iowa DNA Core and Flow Cytometry Core facilities were used for these studies. Reagents obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health, include as follows: AZT-Intermediate Isolate, Dr. D. Richmond (catalog no. 1073); HTLV-IIIMN/H9, Dr. R. Gallo (no. 317); HIV-1LAI, Dr. J. Mellors (no. 2969); and HIV-1ELI, Drs. J.-.M Bechet and L. Montagnier (no. 2521). This work was supported by Veterans Affairs Merit Review grants (to J.X. and J.T.S.) and a NIAID Grant AI58740 and subcontract from the Women's Interagency Health Survey to Iowa City Veteran Affairs (to J.T.S.).
Abbreviations
- GBV-C
GB Virus type C
- HCV
hepatitis C virus
- IFN-R
IFN-resistant
- IFN-S
IFN-sensitive
- PKR
dsRNA-activated protein kinase
- VC
vector control
- VC-GFP
vector control expressing GFP
- X4
HIV isolates that utilize CXCR4 as their entry coreceptor.
Footnotes
Conflict of interest statement: Potential conflicts of interest are present for Drs. J.X. and J.T.S., who hold a patent on an infectious molecular clone of GB virus C, and J.X., J.H.M., and J.T.S., who have a provisional patent application pending related to HIV inhibition by GB virus C NS5A protein.
This article is a PNAS direct submission.
References
- 1.Stapleton JT, Williams CF, Xiang J. J Clin Microbiol. 2004;42:3915–3919. doi: 10.1128/JCM.42.9.3915-3919.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xiang J, Wunschmann S, Diekema DJ, Klinzman D, Patrick KD, George SL, Stapleton JT. N Engl J Med. 2001;345:707–714. doi: 10.1056/NEJMoa003364. [DOI] [PubMed] [Google Scholar]
- 3.Thomas DL, Vlahov D, Alter HJ, Marshall R, Astemborski J, Nelson KE. J Infect Dis. 1998;177:539–542. doi: 10.1086/514245. [DOI] [PubMed] [Google Scholar]
- 4.Tillmann HL, Heringlake S, Trauwein C, Meissner D, Nashan B, Schlitt HJ, Kratochvil J, Hunt J, Qiu X, Lou C, et al. Hepatology. 1998;28:379–384. doi: 10.1002/hep.510280213. [DOI] [PubMed] [Google Scholar]
- 5.Alter HJ. Transfusion. 1997;37:569–572. doi: 10.1046/j.1537-2995.1997.37697335149.x. [DOI] [PubMed] [Google Scholar]
- 6.Zhang W, Chaloner K, Tillmann HL, Williams CF, Stapleton JT. HIV Med. 2006;7:173–180. doi: 10.1111/j.1468-1293.2006.00366.x. [DOI] [PubMed] [Google Scholar]
- 7.Linnen J, Wages J, Zhang-Keck Z-Y, Fry KE, Krawczynski KZ, Alter H, Koonin E, Gallagher M, Alter M, Hadziyannis S, et al. Science. 1996;271:505–508. doi: 10.1126/science.271.5248.505. [DOI] [PubMed] [Google Scholar]
- 8.Simons JN, Leary TP, Dawson GJ, Pilot-Matias TJ, Muerhoff AS, Schlauder GG, Desai SM, Mushahwar IK. Nat Med. 1995;1:564–569. doi: 10.1038/nm0695-564. [DOI] [PubMed] [Google Scholar]
- 9.Simons JN, Desai SM, Schultz DE, Lemon SM, Mushahwar IK. J Virol. 1996;70:6126–6135. doi: 10.1128/jvi.70.9.6126-6135.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiang J, Wunschmann S, Schmidt WN, Shao J, Stapleton JT. J Virol. 2000;74:9125–9133. doi: 10.1128/jvi.74.19.9125-9133.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Hepatology. 2004;39:5–19. doi: 10.1002/hep.20032. [DOI] [PubMed] [Google Scholar]
- 12.Pessoa MG, Terrault NA, Detmer J, Kolberg J, Colllins M, Hassoba HM, Wright TL. Hepatology. 1998;27:877–880. doi: 10.1002/hep.510270335. [DOI] [PubMed] [Google Scholar]
