

Abstract
Chlamydia trachomatis entry into host cells results from a parasite-directed remodeling of the actin cytoskeleton. A type III secreted effector, TARP (translocated actin recruiting phosphoprotein), has been implicated in the recruitment of actin to the site of internalization. To elucidate the role of TARP in actin recruitment, we identified host cell proteins that associated with recombinant GST-TARP fusions. TARP directly associated with actin, and this interaction promoted actin nucleation as determined by in vitro polymerization assays. Domain analysis of TARP identified an actin-binding domain that bears structural and primary amino acid sequence similarity to WH2 domain family proteins. In addition, a proline-rich domain was found to promote TARP oligomerization and was required for TARP-dependent nucleation of new actin filaments. Our findings reveal a mechanism by which chlamydiae induce localized cytoskeletal changes by the translocated effector TARP during entry into host cells.
Keywords: Chlamydia, type III secretion, cytoskeleton, endocytosis
A Gram-negative obligate intracellular bacterium, Chlamydia trachomatis, is the leading cause of preventable blindness worldwide and the most prevalent bacterial pathogen causing sexually transmitted disease in the western world (1). Chlamydiae initiate their intracellular developmental cycle by actively gaining entry into host cells. The extracellular infectious form of the developmental cycle is referred to as an elementary body (EB). Once engulfed by the host cell, the EB differentiates into the replicative reticulate body within the protective confines of a membrane-bound parasitophorous vacuole called an inclusion (2).
The ligands on the EB surface and cognate host cell receptor have not been definitively identified; however, it appears that chlamydiae use an entry mechanism that involves several distinct levels of interaction (3, 4). EB invasion of nonphagocytic cells is thus the product of coordinated cytoskeletal remodeling characterized by the formation of pedestal-like structures and hypertrophic microvilli that are directly triggered by the invading chlamydiae (5).
A recently identified C. trachomatis type III secretion system secreted protein called TARP (for translocated actin recruiting phosphoprotein) is tyrosine-phosphorylated by a host cell kinase and is spatially and temporally associated with the recruitment of actin at the site of EB invasion (6). TARP is present in all pathogenic Chlamydia species examined to date. Analysis of TARP orthologs from C. trachomatis, Chlamydia muridarium, Chlamydia caviae, and Chlamydia pneumoniae indicates that only C. trachomatis serovars are phosphorylated, despite all Chlamydia strains demonstrating the recruitment of actin to the site of entry (6, 7).
We demonstrate here that TARP associates directly with actin by a small domain contained within the C-terminal region of the protein. Furthermore, TARP independently nucleates new actin filaments by forming a large homogenous multimeric protein complex mediated by a proline-rich domain. We propose that TARP participates in the observed “parasite-specified phagocytosis” (8) by promoting the rapid polymerization of actin filaments required for EB uptake.
Results
TARP Binds to Actin but Not the Arp2/3 Complex from HeLa Extracts.
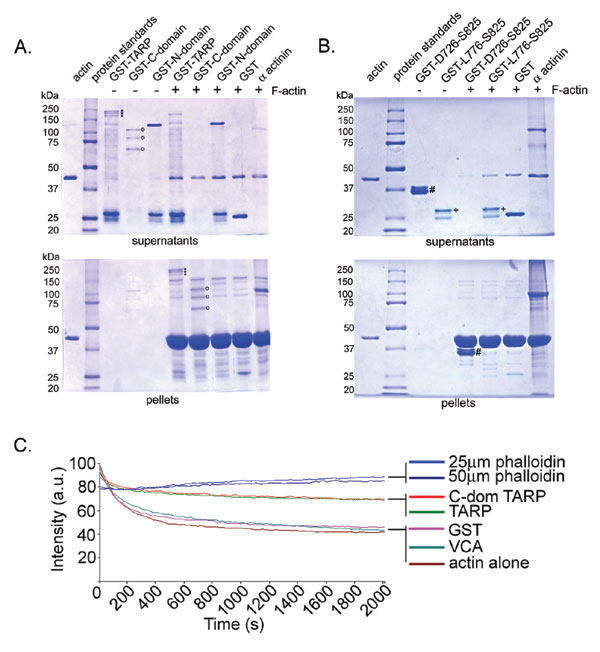
GST-TARP fusions were used in pull-down experiments to identify interacting proteins. The C-terminal domain of TARP or full-length TARP expressed as a GST fusion specifically precipitated a host cell protein of ≈40,000 Da, which was identified as actin (Fig. 1A). Actin was also precipitated by a GST fusion to the verprolin, cofilin, and acidic (VCA) domain of the human adapter protein N-WASP (WASP, Wiskott–Aldrich syndrome protein), known to mediate actin dynamics through direct association of actin monomers and the Arp2/3 complex (9, 10). No proteins were found to specifically interact with the GST protein or uncoated bead controls, and likewise no proteins were precipitated by GST fusion to the N-terminal domain of TARP (Fig. 1A).
Fig. 1.

