

Abstract
The retinal l-type Ca2+ channel Cav1.4 is distinguished from all other members of the high voltage-activated (HVA) Ca2+ channel family by lacking Ca2+-calmodulin-dependent inactivation. In synaptic terminals of photoreceptors and bipolar cells, this feature is essential to translate graded membrane depolarizations into sustained Ca2+ influx and tonic glutamate release. The sequences conferring Ca2+-dependent inactivation (CDI) are conserved throughout the HVA calcium channel family, raising the question of how Cav1.4 manages to switch off CDI. Here, we identify an autoinhibitory domain in the distal C terminus of Cav1.4 that serves to abolish CDI. We show that this domain (ICDI, inhibitor of CDI) uncouples the molecular machinery conferring CDI from the inactivation gate by binding to the EF hand motif in the proximal C terminus. Deletion of ICDI completely restores Ca2+-calmodulin-mediated CDI in Cav1.4. CDI can be switched off again in the truncated Cav1.4 channel by coexpression of ICDI, indicating that ICDI works as an autonomous unit. Furthermore, we show that in the Cav1.2 l-type Ca2+-channel replacement of the distal C terminus by the corresponding sequence of Cav1.4 is sufficient to block CDI. This finding suggests that autoinhibition of CDI can be introduced principally into other Ca2+ channel types. Our data provide a previously undescribed perspective on the regulation of HVA calcium channels by Ca2+.
Keywords: calmodulin, Cav1.4, retina, Ca channels
Synaptic release of neurotransmitters critically depends on the influx of Ca2+ through voltage-gated Ca2+ channels. In most neurons of the CNS, synaptic Ca2+ influx has to decay rapidly to faithfully translate trains of action potentials into corresponding quantal release of synaptic vesicles (1). Fast channel inactivation is principally conferred by voltage but is enhanced by an intrinsic feedback mechanism designated as Ca2+-dependent inactivation (CDI) by which Ca2+ limits its own influx (2). In contrast to most neurons, retinal photoreceptors and bipolar cells generate graded electrical responses (3, 4) and, hence, require a sustained rather than a transient Ca2+ influx for neurotransmitter release (5, 6). Indeed, calcium currents characterized in retinal cells reveal slow inactivation kinetics and are virtually devoid of CDI (7–9). Two members of the l-type subclass of high voltage-activated calcium channels have been described in ribbon synapses of photoreceptors and bipolar cells, Cav1.4 (10–12) and Cav1.3 (12, 13). Genetic studies indicate that the major channel mediating synaptic Ca2+ influx in rod photoreceptors and rod bipolar cells is Cav1.4 (10, 11). The electrophysiological properties of heterologously expressed Cav1.4 are consistent with those of native retinal calcium currents (14–16). In particular, Cav1.4 inactivates slowly and lacks CDI. At first glance, this finding is surprising given that Cav1.4 shares high sequence homology with other high voltage-activated calcium channels that display profound CDI. In this study, we set out to analyze the mechanism and the structural basis underlying this important functional difference.
Results
Fig. 1A shows current traces of the CDI-displaying calcium channel, Cav1.2, expressed in HEK293 cells. In the presence of Ba2+ inactivation is relatively slow and depends only on the membrane potential (voltage-dependent inactivation). If Ca2+ is the permeating ion, the channel inactivates much faster because of the presence of CDI. Pure CDI can be quantified by the f value, which corresponds to the difference in normalized IBa and ICa remaining after 300 ms of depolarization. In Cav1.2, like in other channels with CDI, f values reveal a U-shaped dependence on the test pulse voltage (17), reaching a maximal value (fmax) of ≈0.3 (Fig. 1B). The Ca2+-sensing apparatus of Cav1.2 resides in the proximal C terminus of the channel (17–25) (Fig. 6, which is published as supporting information on the PNAS web site). It comprises a sequence (pre-IQ and IQ segment) that binds calmodulin (CaM), which is the primary Ca2+ sensor of the channel. In addition, it contains an EF hand motif that allosterically couples the Ca2+-sensing apparatus with the inactivation gate of the channel. These domains are highly conserved in the retinal Cav1.4 channel. Surprisingly, however, this channel does not display CDI. IBa and ICa of Cav1.4 show the same kinetics (Fig. 1C), and the f value is zero over the whole voltage range tested (Fig. 1D).
Fig. 1.