- 13.George SL, Varmaz D, Stapleton JT. J Infect Dis. 2006;193:451–454. doi: 10.1086/499435. [DOI] [PubMed] [Google Scholar]
- 14.Fogeda M, Navas S, Martin J, Casqueiro M, Rodriguez E, Arocena C, Carreno V. J Virol. 1999;73:4052–4061. doi: 10.1128/jvi.73.5.4052-4061.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Radkowski M, Kubicka J, Kisiel E, Cianciara J, Nowicki M, Rakela J, Laskus T. Transfusion Med Rev. 2000;95:3986–3989. [PubMed] [Google Scholar]
- 16.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, et al. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 17.Berger EA, Doms RW, Fenyo EM, Korber BT, Littman DR, Moore JP, Sattentau QJ, Schuitemaker H, Sodroski J, Weiss RA. Nature. 1998;391:240. doi: 10.1038/34571. [DOI] [PubMed] [Google Scholar]
- 18.Scarlatti G, Tresoldi E, Bjorndal A, Fredriksson R, Colognesi C, Deng HK, Malnati MS, Plebani A, Siccardi AG, Littman DR, et al. Nat Med. 1997;3:1259–1265. doi: 10.1038/nm1197-1259. [DOI] [PubMed] [Google Scholar]
- 19.Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Science. 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 20.Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, Springer TA. Nature. 1996;382:829–833. doi: 10.1038/382829a0. [DOI] [PubMed] [Google Scholar]
- 21.Lin YL, Mettling C, Portales P, Reynes J, Clot J, Corbeau P. Proc Natal Acad Sci USA. 2002;99:15590–15595. doi: 10.1073/pnas.242134499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Copeland KF. Mini Rev Med Chem. 2005;5:1093–1101. doi: 10.2174/138955705774933383. [DOI] [PubMed] [Google Scholar]
- 23.Amella CA, Sherry B, Shepp DH, Schmidtmayerova H. J Virol. 2005;79:5625–5631. doi: 10.1128/JVI.79.9.5625-5631.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiang J, George SL, Wunschmann S, Chang Q, Klinzman D, Stapleton JT. Lancet. 2004;363:2040–2046. doi: 10.1016/S0140-6736(04)16453-2. [DOI] [PubMed] [Google Scholar]
- 25.Jung S, Knauer O, Donhauser N, Eichenmuller M, Helm M, Fleckenstein B, Reil H. AIDS. 2005;19:1267–1272. doi: 10.1097/01.aids.0000180097.50393.df. [DOI] [PubMed] [Google Scholar]
- 26.Xiang J, Sathar MA, McLinden JH, Klinzman D, Chang Q, Stapleton JT. J Infect Dis. 2005;192:2147–2151. doi: 10.1086/498170. [DOI] [PubMed] [Google Scholar]
- 27.Nattermann J, Nischalke HD, Kupfer B, Rockstroh J, Hess L, Sauerbruch T, Spengler U. AIDS. 2003;17:1457–1462. doi: 10.1097/00002030-200307040-00006. [DOI] [PubMed] [Google Scholar]
- 28.Pawlotsky JM, Germanidis G. J Viral Hepat. 1999;6:343–356. doi: 10.1046/j.1365-2893.1999.00185.x. [DOI] [PubMed] [Google Scholar]
- 29.Polyak SJ. Clin Liver Dis. 2003;7:67–88. doi: 10.1016/s1089-3261(02)00075-2. [DOI] [PubMed] [Google Scholar]
- 30.Macdonald A, Harris M. J Gen Virol. 2004;85:2485–2502. doi: 10.1099/vir.0.80204-0. [DOI] [PubMed] [Google Scholar]
- 31.Tellinghuisen TL, Marcotrigiano J, Gorbalenya AE, Rice CM. J Biol Chem. 2004;279:48576–48587. doi: 10.1074/jbc.M407787200. [DOI] [PubMed] [Google Scholar]
- 32.Xiang J, Martinez-Smith C, Gale M, Jr, LaBrecque DR, Schmidt WN, Stapleton JT. J Interferon Cytokine Res. 2005;25:261–270. doi: 10.1089/jir.2005.25.261. [DOI] [PubMed] [Google Scholar]