TARP associates with actin but not the Arp2/3 complex. (A) Extracts from HeLa cells were incubated with GST or GST fusions to TARP and specifically bound proteins were resolved by SDS/PAGE and visualized by Coomassie blue staining. In this experiment, Tarp and Tarp-C-domain GST fusion constructs (GST-TARPΔ and GST-C-domainΔ, respectively) were truncated at the C terminus by 150 amino acids to prevent proteolytic breakdown (see Fig. 4A). GST fusions are indicated by open circles. Actin (arrow) from HeLa lysates was specifically bound by GST-TARPΔ or GST-C-domainΔ. Full-length Tarp and Tarp–C domain showed identical results (data not shown). The HeLa lysate shown in the first lane represents 1% of the material used in the +lysate pull-down lanes. Samples identical to those shown in the Coomassie-stained gel were subjected to immunoblotting with actin (α actin), actin-related protein 3 (α Arp3), or formin (α Dia1)-specific antibodies. Molecular mass of protein standards is in kDa. (B) GST-TARP associates with monomeric actin (G actin) directly. Immobilized GST-fusion proteins to full-length TARP, the C-terminal domain of TARP, and the N-terminal domain of TARP were incubated with monomeric actin in G buffer. Bound proteins eluted from glutathione beads were resolved by SDS/PAGE and subjected to immunoblotting with an actin (α actin)-specific antibody.
The Arp2/3 complex, a complex of seven proteins known to nucleate new actin filaments within eukaryotic cells, was also readily precipitated by the GST-VCA fusion but not by TARP (Fig. 1A). Probes for diaphanous family formins, Dia1 (Fig. 1A) and Dia2 (not shown), were similarly negative. Collectively, the results suggest that cellular actin-binding proteins do not mediate the association between actin and TARP.
In agreement with previous findings (7), GST fusions to the full-length TARP protein or N-terminal domain containing the tyrosine-rich repeat region of TARP were phosphorylated when incubated in the presence of HeLa extracts (data not shown). These findings confirm that TARP phosphorylation and actin recruitment involve distinct domains of the protein.
TARP Binds to Actin Directly.
Because actin was the only observed protein precipitated from HeLa extracts by TARP, we analyzed the capacity of TARP to bind to actin directly. The two recombinant TARP proteins that associate with actin from HeLa extracts, GST-TARP and GST-C-domain, were found to cosediment with purified filamentous (F) actin (see Fig. 6, which is published as supporting information on the PNAS web site). The assay was validated by the observed binding of the F actin-binding protein α actinin to actin filaments and the absence of actin binding to the N-terminal domain of TARP and GST control. To investigate whether monomeric actin (G actin) similarly associated directly with the TARP C domain, purified rabbit actin was used in GST pull-down experiments performed under monomeric actin-supporting conditions. Again, both the full-length TARP and C- terminal domain of TARP precipitated G actin, whereas the N-terminal domain of TARP and GST control did not (Fig. 1B). The direct association between mammalian actin and TARP protein suggests that chlamydiae directly manipulate the host cell's microfilament network.
TARP Promotes Actin Polymerization.
To investigate whether TARP influences actin polymerization, an in vitro assay using pyrene-conjugated actin was used to analyze the kinetics of actin polymerization. Pyrene-actin fluorescence intensity increases up to 20-fold as monomeric actin is organized into filaments (11). In physiological buffers, G actin spontaneously organizes into filaments (12). This assembly of actin monomers is limited by the instability of actin dimers and trimers that serve as a nucleus for the rapid elongation of actin filaments (13). The kinetic barrier of nucleation is reflected in a characteristic lag phase in pyrene-actin polymerization assays and is followed by a rapid increase in fluorescence, which represents the elongation phase. Nucleation is the rate-limiting step in actin polymerization; thus, actin nucleators display a shortened or virtually nonexistent lag phase (11).
Both full-length TARP and the C-terminal domain of TARP promoted actin polymerization at a greater rate compared with the N-terminal domain of TARP, GST, or pyrene actin alone controls (Fig. 2A and D). The pyrene assay was also performed in the presence of HeLa extracts (Fig. 2B). Interestingly, the rate of actin polymerization in the presence of extract was less than that of TARP alone, presumably because of the presence of actin-depolymerizing and/or sequestering agents in the extract. The observation that actin polymerization accelerates in the absence of Arp2/3 complex or other cellular factors suggests that TARP directly nucleates actin polymerization. The dramatic effect of TARP on actin polymerization kinetics occurred despite minimal effects of TARP on the actin critical concentration (see Fig. 7 and Table 1, which are published as supporting information on the PNAS web site).
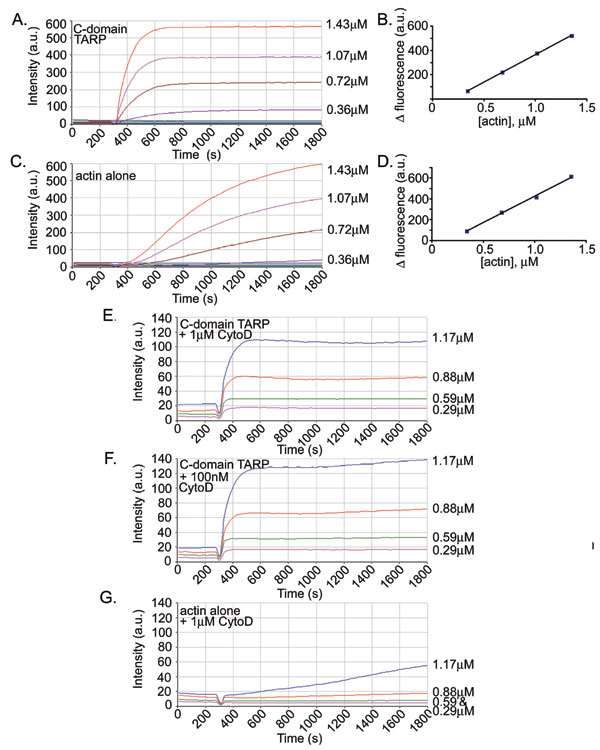
Fig. 2.