Cav1.4 binds CaM but does not reveal CDI. (A) Representative traces of ICa (red traces) and IBa (black traces) through Cav1.2 evoked by stepping from a holding potential of −80 mV to +10 mV. Current traces were normalized to peak current. Throughout, Ba2+ traces for CDI are scaled to match Ca2+ traces (scale bar) (B) Strength of CDI, f, in relation to voltage for Cav1.2. fmax is the maximal f value, n number of cells. (C and D) Representative ICa and IBa currents (C) and voltage-dependence of f for Cav1.4 (D). (E) GST fusion proteins containing fragments corresponding to the proximal and distal C terminus of Cav1.2 (CT1.2-1667Stop and CT1.2-1667–2166) or Cav1.4 (CT1610Stop and CT1610–1984) were incubated with CaM in the presence (+) or absence (−) of Ca2+. Immunoblotting was performed with an anti-CaM antibody. In the last lane, 1 μg of CaM was blotted to demonstrate specificity of the assay. (F) Co-IP of HEK293 cells coexpressing triple flag-tagged CaM1234 together with the myc-tagged full-length C terminus of Cav1.2 (CT1.2) or Cav1.4 (CT1.4). Lysates were immunoprecipitated with anti-myc (lanes 5 and 7) or anti-GST (control, lanes 6 and 8), blotted and probed with anti-flag. Lane 3 and 4: negative control.
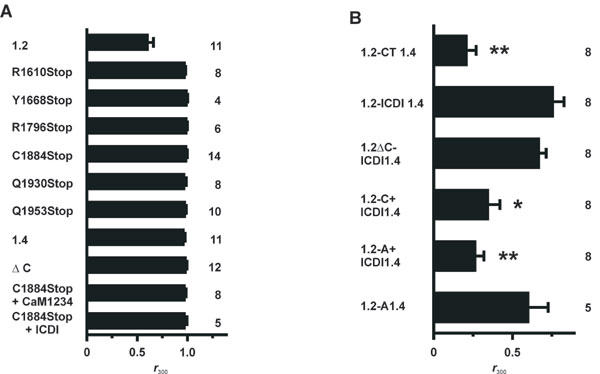
We first considered that Cav1.4 may lack CDI because it is deficient in binding CaM. To test for this option, we performed pulldown assays with GST fusion proteins containing C-terminal fragments of Cav1.2 and Cav1.4 and purified CaM (Figs. 1E; see Fig. 7A, which is published as supporting information on the PNAS web site). CaM bound to the proximal C terminus of both channels (CT1.2-1667Stop and CT1610Stop) in a Ca2+-dependent fashion. Binding of CaM also was observed to the full-length C terminus of Cav1.4 (data not shown). By contrast, no interaction was seen with the distal C termini, comprising sequences downstream of the IQ segment (CT1.2-1667–2166 and CT1610–1984). The absence of signals in the presence of EGTA did not necessarily mean that Ca2+-free CaM (apocalmodulin) is principally insufficient to bind to Cav1.4 and Cav1.2 because some basal Ca2+ may be transiently required for the proper folding of a binding pocket for apocalmodulin (23, 24). To explore this issue, we performed coimmunoprecipitations (Co-IPs) in HEK293 cells by using a CaM mutant (CaM1234) that is deficient for Ca2+ binding serving as an apocalmodulin surrogate (26). In these assays, CaM1234 bound to the C termini of either Cav1.2 or Cav1.4 channels strongly supports the hypothesis that apocalmodulin indeed can bind to both channels at basal Ca2+ conditions (Fig. 1F). Together, these findings excluded loss of the calcium sensor CaM as a mechanism explaining Ca2+ insensitivity of Cav1.4 but rather suggested that CDI may be actively masked by an inhibitory channel domain. Indeed, when the last 100 aa were removed from the C terminus of Cav1.4 (C1884Stop), CDI was recovered fully (Fig. 2A and B). Like the Cav1.2 channel C1884Stop showed fast inactivation in the presence of Ca2+ that was best fitted with two time constants, whereas there was no inactivation with Ba2+ (Table 1 and Fig. 8A, which are published as supporting information on the PNAS web site). CDI also was present in other Cav1.4 channels with truncations between the IQ segment and C1884 (R1610Stop, Y1668Stop, and R1796Stop) (Fig. 2 A and F). The f value of C1884Stop showed the typical U-shaped voltage dependence, and fmax was virtually identical to that of Cav1.2 [0.3 ± 0.03 (n = 14); Figs. 2 C and F and 8A and Table 1]. To demonstrate that CDI in C1884Stop was mediated by CaM, we coexpressed the channel together with the negative-dominant CaM mutant CaM1234. Under these conditions, no CDI was observed (Figs. 2 D and F and 8A and Table 1). Finally, we coexpressed C1884Stop with a peptide corresponding to the last 100 aa of Cav1.4 (ICDI, inhibitor of CDI) in HEK293 cells. Again, CDI was totally absent (Figs. 2 E and F and 8A), indicating that ICDI was acting as an independent protein unit and was sufficient to block CDI in Cav1.4. In support of this model, a Cav1.4 mutant in which ICDI was directly attached to the IQ motif also showed no CDI (ΔC, Figs. 2F and 8A). We constructed two additional Cav1.4 truncations to further narrow down the minimal sequence required for block of CDI (Figs. 2F and 8A). Q1930Stop showed CDI, whereas Q1953Stop did not, indicating that the sequence stretch between amino acids 1930–1953 is of particular importance in blocking CDI.