- 33.Gale M, Jr, Blakely CM, Kwieciszewski B, Tan SL, Dossett M, Tang NM, Korth MJ, Polyak SJ, Gretch DR, Katze MG. Mol Cell Biol. 1998;18:5208–5218. doi: 10.1128/mcb.18.9.5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gale M, Katze MG., Jr Methods. 1997;11:383–401. doi: 10.1006/meth.1996.0436. [DOI] [PubMed] [Google Scholar]
- 35.Gale M, Jr, Kwieciszewski B, Dossett M, Nakao H, Katze MG. J Virol. 1999;73:6506–6516. doi: 10.1128/jvi.73.8.6506-6516.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gong G, Waris G, Tanveer R, Siddiqui A. Proc Natl Acad Sci USA. 2001;98:9599–9604. doi: 10.1073/pnas.171311298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Polyak SJ, Khabar KS, Paschal DM, Ezelle HJ, Duverlie G, Barber GN, Levy DE, Mukaida N, Gretch DR. J Virol. 2001;75:6095–6106. doi: 10.1128/JVI.75.13.6095-6106.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ezelle HJ, Balachandran S, Sicheri F, Polyak SJ, Barber GN. Virol. 2001;281:124–137. doi: 10.1006/viro.2001.0815. [DOI] [PubMed] [Google Scholar]
- 39.George SL, Xiang J, Stapleton JT. Virology. 2003;316:191–201. doi: 10.1016/s0042-6822(03)00585-3. [DOI] [PubMed] [Google Scholar]
- 40.Ioannidis JP, Rosenberg PS, Goedert JJ, Ashton LJ, Benfield TL, Buchbinder SP, Coutinho RA, Eugen-Olsen J, Gallart T, Katzenstein TL, et al. Ann Int Med. 2001;135:782–795. doi: 10.7326/0003-4819-135-9-200111060-00008. [DOI] [PubMed] [Google Scholar]
- 41.Polgreen PM, Xiang J, Chang Q, Stapleton JT. Microbes Infect. 2003;5:1255–1261. doi: 10.1016/j.micinf.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 42.Pomerantz RJ, Nunnari G. N Engl J Med. 2004;350:963–965. doi: 10.1056/NEJMp048004. [DOI] [PubMed] [Google Scholar]
- 43.Muerhoff AS, Tillmann HL, Manns M, Dawson GJ, Desai SM. J Med Virol. 2003;70:141–149. doi: 10.1002/jmv.10375. [DOI] [PubMed] [Google Scholar]
- 44.Watt G, Kantipong P, Jongsakul K. Clin Infect Dis. 2003;36:1067–1069. doi: 10.1086/374600. [DOI] [PubMed] [Google Scholar]
- 45.Moss WJ, Ryon JJ, Monze M, Cutts F, Quinn TC, Griffin DE. J Infect Dis. 2002;185:1035–1042. doi: 10.1086/340027. [DOI] [PubMed] [Google Scholar]
- 46.Grivel JC, Garcia M, Moss WJ, Margolis LB. J Infect Dis. 2005;192:71–78. doi: 10.1086/430743. [DOI] [PubMed] [Google Scholar]
- 47.Medin CL, Fitzgerald KA, Rothman AL. J Virol. 2005;79:11053–11061. doi: 10.1128/JVI.79.17.11053-11061.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen CJ, Chen JH, Chen SY, Liao SL, Raung SL. J Virol. 2004;78:12107–12119. doi: 10.1128/JVI.78.22.12107-12119.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Apolinario A, Majano PL, Alvarez-Perez E, Saez A, Lozano C, Vargas J, Garcia-Monzon C. Am J Gastroenterol. 2002;97:2861–2870. doi: 10.1111/j.1572-0241.2002.07054.x. [DOI] [PubMed] [Google Scholar]
- 50.Belyaev AS, Chong S, Novikov A, Kongpachith A, Masiarz FR, Lim M, Kim JP. J Virol. 1998;72:868–872. doi: 10.1128/jvi.72.1.868-872.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wunschmann S, Klinzman D, Medh J, Klinzman D, Schmidt WN, Stapleton JT. J Virol. 2000;74:10055–10062. doi: 10.1128/jvi.74.21.10055-10062.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}