TARP promotes actin polymerization. (A) Pyrene actin polymerization in the presence of GST-TARP fusions. GST-TARP fusions representing the entire protein (TARP), C-terminal domain of TARP (C-dom TARP), or N-terminal domain of TARP (N-dom TARP) were incubated with 1 μM monomeric pyrene-labeled actin. A TARP-mediated increase in actin polymerization after the addition of polymerization buffer at 600 sec was measured as arbitrary fluorescence intensity (Intensity a.u) over time (seconds) with excitation and emission wavelengths of 365 and 407 nm, respectively. Purified GST (GST) and pyrene actin alone (actin alone) served as negative controls. (B) TARP-mediated actin polymerization occurs in the presence of HeLa extracts. Pyrene actin polymerization as described in A with the addition of 10 μg of HeLa extracts. (C) CD inhibits actin filament growth initiated by TARP. Pyrene assay as in A with the addition of 1 μM CD added to the C-terminal domain of TARP (C-dom TARP + CD) and actin control (actin alone + CD). (D) Pyrene-conjugated actin was visualized by transmission electron microscopy after initiation of polymerization. Actin filaments generated in the presence of the C-terminal domain of TARP (C-dom TARP) or GST control (GST) were collected at 5, 10, and 30 min postpolymerization initiation and stained with 1% uranyl-acetate. (Scale bar, 0.1 μm.)
TARP Nucleates New Actin Filaments.
Actin polymerization is prevented by the addition of the fungal metabolite cytochalasin D (CD), which caps the fast-growing or barbed end of an actin filament (14). CD (1 μM) inhibited TARP-induced actin filament growth, indicating that TARP promotes actin filament elongation from the barbed end (Fig. 2C). Interestingly, a concentration-dependent increase in pyrene actin fluorescence was observed with increased TARP concentrations in the presence of 1 μM CD (Fig. 3A). Actin filaments produced in the presence of TARP and CD were significantly shorter (≈0.07 microns) compared with untreated actin filaments (≈0.65 microns) when examined by transmission electron microscopy (Fig. 3B). Actin filament growth was arrested shortly after the initiation of polymerization, producing short actin filaments of similar length despite an 8-fold increase in TARP concentration (Fig. 3C). TARP reduced the critical concentration of actin in the presence of 100 nM or 1 μM CD to approximately that of actin alone, although the kinetics of polymerization resembled the initial burst of TARP-induced actin nucleation rather than actin alone (see Fig. 7 and Table 2, which are published as supporting information on the PNAS web site). Because the TARP-mediated rise in pyrene actin fluorescence did not correspond significantly with an increase in actin filament length, we determined whether increased fluorescence was because of the production of a greater quantity of similar-length actin filaments. An increase of pelleted filamentous actin was found to correlate with an increase in TARP concentration (data not shown). These data indicate that increased concentrations of TARP produced a greater number of actin nucleation events but ultimately generated actin filaments of a similar length in the presence of CD.
Fig. 3.

TARP nucleates new actin filaments. (A) Nucleation of new actin filaments is TARP concentration-dependent in the presence of 1 μM CD (+CD). An increase in pyrene actin fluorescence was observed as actin polymerization was assessed in the presence of 150, 300, 600, and 1,200 nM GST fusions to the C-terminal domain of TARP (C-dom TARP). (B) Pyrene-conjugated actin filaments generated in A were visualized by transmission electron microscopy after initiation of polymerization. Actin filaments generated in the presence of 150 nM C-terminal domain of TARP (150 nM C-dom TARP + CD) and (150 nM C-dom TARP − CD) or actin alone control (actin alone + CD) were collected 30 min postpolymerization initiation and stained with 1% uranyl-acetate. (Scale bar, 0.1 μm.) (C) Actin filament length was measured from micrographs collected in B. Data are represented as the average length of 40 individual actin filaments per condition. Error bars depict one standard deviation from the mean.
Identification of the Actin-Binding Domain of TARP.