Fig. 2.

The ICDI domain is required to block CDI in Cav1.4. (A) Scheme of the Cav1.4 C terminus starting after the IVS6 segment. Numbers delineate the borders of peptides A, C, and ICDI. (B) Representative traces of ICa (red trace) and IBa (black trace) through a truncated Cav1.4 channel lacking ICDI (C1884Stop). (C) Voltage-dependence of f for C1884Stop. (D and E) Coexpression of C1884Stop and a dominant negative CaM1234 (D) or ICDI (E). (F) fmax for wild-type and mutant channels and for coexpression experiments as indicated.
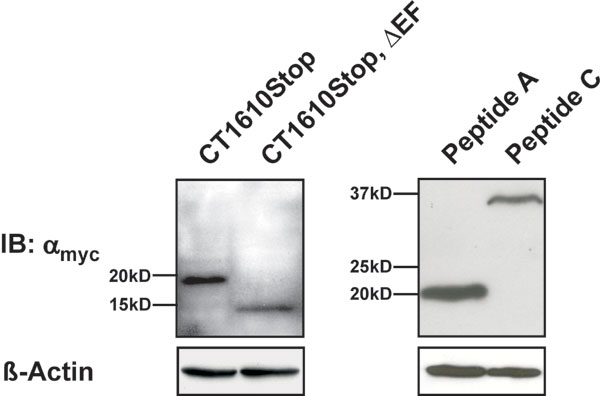
We next asked whether the ICDI domain could interact with the Ca2+-sensing apparatus of Cav1.4. To explore this issue, we performed Co-IPs with myc-tagged C-terminal fragments of Cav1.4 and triple-flag-tagged ICDI (Figs. 3A and B; see Fig. 9, which is published as supporting information on the PNAS web site). As proposed, ICDI specifically bound to the proximal C terminus of Cav1.4 (Fig. 3B Upper, lane 4). However, ICDI no longer interacted with this domain when the EF hand motif was deleted (Fig. 3B Upper, lane 5). Moreover, no interaction was observed between ICDI and peptides A and C that were derived from the distal C terminus of Cav1.4 (Fig. 3 A and B Lower). This important finding was confirmed by a series of pulldown assays with bacterial GST fusion proteins (Fig. 7). Finally, to prove directly that ICDI binds to the EF hand, GST pulldowns were performed with peptides containing the EF hand or the EF hand and the N-terminal half of the PreIQ (Fig. 3C, CT1493Stop and CT1535Stop). Both peptides bound the ICDI, indicating that the EF hand serves as target sequence of ICDI. Analysis of the primary sequence indicates that ICDI has an α helical structure [predicted by PhD analysis (27), confidence level >82%]. Given that the EF hand also contains helices (25), it is tempting to speculate that ICDI and EF hand can form a paired helix complex. Considering the central role of the EF hand as a general downstream transduction element for CDI (19, 24, 28–30), we propose that the interaction between ICDI and EF hand uncouples the Ca2+-sensing apparatus from the inactivation gate of Cav1.4.
Fig. 3.

The ICDI domain binds to the EF hand motif of Cav1.4. (A) Scheme of the Cav1.4 C terminus starting after the IVS6 segment. (B Upper) Co-IP of HEK293 cells coexpressing triple flag-tagged ICDI together with the myc-tagged CT1610Stop or CT1610Stop,ΔEF. Lysates were immunoprecipitated with anti-myc (lanes 4 and 5) or anti-ras (control, lanes 6 and 7), blotted and probed with anti-flag. Lane 8: negative control. (B Lower) Immunoprecipitation of HEK293 cells expressing ICDI and myc-tagged peptides C or A. Co-IP has been carried out as in Upper. (C) ICDI interaction assay with GST-tagged fragments of the proximal C terminus. GST fusion proteins were incubated with bacterial lysates containing flag-tagged ICDI. Immunoblotting was performed with an anti-flag antibody. In the last lane, pure ICDI was blotted to demonstrate specificity of the assay. Each experiment was repeated at least three times.
