

Abstract
Using a selection for Dictyostelium mutants that preferentially form spores, we have recovered a mutant called CheaterA. In chimeras with isogenic wild-type cells, the CheaterA mutant preferentially forms viable spores rather than inviable stalk cells. The mutant causes wild-type cells that have begun to express spore-specific genes to accumulate in the prestalk compartment of the developing organism. In the wild-type cells, the chtA transcript is absent in growing cells and appears early in development. No transcript was detected in the mutant by Northern blot. The chtA gene codes for a protein with an F-box and WD40 domains. This class of protein usually forms part of an Skp1, cullin, F-box (SCF) complex that targets specific protein substrates for ubiquitination and degradation.
Keywords: SCF complex, proteolysis, ubiquitination, somatic cell parasite, kin selection
Dictyostelium amoebae feed on bacteria during growth, but when the food supply is exhausted, they aggregate to form a mound and then a fruiting body made of stalk and spore cells. In the mound, cells transform to prestalk and prespore cells, and the prestalk cells sort to the apex, forming a tip. The tip elongates to form a cylindrical slug with a posterior zone composed of cells that will become spores and an anterior zone made up of cells that will give rise to the stalk. Under the appropriate conditions, the slug transforms into a fruiting body by a process called culmination. In the fruiting bodies, about 80% of the amoebae form spore cells, whereas 20% form stalk cells. Spore cells germinate to give rise to the next generation, but stalk cells die. The stalk cells have been described as altruistic, sacrificing themselves for the improved dispersal of spores (1, 2).
How this developmental process evolved has been widely discussed but is not simple to explain (3). One of the problems, as pointed out by Buss and others, is that in the wild, parasitic forms of Dictyostelium should arise that form only spore cells (4–6). There is genetic diversity in wild-type populations of Dictyostelium from which such variants would be expected to arise over evolutionary time (7). Such parasites, not having to tithe 20% to the stalk cells, would increase in the population and eventually impede the cooperativeness that leads to fruiting body formation. There have been two reports that in freshly isolated uncloned populations of cells, variants exist that preferentially make spores in chimeras with wild-type cells (4, 8). Buss found a single soil sample which, when plated clonally, gave rise to normal colonies and colonies that only formed spores and competed in chimeras. See the PNAS commentary by Buss, which compares this behavior to organisms in other phyla (9).
The mechanisms that these cells, which we call cheaters, use to avoid the stalk cell fate in a chimera could be based on disruption of cell–cell communication pathways that establish the strict 80:20 proportion of spores and stalk cells. Although studied for many years, these mechanisms have remained elusive (10). If mutant strains that preferentially form spores in chimeras and also have defects in proportionality when developing alone can be produced in the laboratory, the basis of the competition and the communication among cells during normal development can be studied in a way that is independent of previous approaches. Dictyostelium offers powerful molecular genetic tools for this endeavor.
The chtA gene was recovered after a selection based on these ideas and codes for a molecule that has the characteristic motifs of F-box proteins, including an F-box and WD40 domains. F-box proteins form part of the SCF complex (Skp1, cullin, F-box proteins) and function to bring target proteins into contact with the ubiquitination machinery. WD40 domains, of which there are usually five to seven in F-box proteins, are variable, and each F-box protein may bind a different target. The F-box domain in the protein interacts with Skp1 and cullin, whereas the WD40 domains bind one or more target proteins (11–13). The F-box protein described here is one of several involved in cell-fate decisions (14). We speculate that by not degrading one or more protein regulators of development, the mutant cells are forced down the prespore pathway and, as a consequence, produce a signal to convert the wild-type cells to a prestalk fate.
Materials and Methods
Strains.
The mutant was created by restriction enzyme-mediated integration (REMI), as described (15), by using the blasticidin resistance plasmid pBSR3 (16). The chtA mutation isolated in our laboratory was recovered in strain AX3K after a series of selections for spore-forming cells. Another mutation with the chtA phenotype was recovered after REMI, but without the selection for spores, by using uracil prototrophy in strain DH1 as a selection. This mutant was called fbxA (F-box A) and was independently isolated by M. K. Nelson, J. G. Williams (University of Dundee), and R. A. Firtel (University of California at San Diego, La Jolla) (accession no. AF151733). These authors have graciously supplied their mutant. In both strains, the plasmids integrated into the same site in the chtA gene (see below). A list of strains and their marked progeny can be found at: http://cpmcnet.columbia.edu/dept/gsas/anatomy/Faculty/Kessin/index.html.