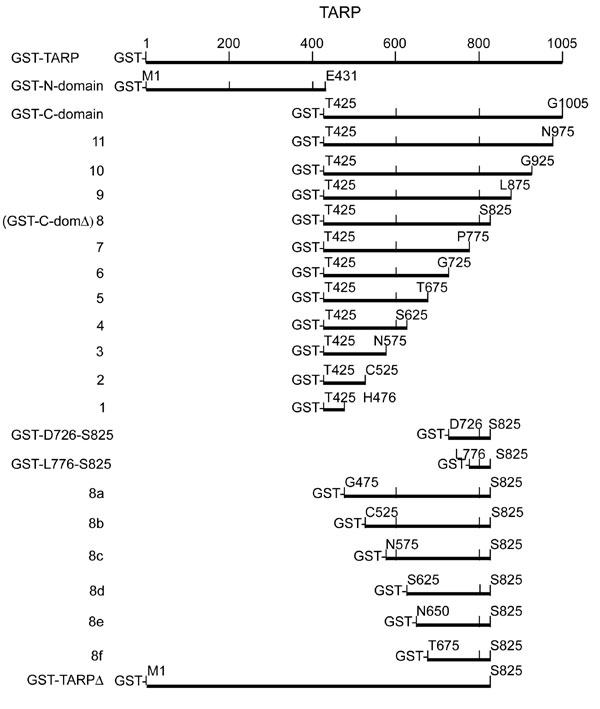
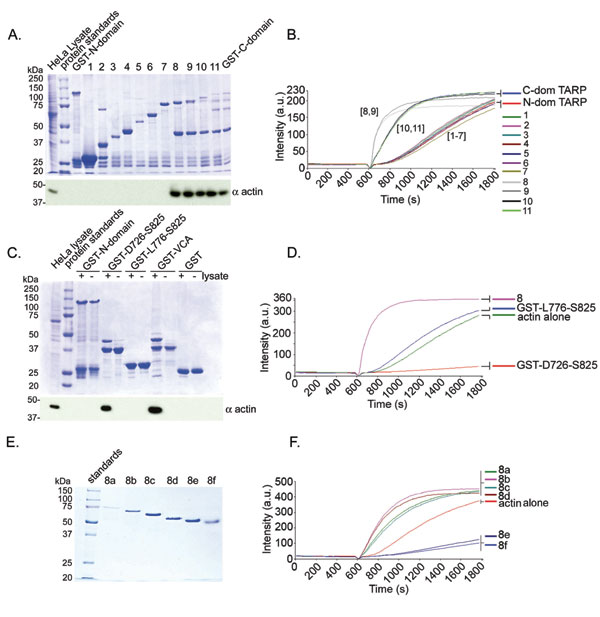
To define the residues within TARP responsible for actin polymerization, a series of truncated GST fusions (1–11) with the TARP C domain were generated (see Fig. 8, which is published as supporting information on the PNAS web site). The GST-TARP fusions were tested for their ability to bind actin in HeLa pull-down experiments and for their ability to polymerize actin in pyrene actin polymerization assays. The smallest GST-TARP fusion capable of binding to and polymerizing actin encompassed residues T425-S825. To define the minimal actin-binding domain of TARP, an additional overlapping GST fusion was generated containing a 100-aa fragment, residues D726-S825 (GST-D726-S825). This 100-aa fragment (GST-D726-S825) associated with actin but failed to induce polymerization in pyrene assays (see Fig. 9, which is published as supporting information on the PNAS web site). The rate of polymerization was below that of pyrene actin alone, resembling rates observed for typical G actin-binding proteins that depress rates of actin polymerization by sequestering G actin (11). These data indicate that the TARP actin-binding domain is contained within residues D726-S825. This fragment also binds F actin, suggesting that the G and F actin-binding domains are the same (see Fig. 6).
The 100-aa fragment of TARP, which binds actin, was found to contain a short sequence of amino acids (A748-K758) predicted to form a helical secondary structure. This helical domain harbors striking identity and similarity to the helical actin-binding domain of WH2 domain family proteins (ref. 15; see Fig. 4). The separation of an actin-binding domain from a polymerization domain suggests that TARP-mediated actin polymerization is not simply the product of a stable TARP–actin compound but a more complex association involving other domains of TARP.
Fig. 4.

Schematic of C. trachomatis TARP and GST-TARP fusions used in this study. (A) L2 TARP harbors an N-terminal tyrosine-rich repeat region (green boxes), which are contained within the GST fusions: GST-TARP, GST-TARPΔ, and the GST-N domain. Proline-dense (blue box) and actin-binding domains (red box) are contained within the C-terminal half of TARP located in the GST fusions: GST-C-domain, GST-C-domainΔ, and 8d. The GST fusion 8e contains the actin-binding domain only. (B) TARP harbors a conserved actin-binding domain. Sequence alignment of the actin-binding helix of WAVE2 and the putative actin-binding helix of TARP. Black bars indicate the residues that fall within the actin-binding helix for TARP (above) and WAVE2 (below). Yellow shading indicates identical residues, and green shading indicates similarity. A consensus sequence is shown under the alignment.
A Proline-Rich Domain Is Required for Actin Polymerization.
To identify the smallest fragment of TARP capable of polymerizing actin, GST-C-dom TARP no. 8, which harbors the actin-binding domain, was further truncated in 25- or 50-aa increments from the N terminus of the fragment to generate GST-TARP fragments 8a-8f. GST-TARP fusions 8a-8d were capable of polymerizing actin, whereas the shorter GST-TARP fusions 8e and 8f did not promote actin polymerization and demonstrated pyrene profiles similar to an actin sequestering protein (see Fig. 9, which is published as supporting information on the PNAS web site). The smallest GST-TARP fragment capable of polymerizing actin, 8d, also polymerized actin with the GST moiety removed (data not shown and Fig. 5C). The minimum actin polymerizing TARP fragment 8d* (* indicates GST is removed) capable of polymerizing actin differs from fragment 8e*, an actin sequestering peptide, by harboring a proline-rich domain contained within TARP amino acids S625-N650. We hypothesize that this region of TARP stimulates actin polymerization by promoting TARP oligomerization, thus bringing multiple actin-binding domains together to nucleate a new actin filament.