We were wondering whether the inactivation of Cav1.2 could be rendered insensitive to Ca2+ by replacing C-terminal portions of this channel by corresponding sequences of Cav1.4. To this end, we constructed a series of Cav1.2-1.4 chimeras and deletion mutants (Fig. 4A) and determined the inactivation parameters of these channels (Fig. 4F and 8B and Table 1). When the complete C terminus of Cav1.2 was replaced by the corresponding sequence of Cav1.4 (1.2-CT1.4) CDI was completely abolished (Fig. 4F). Importantly, a Cav1.2-1.4 chimeric channel in which only the portion downstream of the IQ motif was derived from Cav1.4 (1.2-C+ICDI1.4) also lacked CDI, confirming our concept that the distal C terminus of Cav1.4 is sufficient to block CDI (Fig. 4 B, C, and F and Table 1). However, Cav1.2 channels in which only the last 149 aa were replaced by ICDI of Cav1.4 (1.2-ICDI1.4) or where ICDI was fused directly to the IQ motif (1.2ΔC-ICDI1.4) still displayed CDI (Fig. 4 D and F and Table 1). To abolish CDI, it was necessary to introduce the peptide A of Cav1.4 together with ICDI into the Cav1.2 backbone. The resulting chimera (1.2-A+ICDI1.4) passed a fast inactivating current with kinetics that were in the range of those observed for ICa of wild-type Cav1.2 (Fig. 4 E and F and Table 1). Attempts to further narrow down the borders of peptide A failed, suggesting that the whole sequence is needed for blocking CDI (data not shown). Notably, however, in the absence of ICDI, peptide A was not sufficient to block CDI (chimera 1.2-A1.4; Fig. 4F). In conclusion, ICDI is obligatorily required to block CDI in Cav1.2. However, unlike in Cav1.4, in the Cav1.2 backbone, it needs the cooperation of peptide A. Because peptide A does not bind ICDI (Figs. 3B and 7B), we propose that it supports the inhibitory action of ICDI via an indirect mechanism.
Fig. 4.

The distal C terminus of Cav1.4 suppresses CDI in Cav1.2. (A) Schematic representation of Cav1.2-1.4 chimeric channels. Channel portions derived from Cav1.2 and Cav1.4 are displayed in red and black, representatively. Deletions are marked by dotted lines. (B) Representative ICa (red) and IBa (black) through 1.2-C+ICDI1.4. (C) Voltage-dependence of f for 1.2-C+ICDI1.4. (D and E) Representative ICa and IBa through 1.2ΔC-ICDI1.4 (D) and 1.2-A+ICDI1.4 (E). (F) fmax values for channels are shown in A.
Discussion
Here, we demonstrate that the retinal Cav1.4 channel utilizes the intramolecular interaction between an autoinhibitory domain present in the distal C terminus (ICDI) and the EF hand in the proximal C terminus to functionally block CaM-mediated CDI. Our data have profound mechanistic consequences with regard to the regulation of l-type calcium channels by Ca2+. According to established models, CDI of high voltage-activated Ca2+ channels requires functional interaction between the channel core and the proximal C terminus that is transduced by the EF hand (refs. 24, 28, and 29; Fig. 5A). At rest, when cytosolic Ca2+ levels are low, the EF hand is presumed to interact weakly with the cytoplasmic gate in a manner that permits ready opening of the gate (29), stabilizes the open state, or helps to create a deceleratory signal to retard channel inactivation (24, 30). Voltage induces a slow conformational change that induces closure of the pore (voltage-dependent inactivation). Interaction of Ca2+ with prebound CaM induces a conformational change in the EF hand (17, 29) that promotes an interaction between the EF hand motif and its target region(s) on the channel, thus conferring fast CDI (17, 29, 30). It is a matter of controversy where the interaction site of the EF hand is localized, but it may lie within the core (17, 29) or the linker between domains I and II of the channel (30–33). These channel domains are represented as a latch in Fig. 5. Our data extend this model to channels lacking CDI (Fig. 5B). We propose that in Cav1.4 and Cav1.2 carrying the distal C terminus of Cav1.4, ICDI constitutively binds to the EF hand motif. Hence, the conformational change in the EF hand is impaired, and inactivation occurs in a strictly voltage-dependent fashion with kinetics that are intrinsic to the respective channel core (very slow in Cav1.4 and fast in Cav1.2). Because the EF hand motif is uncoupled from the Ca2+-sensing apparatus, the C terminus no longer can interact with its target regions. As a result, there is no CDI and inactivation is purely determined by voltage. This model is supported by the properties of a Cav1.2 deletion mutant lacking the EF hand motif (mutant 77dEF) (19). This channel completely lacks CDI and has inactivation time constants that are virtually identical to those of Cav1.2 carrying the distal C terminus of Cav1.4. The molecular details underlying the action of the A peptide of Cav1.4 are not yet known. This peptide is not sufficient but is required to block CDI in Cav1.2, suggesting that it supports the primary action of ICDI via an independent pathway. It is noteworthy to mention that unlike in Cav1.1 (34) and Cav1.2 (35), peptide A is not binding to the distal C terminus in Cav1.4. Quantitatively, the impact of peptide A is likely to be of minor importance because in Cav1.4, the peptide is not essential for blocking CDI.
Fig. 5.

Current (A) and extended (B) model of CDI. CaM and Ca2+ are displayed as a gray circle and black triangle, respectively. See Discussion for additional details.
Our findings also may have pathophysiological relevance. Loss-of-function mutations in the human Cav1.4 channel cause incomplete congenital stationary night blindness type 2 (CSNB2), an X-linked recessive disorder with symptoms of low visual acuity, myopia, nystagmus, and night blindness (10, 11, 36). In addition, two mutations in CSNB2 patients have been discovered, leading to channels with truncations in the distal C terminus that closely match truncations of murine Cav1.4 analyzed in the present study (K1591X and R1816X corresponding to murine K1605X and R1834X, respectively; Fig. 6) (11, 37). Moreover, a frameshift mutation (5665delC; ref. 36) has been identified, resulting in the deletion of the sequence downstream of the first 10 aa of ICDI (Fig. 6). All three mutations leave the channel core and the Ca2+-sensing apparatus intact but selectively remove ICDI. Our data strongly suggest that these human Cav1.4 mutants, if produced in vivo, very likely will display CDI and, hence, will cause a profound impairment of synaptic transmission in retinal cells, which explains the clinical phenotype.
Methods
Constructs for Electrophysiology.
The rabbit Cav1.2b subunit (38) (GenBank accession no. X55763), chimeric channels based on Cav1.2 backbone, and a Ca2+-insensitive CaM mutant harboring aspartate-to-alanine mutations in all four EF hands (CaM1234) (26) were cloned into the pcDNA3 expression vector (Invitrogen, Carlsbad, CA). For expression of murine Cav1.4 (14) (accession no. AJ579852) and channels containing the Cav1.4 backbone, the bicistronic pIRES2-EGFP expression vector (Clontech, Mountain View, CA) was used. For the construction of truncated Cav1.4 channels (R1610Stop, Y1668Stop, R1796Stop, C1884Stop, Q1930Stop, and Q1953Stop), a fragment of the wild-type Cav1.4 expression plasmid was replaced by DNA fragments that are carrying the required stop codon and a restriction site introduced by 3′ primers. The ΔC channel was cloned by deleting amino acids R1610–C1884 of Cav1.4 by using overlap PCR. The following chimeric Cav1.2-1.4 channels also were generated by using overlap PCR (see also Fig. 4A): In 1.2CT1.4, D1502–L2166 of Cav1.2 was replaced by D1445–L1984 of Cav1.4; in 1.2-ICDI1.4, G2018–L2166 of Cav1.2 was replaced by L1885–L1984 of Cav1.4; in 1.2ΔC-ICDI1.4, K1667–L2166 of Cav1.2 was replaced by L1885–L1984 of Cav1.4; in 1.2-C+ICDI1.4, K1667–L2166 was replaced by R1610–L1984 of Cav1.4; in 1.2-A+ICDI1.4, K1667–L2166 of Cav1.2 was replaced by R1610–L1984 with a deletion of I1742–C1884 of Cav1.4. In 1.2-A1.4, K1667–L2166 of Cav1.2 was replaced by R1610–S1741 and a stop codon was introduced after S1741. See also Fig. 6 for alignment of the C termini of Cav1.2 and Cav1.4.