Fig. 5.

The actin-binding domain of TARP oligomerizes to form an actin nucleator. TARP fragments 8d* (* indicates GST is removed) capable of polymerizing actin and 8e* missing a proline-rich domain were analyzed by gel filtration. (A) An additional peak representing oligomerized Tarp (arrow) was detected in the A280 trace of eluted fragment 8d*. Elution times are indicated above the major peaks. The protease used to remove the GST moiety eluted at the 31.5-min peak. (B) Protein fractions were collected in 2-min intervals from the gel filtration column. Protein fractions were resolved by SDS/PAGE and were subjected to immunoblotting with a TARP-specific antibody. Dextran Blue 2000, polyacrylic sizing beads, and protein standards are indicated above the immunoblot with respective molecular weight and peak elution times. (C) Oligomerized TARP peptide polymerizes pyrene actin. GST-TARP peptide 8d* fraction 17 increased actin polymerization compared with the smaller 8d* fraction 27, which polymerized actin to similar levels as the actin alone control. 8e* fraction 29 reduced the rate of actin polymerization.
TARP Oligomerizes to Form an Actin Nucleator.
Gel filtration was used to determine the size of a potential TARP complex. Absorbance (280 nm) of protein standards demonstrated separation of proteins ranging in size from 1.1 mDa to 15 kDa. Fragment 8d* eluted as a symmetrical peak at ≈17.5 min (Fig. 5A), suggesting that the TARP peptide oligomerized to form a species of ≈800 kDa. Minor amounts of TARP were also detected in fractions 27 and 29 by Western blot analysis (Fig. 5B). When the smaller fragment of TARP not capable of actin polymerization, 8e*, was placed over the gel filtration column, no protein was observed in this size range (Fig. 5 A and B), suggesting the inability to form a large complex. The actin polymerization activity from the fractions collected harboring the major absorbance peaks for TARP fragments 8d* and 8e* demonstrates that the polymerization activity is contained in oligomerized 8d* (Fig. 5C). All TARP constructs examined that were capable of nucleating actin polymerization similarly oligomerized (data not shown).
TARP is known to migrate aberrantly on SDS/PAGE (6). Depending upon the values used in the calculations (predicted mass 20,520 Da vs. Mr 37,000), the 800-kDa homomeric complex represents 20–40 TARP monomers. These data taken together indicate that TARP has the capacity to form large homogenous complexes, and that this ability to oligomerize is essential for the actin-nucleating activity.
Discussion
Chlamydial attachment to the surface of epithelial cells induces rapid cytoskeletal rearrangements that culminate in the endocytosis of the invading bacterium. To investigate a role for TARP in chlamydial internalization, GST-TARP fusion proteins were used to identify interacting host cell proteins. Both monomeric actin (G actin) and filamentous actin (F actin) associated with TARP directly, and this binding occurred independently of TARP phosphorylation. In pyrene actin polymerization assays, TARP induced rapid and dose-dependent actin polymerization in the absence of additional host or chlamydial factors. The data suggest that TARP independently nucleates new actin filaments.
Three classes of actin nucleation factors have been described. These include the Arp2/3 complex, the formins, and the recently described Drosophila Spire protein (16–21). Each protein or protein complex nucleates actin with distinct features and polymerization kinetics. Other than the 9-aa sequence similarity to the actin-binding domain of WH2-family proteins, TARP displays no sequence similarity to components of the Arp2/3 complex, the formins, or Spire, and TARP-induced actin polymerization differs from these known actin nucleators in several properties. Unlike the Arp2/3 complex, TARP induces the formation of long unbranched actin filaments and does not require preexisting actin filaments from which to nucleate new filaments. Furthermore, Arp2/3 requires additional host factors, such as WASP-family proteins (themselves activated by Rho-family GTPases; refs. 22 and 23), whereas TARP does not. Unlike formins, TARP does not contain formin homology domains (FH1 and FH2) and binds monomeric actin in the absence of host proteins. In contrast to the four actin-binding domains of Spire, TARP requires only a single actin-binding domain to promote nucleation of actin filaments.
Like the Arp2/3 complex and Spire, actin filament formation by TARP is strongly inhibited by CD, an inhibitor of elongation that acts by capping the fast-growing barbed end of actin filaments (14, 24). TARP, like all actin-nucleating factors, thus stimulates filament formation by promoting elongation from the barbed end. Although TARP lacks the characteristic FH2 domain of formins, oligomerization appears to be a requirement for actin polymerization in the same sense that dimerization is a prerequisite for formins (25). TARP binds monomeric G actin, but oligomerization of TARP is required, presumably bringing multiple actin monomers into close proximity to nucleate a new actin filament. The effects of CD on TARP-induced filament formation are suggestive of a very rapid formation of an actin nucleus that is inhibited from elongation by the capping activity of cytochalasins. TARP thus shares characteristics with other known actin-nucleating agents but also demonstrates key differences that indicate actin nucleation likely occurs by means of a novel mechanism.