Electrophysiology.
HEK293 cells were transiently transfected with expression vectors encoding calcium channel α subunits together with equimolar amounts of vectors encoding β2a and α2δ1 as described in ref. 14. ICa and IBa were measured by using the following solutions: (pipette solution) 112 mM CsCl/3 mM MgCl2/3 mM MgATP/10 mM EGTA/5 mM Hepes, adjusted to pH 7.4 with CsOH; (bath solution) 82 mM NaCl/30 mM BaCl2/5.4 mM CsCl2/1 mM MgCl2/20 mM tetraethylammonium/5 mM Hepes/10 mM glucose, adjusted to pH 7.4 with NaOH. For experiments with 10 mM Ba2+ or 10 mM Ca2+ in the bath solution the NaCl concentration was increased to 102 mM. ICa and IBa was measured from the same cell. Bath solution was changed by a local solution exchanger. Currents were recorded at room temperature 2–4 days after transfection by using whole-cell patch-clamp technique. Data were analyzed by using Origin 6.1 software (OriginLab, Northampton, MA).
The peak I–V relationship was determined by applying 350-ms voltage pulses to potentials between −80 and + 70 mV in 10-mV increments from a holding potential of –80 mV. The time course of current inactivation was fitted by the biexponential function: I(t) = Afast × exp(−t/τfast) + Aslow × exp(−t/τslow) + C. τslow and τfast represent slow and fast time constants of inactivation, respectively. Aslow and Afast are the amplitudes of the current components. Afast (%) has been calculated as Afast/(Afast + Aslow) × 100. IBa of Cav1.2, 1.2-ICDI1.4, and 1.2ΔC-ICDI1.4 was fitted best by the monoexponential function: I(t) = A0 × exp(−t/τ) + C, where I(t) is the current at time t after a voltage pulse to Vmax, A0 the steady-state current amplitude with the respective time constant of inactivation, τ, and C, the remaining steady-state current. For IBa and ICa of Cav1.4α1, C1884Stop + CaM1234 and C1884Stop +ICDI and IBa of C1884Stop time course of inactivation was linear and, therefore, not fitted.
CDI was quantified by determining the f value essentially as described in ref. 39. Briefly, the fraction of peak Ba2+ and Ca2+ currents remaining after 300 ms of depolarization (r300) was calculated. f is defined as the difference (r300 of IBa) − (r300 of ICa) and, hence, quantifies pure CDI. f can vary between 0 (no CDI) and 1 (complete CDI.) f was determined for various test potentials, and maximal f was taken for comparison of different channels.
Statistics.
All values are given as mean ± SEM. n is the number of experiments. An unpaired t test was performed for the comparison between two groups. Significance was tested by ANOVA followed by Dunett test if multiple comparisons were made. Values of P < 0.05 were considered significant.
GST Pulldown of CaM.
Sequences corresponding to C-terminal fragments of Cav1.2 [CT1.2-1667Stop (D1502–G1666); CT1.2-1667–2166 (K1667–L2166)] or Cav1.4 [CT1610Stop (D1445–G1609); CT1610–1984 (R1610–L1984)] were amplified by PCR and cloned into the pET41a bacterial expression vector (Novagen, Darmstadt, Germany) in frame with a N-terminal GST sequence. GST pulldowns were performed as described in ref. 17 with modifications specified in Supporting Materials and Methods, which is published as supporting information on the PNAS web site. Each GST pulldown was repeated at least three times.
ICDI Interaction Assay.
Sequences corresponding to C-terminal fragments of Cav1.4 [CT1493Stop (D1445–I1492); CT1535Stop (D1445–V1534); CT1610Stop (D1445–G1609); and CT1610Stop,ΔEF (D1445–G1609, Δ P1459–I1492)] were amplified by PCR and cloned into the pET41a bacterial expression vector (Novagen) in frame with a N-terminal GST sequence. Flag-tagged ICDI-peptide (L1885-L1984) was cloned into pQE30 bacterial expression vector (Qiagen, Valencia, CA) by using primers containing the N-terminal flag sequence and the required restriction sites. GST pulldowns were performed as specified in Supporting Text. Each GST pulldown was repeated at least three times.
Co-IP in HEK293 Cells.
The complete C terminus of Cav1.2 [CT1.2 (D1502–L2166)] or Cav1.4 [CT1.4 (D1445–L1984)] or C-terminal fragments of Cav1.4 [CT1610Stop (D1445–G1609); CT1610Stop,ΔEF (D1445–G1609, Δ P1459–I1492); peptide C (R1610–C1884); peptide A (R1610–S1741)] were amplified by PCR and cloned into the pcDNA3 expression vector. All sequences were fused with a myc tag at the N terminus. The triple flag-tagged ICDI peptide (L1885–L1984) and CaM1234 was constructed in the same manner by using a 5′ primer containing the triple flag sequence. For expression of recombinant proteins, HEK293 cells were transfected by using the calcium phosphate method. Immunoprecipitation was performed 3 days after transfection. A detailed protocol is available in Supporting Materials and Methods. Each Co-IP was repeated at least three times.
Supplementary Material
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft.
Abbreviations
- CaM
calmodulin
- CDI
calcium-dependent inactivation
- Co-IP
coimmunicipation
- ICDI
inhibitor of CDI.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
References
- 1.Wheeler DB, Randall A, Tsien RW. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- 2.Liang H, DeMaria CD, Erickson MG, Mori MX, Alseikhan BA, Yue DT. Neuron. 2003;39:951–960. doi: 10.1016/s0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- 3.Knapp AG, Dowling JE. Nature. 1987;325:437–439. doi: 10.1038/325437a0. [DOI] [PubMed] [Google Scholar]
- 4.Parsons TD, Sterling P. Neuron. 2003;37:379–382. doi: 10.1016/s0896-6273(03)00062-x. [DOI] [PubMed] [Google Scholar]
- 5.Tachibana M, Okada T, Arimura T, Kobayashi K. Ann NY Acad Sci. 1993;707:359–361. doi: 10.1111/j.1749-6632.1993.tb38070.x. [DOI] [PubMed] [Google Scholar]
- 6.Schmitz Y, Witkovsky P. Neuroscience. 1997;78:1209–1216. doi: 10.1016/s0306-4522(96)00678-1. [DOI] [PubMed] [Google Scholar]
- 7.Yagi T, Macleish PR. J Neurophysiol. 1994;71:656–665. doi: 10.1152/jn.1994.71.2.656. [DOI] [PubMed] [Google Scholar]
- 8.Protti DA, Llano I. J Neurosci. 1998;18:3715–3724. doi: 10.1523/JNEUROSCI.18-10-03715.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berntson A, Taylor WR, Morgans CW. J Neurosci Res. 2003;71:146–151. doi: 10.1002/jnr.10459. [DOI] [PubMed] [Google Scholar]
- 10.Bech-Hansen NT, Naylor MJ, Maybaum TA, Pearce WG, Koop B, Fishman GA, Mets M, Musarella MA, Boycott KM. Nat Genet. 1998;19:264–267. doi: 10.1038/947. [DOI] [PubMed] [Google Scholar]
- 11.Strom TM, Nyakatura G, Apfelstedt-Sylla E, Hellebrand H, Lorenz B, Weber BH, Wutz K, Gutwillinger N, Ruther K, Drescher B, et al. Nat Genet. 1998;19:260–263. doi: 10.1038/940. [DOI] [PubMed] [Google Scholar]
- 12.Heidelberger R, Thoreson WB, Witkovsky P. Prog Retin Eye Res. 2005;24:682–720. doi: 10.1016/j.preteyeres.2005.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgans CW, Bayley PR, Oesch NW, Ren G, Akileswaran L, Taylor WR. Vis Neurosci. 2005;22:561–568. doi: 10.1017/S0952523805225038. [DOI] [PubMed] [Google Scholar]
- 14.Baumann L, Gerstner A, Zong X, Biel M, Wahl-Schott C. Invest Ophthalmol Vis Sci. 2004;45:708–713. doi: 10.1167/iovs.03-0937. [DOI] [PubMed] [Google Scholar]