Actin filament formation is promoted by at least three distinct mechanisms. Severing of actin filaments can create new ends from which to extend actin filaments, rates of elongation may change by uncapping of barbed ends, or actin may be nucleated de novo (26). TARP appears to promote actin filament assembly by direct nucleation. The kinetics of actin polyermerization in the presence of TARP suggests de novo nucleation activity and is inconsistent with a severing action, because addition of TARP to preformed actin filaments did not result in changes to filaments, as observed by pyrene-actin fluorescence or transmission electron microscopy (data not shown). Furthermore, actin-severing proteins typically demonstrate a lag phase in pyrene actin polymerization assays that was not observed when performed in the presence of TARP. The almost immediate nucleation of actin is illustrated by the TARP concentration-dependent increase in pyrene-actin fluorescence in the presence of CD. This increase is very rapid and plateaus quickly when elongation is inhibited by CD. We have not ruled out the possibility that TARP also increases the rate of elongation, but that function would most likely be in addition to the observed nucleation function.
C. trachomatis L2 TARP contains a single actin-binding domain that bears sequence similarity to the WH2 domain of WASP-family protein member 2 (WAVE2). This region of TARP was unable to promote actin polymerization without additional TARP residues. Further analysis identified a proline-rich domain whose presence correlated with homooligomerization of TARP and, in conjunction with the actin-binding domain, the ability to nucleate actin. TARP multimerization likely occurs in the host cell after translocation through the type III secretion system, because complexes >800 kDa, the observed size of oligomerized TARP peptide (200 aa) by gel filtration are too large for secretion. TARP complexes in vitro appear to be stable; however, it is possible that complex formation in vivo is more transient given the dynamic regulation of actin recruitment and pedestal formation associated with EB internalization (5, 27).
The TARP orthologs demonstrate striking sequence diversity among C. trachomatis strains and species of Chlamydophila (7, 28). Interestingly, the tyrosine-phosphorylated tandem repeat domain of TARP is not conserved between species, yet both the actin-binding helix and proline-rich oligomerization domains are conserved among the sequenced strains, suggesting that these domains are integral to the function of TARP (7, 28). C. trachomatis L2 TARP contains a single actin-binding domain that is sufficient to promote actin polymerization. C. trachomatis serovars D and A (28, 29), respectively, contain two or three sequences nearly identical to the single L2 actin-binding domain. It will be of interest to compare the kinetics of actin polymerization among these TARP orthologs to determine whether polymerization rates correlate with observed differences in tissue tropism and/or disease type.
Many bacterial pathogens subvert actin cytoskeletal dynamics to their own ends. Bacteria such as Listeria, Shigella, Rickettsia, and Burkholderia, which replicate freely within the cytoplasm of the host cell, use an actin-based motility mechanism to spread directly from cell to cell (30–37). This mechanism typically involves a bacterial outer membrane protein that recruits components of the host actin machinery, including the Arp2/3 actin nucleation complex (38). Microbial pathogens also manipulate actin dynamics to promote, or avoid, internalization. Intracellular pathogens invade host cells by interactions between a bacterial cell surface ligand and cognate host cell receptor to initiate a cascade of signals that culminate in cytoskeletal rearrangements and phagocytosis. Alternatively, they may directly secrete effector proteins into the cell that interact with the actin machinery to promote internalization (39, 40). Generally, these processes are also accomplished by mechanisms that involve Rho-family GTPases and ultimately lead to recruitment of the Arp2/3 complex. Only one bacterial effector protein, SipC of Salmonella, has been proposed to directly nucleate actin filament formation (32). Chlamydial TARP also appears to directly nucleate actin filament formation during the entry process yet shares no sequence similarity with SipC.
Although TARP appears to directly nucleate actin, the internalization of chlamydiae is clearly multifaceted and likely involves multiple actin regulators. The Rho family GTPase, Rac, is activated and required for C. trachomatis entry, whereas both Rac and Cdc42 appear to be required for C. caviae internalization (27, 41). Rac activation is associated with lamellipodia formation in a process that involves WAVE2 and Arp2/3 (23). Preliminary data suggest that WAVE2, Abi1, and the Arp2/3 complex are recruited to the site of entry in a Rac-dependent manner and are required for efficient entry of chlamydial EBs.¶ The actin filaments induced by TARP may serve as a scaffold for the branching activity of activated Arp2/3 complex, synergistically altering the cytoskeleton of the host cell to promote chlamydial invasion. Current studies are directed toward elucidating the cooperative nature of this interaction during EB uptake. In addition to the significant contribution TARP makes in bacterial invasion as a novel nucleator, further studies on TARP-induced polymerization of actin will provide new insights into actin dynamics.
Materials and Methods
Cloning and Protein Expression.
In-frame GST fusion proteins for full-length TARP or truncation mutants were generated by PCR, amplifying the corresponding coding regions from C. trachomatis L2 genomic DNA using custom synthesized oligonucleotide primers (Integrated DNA Technologies, Coralville, IA). pGEX-6P-1 plasmids encoding the GST-TARP fusion proteins were transformed into the BL21 strain of Escherichia coli (Novagen, Madison, WI). Protein expression and purification were performed according to the procedures outlined in the Bulk GST Purification Module (Amersham Biosciences, Piscataway, NJ).
GST Fusion Protein Pull-Down Experiments.