- 15.Koschak A, Reimer D, Walter D, Hoda JC, Heinzle T, Grabner M, Striessnig J. J Neurosci. 2003;23:6041–6049. doi: 10.1523/JNEUROSCI.23-14-06041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McRory JE, Hamid J, Doering CJ, Garcia E, Parker R, Hamming K, Chen L, Hildebrand M, Beedle AM, Feldcamp L, et al. J Neurosci. 2004;24:1707–1718. doi: 10.1523/JNEUROSCI.4846-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 18.Zhou J, Olcese R, Qin N, Noceti F, Birnbaumer L, Stefani E. Proc Natl Acad Sci USA. 1997;94:2301–2305. doi: 10.1073/pnas.94.6.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zuhlke RD, Reuter H. Proc Natl Acad Sci USA. 1998;95:3287–3294. doi: 10.1073/pnas.95.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qin N, Olcese R, Bransby M, Lin T, Birnbaumer L. Proc Natl Acad Sci USA. 1999;96:2435–2438. doi: 10.1073/pnas.96.5.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 22.Pate P, Mochca-Morales J, Wu Y, Zhang JZ, Rodney GG, Serysheva II, Williams BY, Anderson ME, Hamilton SL. J Biol Chem. 2000;275:39786–39792. doi: 10.1074/jbc.M007158200. [DOI] [PubMed] [Google Scholar]
- 23.Romanin C, Gamsjaeger R, Kahr H, Schaufler D, Carlson O, Abernethy DR, Soldatov NM. FEBS Lett. 2000;487:301–306. doi: 10.1016/s0014-5793(00)02361-9. [DOI] [PubMed] [Google Scholar]
- 24.Pitt GS, Zuhlke RD, Hudmon A, Schulman H, Reuter H, Tsien RW. J Biol Chem. 2001;276:30794–30802. doi: 10.1074/jbc.M104959200. [DOI] [PubMed] [Google Scholar]
- 25.Erickson MG, Liang H, Mori MX, Yue DT. Neuron. 2003;39:97–107. doi: 10.1016/s0896-6273(03)00395-7. [DOI] [PubMed] [Google Scholar]
- 26.Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP. Nature. 1998;395:503–507. doi: 10.1038/26758. [DOI] [PubMed] [Google Scholar]
- 27.Rost B, Sander C. J Mol Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- 28.Bernatchez G, Talwar D, Parent L. Biophys J. 1998;75:1727–1739. doi: 10.1016/S0006-3495(98)77614-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peterson BZ, Lee JS, Mulle JG, Wang Y, de Leon M, Yue DT. Biophys J. 2000;78:1906–1920. doi: 10.1016/S0006-3495(00)76739-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J, Ghosh S, Nunziato DA, Pitt GS. Neuron. 2004;41:745–754. doi: 10.1016/s0896-6273(04)00081-9. [DOI] [PubMed] [Google Scholar]
- 31.Cens T, Restituito S, Galas S, Charnet P. J Biol Chem. 1999;274:5483–5490. doi: 10.1074/jbc.274.9.5483. [DOI] [PubMed] [Google Scholar]
- 32.Stotz SC, Hamid J, Spaetgens RL, Jarvis SE, Zamponi GW. J Biol Chem. 2000;275:24575–24582. doi: 10.1074/jbc.M000399200. [DOI] [PubMed] [Google Scholar]
- 33.Bernatchez G, Berrou L, Benakezouh Z, Ducay J, Parent L. Biochim Biophys Acta. 2001;1514:217–229. doi: 10.1016/s0005-2736(01)00373-x. [DOI] [PubMed] [Google Scholar]
- 34.Hulme JT, Konoki K, Lin TW, Gritsenko MA, Camp DG II, Bigelow DJ, Catterall WA. Proc Natl Acad Sci USA. 2005;102:5274–5279. doi: 10.1073/pnas.0409885102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao T, Cuadra AE, Ma H, Bunemann M, Gerhardstein BL, Cheng T, Eick RT, Hosey MM. J Biol Chem. 2001;276:21089–21097. doi: 10.1074/jbc.M008000200. [DOI] [PubMed] [Google Scholar]
- 36.Boycott KM, Maybaum TA, Naylor MJ, Weleber RG, Robitaille J, Miyake Y, Bergen AA, Pierpont ME, Pearce WG, Bech-Hansen NT. Hum Genet. 2001;108:91–97. doi: 10.1007/s004390100461. [DOI] [PubMed] [Google Scholar]
- 37.Wutz K, Sauer C, Zrenner E, Lorenz B, Alitalo T, Broghammer M, Hergersberg M, de la Chapelle A, Weber BH, Wissinger B, et al. Eur J Hum Genet. 2002;10:449–456. doi: 10.1038/sj.ejhg.5200828. [DOI] [PubMed] [Google Scholar]
- 38.Biel M, Ruth P, Bosse E, Hullin R, Stuhmer W, Flockerzi V, Hofmann F. FEBS Lett. 1990;269:409–412. doi: 10.1016/0014-5793(90)81205-3. [DOI] [PubMed] [Google Scholar]
- 39.Mori MX, Erickson MG, Yue DT. Science. 2004;304:432–435. doi: 10.1126/science.1093490. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}