HeLa 229 cells were suspended in 100 mM KCl/10 mM Hepes (pH 7.7)/2 mM MgCl2/2 mM ATP (buffer A) and disrupted by ultrasonication (Misonix, Farmingdale, NY). Insoluble material was removed by centrifugation (12,000 × g; 25 min, 4°C). Glutathione-Sepharose beads were incubated with 10 μg of GST fusion proteins or GST for 1 h at 4°C in PBS (Amersham Biosciences). GST-fusion protein-coated Sepharose beads were washed twice with PBS and once with buffer A before the addition of ≈100 μg of HeLa extracts. Extracts and beads were incubated together for 2 h at 4°C, washed three times with fresh buffer A, and bound proteins were eluted using Laemmli sample buffer.
Actin Binding.
F actin binding was performed according to the procedures outlined in the Actin Binding Protein Biochem Kit (Cytoskeleton, Denver, CO). Details are provided in Supporting Text, which is published as supporting information on the PNAS web site, as are protocols used for analysis of G actin binding.
SDS/PAGE and Immunoblotting.
SDS/PAGE and immunoblotting were as described (6). Polyclonal rabbit antiactin-related protein3 (Arp3) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-Diaphanous1 and Rabbit anti-Diaphanous 2 were purchased from Bethyl Laboratories (Montgomery, TX). The antiactin C4 monoclonal antibody was purchased from Chemicon International (Hampshire, U.K.). Polyclonal rabbit antibodies to TARP have been described (6).
Pyrene Assay.
The rate of actin polymerization in the presence of GST fusions was monitored according to the methods outlined in the Actin Polymerization Biochem Kit from Cytoskeleton. Details are provided in Supporting Text.
Transmission Electron Microscopy.
Specimens were obtained directly from the pyrene assay, and 5-μl droplets were placed on 200 mesh copper grids having a carbon-coated formvar support film and negatively stained for 1 min with 1% (wt/vol) aqueous uranyl acetate. Samples were viewed on a Philips CM-10 TEM (FEI, Hillsboro, OR) at 60 kV. Digital images were processed with Adobe Photoshop, Ver. 7.0 (Adobe Systems, Mountain View, CA).
Gel Filtration.
TARP peptides suspended in 1× PBS or buffer A (see above) were added to a Superdex 200 10/300 GL gel filtration column (Amersham Biosciences) controlled by a BioCAD Sprint Perfusion Chromatography System (PerSeptive Biosystems, Framingham, MA). Eluted proteins were monitored by A280 absorbance, and peak fractions harboring eluted TARP were confirmed by immunoblot analysis. Protein standards were also run to determine retention times and plotted against the log of their respective molecular weight. Linear regression from these data provided estimates for the molecular weight of the TARP peptide complexes.
Supplementary Material
Acknowledgments
We thank Drs. R. Heinzen, J. Gordon, W. Jamieson, C. Dooley, and M. Jewett for critical review of the manuscript. This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases/National Institutes of Health.
Abbreviations
- EB
elementary body
- CD
cytochalasin D.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
Carabeo, R., Grieshaber, S., Dooley, C., Hazenkrug, A., Hackstadt, T. (2004) Abstr. Annu. Mtg. ASM..
References
- 1.Schachter J. In: Chlamydia; Intracellular Biology, Pathogenesis, and Immunity. Stephens RS, editor. Washington, DC: Am Soc Microbiol Press; 1999. pp. 139–169. [Google Scholar]
- 2.Moulder JW. Microbiol Rev. 1991;55:143–190. doi: 10.1128/mr.55.1.143-190.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carabeo RA, Hackstadt T. Infect Immun. 2001;69:5899–5904. doi: 10.1128/IAI.69.9.5899-5904.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang JP, Stephens RS. Cell. 1992;69:861–869. doi: 10.1016/0092-8674(92)90296-o. [DOI] [PubMed] [Google Scholar]
- 5.Carabeo RA, Grieshaber SS, Fischer E, Hackstadt T. Infect Immun. 2002;70:3793–3803. doi: 10.1128/IAI.70.7.3793-3803.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clifton DR, Fields KA, Grieshaber S, Dooley CA, Fischer E, Mead D, Carabeo RA, Hackstadt T. Proc Natl Acad Sci USA. 2004;101:10166–10171. doi: 10.1073/pnas.0402829101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clifton DR, Dooley CA, Grieshaber SS, Carabeo RA, Fields KA, Hackstadt T. Infect Immun. 2005;73:3860–3868. doi: 10.1128/IAI.73.7.3860-3868.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Byrne GI, Moulder JW. Infect Immun. 1978;19:598–606. doi: 10.1128/iai.19.2.598-606.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miki H, Miura K, Takenawa T. EMBO J. 1996;15:5326–5335. [PMC free article] [PubMed] [Google Scholar]
- 10.Miki H, Sasaki T, Takai Y, Takenawa T. Nature. 1998;391:93–96. doi: 10.1038/34208. [DOI] [PubMed] [Google Scholar]
- 11.Cooper JA, Pollard TD. Methods Enzymol. 1982;85:182–210. doi: 10.1016/0076-6879(82)85021-0. [DOI] [PubMed] [Google Scholar]
- 12.Frieden C, Goddette DW. Biochemistry. 1983;22:5836–5843. doi: 10.1021/bi00294a023. [DOI] [PubMed] [Google Scholar]
- 13.Goddette DW, Uberbacher EC, Bunick GJ, Frieden C. J Biol Chem. 1986;261:2605–2609. [PubMed] [Google Scholar]
- 14.Goddette DW, Frieden C. J Biol Chem. 1986;261:15974–15980. [PubMed] [Google Scholar]
- 15.Dominguez R. Trends Biochem Sci. 2004;29:572–578. doi: 10.1016/j.tibs.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Baum B, Kunda P. Curr Biol. 2005;15:R305–R308. doi: 10.1016/j.cub.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 17.Evangelista M, Pruyne D, Amberg DC, Boone C, Bretscher A. Nat Cell Biol. 2002;4:32–41. doi: 10.1038/ncb718. [DOI] [PubMed] [Google Scholar]
- 18.Prehoda KE, Scott JA, Mullins RD, Lim WA. Science. 2000;290:801–806. doi: 10.1126/science.290.5492.801. [DOI] [PubMed] [Google Scholar]
- 19.Pruyne D, Evangelista M, Yang C, Bi E, Zigmond S, Bretscher A, Boone C. Science. 2002;297:612–615. doi: 10.1126/science.1072309. [DOI] [PubMed] [Google Scholar]
- 20.Quinlan ME, Heuser JE, Kerkhoff E, Mullins RD. Nature. 2005;433:382–388. doi: 10.1038/nature03241. [DOI] [PubMed] [Google Scholar]
- 21.Stradal TE, Scita G. Curr Opin Cell Biol. 2006;18:4–10. doi: 10.1016/j.ceb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Miki H, Suetsugu S, Takenawa T. EMBO J. 1998;17:6932–6941. doi: 10.1093/emboj/17.23.6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takenawa T, Miki H. J Cell Sci. 2001;114:1801–1809. doi: 10.1242/jcs.114.10.1801. [DOI] [PubMed] [Google Scholar]
- 24.Cooper JA. J Cell Biol. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Copeland JW, Copeland SJ, Treisman R. J Biol Chem. 2004;279:50250–50256. doi: 10.1074/jbc.M404429200. [DOI] [PubMed] [Google Scholar]
- 26.Condeelis J. Trends Cell Biol. 2001;11:288–293. doi: 10.1016/s0962-8924(01)02008-6. [DOI] [PubMed] [Google Scholar]
- 27.Carabeo RA, Grieshaber S, Hasenkrug A, Dooley CA, Hackstadt T. Traffic. 2004;5:418–425. doi: 10.1111/j.1398-9219.2004.00184.x. [DOI] [PubMed] [Google Scholar]
- 28.Carlson JH, Porcella SF, McClarty G, Caldwell HD. Infect Immun. 2005;73:6407–6418. doi: 10.1128/IAI.73.10.6407-6418.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, et al. Science. 1998;282:754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- 30.Gouin E, Gantelet H, Egile C, Lasa I, Ohayon H, Villiers V, Gounon P, Sansonetti PJ, Cossart P. J Cell Sci. 1999;112:1697–1708. doi: 10.1242/jcs.112.11.1697. [DOI] [PubMed] [Google Scholar]
- 31.Gouin E, Welch MD, Cossart P. Curr Opin Microbiol. 2005;8:35–45. doi: 10.1016/j.mib.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 32.Hayward RD, Koronakis V. EMBO J. 1999;18:4926–4934. doi: 10.1093/emboj/18.18.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heinzen RA, Grieshaber SS, Van Kirk LS, Devin CJ. Infect Immun. 1999;67:4201–4207. doi: 10.1128/iai.67.8.4201-4207.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stevens JM, Galyov EE, Stevens MP. Nat Rev Microbiol. 2006;4:91–101. doi: 10.1038/nrmicro1320. [DOI] [PubMed] [Google Scholar]
- 35.Stevens JM, Ulrich RL, Taylor LA, Wood MW, Deshazer D, Stevens MP, Galyov EE. J Bacteriol. 2005;187:7857–7862. doi: 10.1128/JB.187.22.7857-7862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stevens MP, Stevens JM, Jeng RL, Taylor LA, Wood MW, Hawes P, Monaghan P, Welch MD, Galyov EE. Mol Microbiol. 2005;56:40–53. doi: 10.1111/j.1365-2958.2004.04528.x. [DOI] [PubMed] [Google Scholar]
- 37.Welch MD, Rosenblatt J, Skoble J, Portnoy DA, Mitchison TJ. Science. 1998;281:105–108. doi: 10.1126/science.281.5373.105. [DOI] [PubMed] [Google Scholar]
- 38.Cossart P. Cell Microbiol. 2000;2:195–205. doi: 10.1046/j.1462-5822.2000.00053.x. [DOI] [PubMed] [Google Scholar]
- 39.Rottner K, Lommel S, Wehland J, Stradal TE. J Pathol. 2004;204:396–406. doi: 10.1002/path.1638. [DOI] [PubMed] [Google Scholar]
- 40.Rottner K, Stradal TE, Wehland J. Dev Cell. 2005;9:3–17. doi: 10.1016/j.devcel.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 41.Subtil A, Wyplosz B, Balana ME, Dautry-Varsat A. J Cell Sci. 2004;117:3923–3933. doi: 10.1242/jcs.01247. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}