REMI.
The axenic strain AX3K was subjected to electroporation in the presence of the restriction enzyme DpnII and the selectable plasmid pBSR3, which had been linearized with BamHI (15, 17). pBSR3 contains a gene that codes for resistance to blasticidin (16). After 1 wk of selection, nontransformed cells were killed, and the survivors were recovered. The blasticidin-resistant population was subjected to the selection as described in Results.
Isolation of the Gene.
The mutated gene was recovered by digesting the genomic DNA with BclI, ligating, and transforming Escherichia coli strain DH5α (18). The DNA flanking the plasmid was sequenced. Further sequence of chtA was recovered from cDNA clones and from the genomic sequence database (http://www.uni-koeln.de/dictyostelium) and from a cDNA database (http://www.csm.biol.tsukuba.ac.jp/cDNAproject.html). The annotated chtA sequence has been submitted to GenBank, accession nos: AF151111 (genomic sequence) and AF151112 (partial cDNA sequence, including 5′ UTR). The transcriptional start-site of chtA was mapped by using the Ver. 2 (GIBCO/BRL) 5′ rapid amplification of cDNA ends (RACE) system. Total RNA was prepared from axenically grown cells in midlogarithmic phase (4–5 × 106cells/ml), which were starved for 6 hr in SorensenC (SorC) (17 mM KH2PO4/Na2HPO4, 50 μM CaCl2, pH 6.0) at 2 × 107/ml at 22°C. First-strand cDNA was synthesized from 2 μg total RNA by using the primer ctr-1 (5′-ATCTTGAAGAAATATCC-3′). The original RNA template was removed by RNase H treatment, and the first-strand DNA was purified with a spin column [Qiagen (Chatsworth, CA) Qiaquick]. After dC-tailing of the 3′-end of the cDNA, the 5′ extension product was amplified by using the provided 5′ RACE Abridged Anchor Primer and ctr-2 (5′-CTGCAACACCTTTACTTAAATAAATACC-3′), followed by reamplification with the provided primer AUAP and ctr-3 (5′-AATAGGAATGTATTATATGTTTTTCAC-3′). The 5′ extension product that includes a 5′ untranslated region of 229 bp was subcloned into the T/A cloning vector pCR2.1-TOPO (Invitrogen) for sequencing. A full-length cDNA was prepared and sequenced. Northern blot analysis was performed as described (19) with a probe consisting of nucleotides 279–779 of chtA.
Expression Vectors Containing β-Galactosidase (β-gal) and GFP.
Parental and mutant strains were marked with cell-type-specific and constitutively regulated genes. V/PsA-I-s65t contains a prespore promoter, psA, which drives the expression of the s65tGFP (GFP, green fluorescent protein) ubiquitin fusion protein (20). The protein has a half-life of about 4 hr. In V/63-I-s65t, the promoter driving the s65tGFP was replaced with the entire ecmA promoter. The V/PsA-β-gal vector contains the prespore promoter driving the lacZ gene (21). The psA promoter in the V/PsA-β-gal plasmid was replaced with the ecmA promoter to make the V/63-β-gal plasmid. To drive the expression of lacZ constitutively, the actin 6 promoter was fused to the lacZ gene in the V/A6-β-gal plasmid. The V/A15GFP construct permits the constitutive expression of GFP from the actin 15 promoter. All of the plasmids were generously provided by Harry MacWilliams, University of Munich.
Development.
For multicellular development, axenically growing cells in mid-log phase (2–4 × 106 cells/ml) were washed twice in cold SorC buffer. The cells, at a density of 3.3 × 106 cells/cm2 in SorC buffer, were plated on Whatman filters (grade 50) or Millipore HAWP nitrocellulose filters, which rested on SorC-saturated Whatman filter pads (grade 17) (22). For chimeric studies, chtA transformed with the plasmids containing lacZ driven by the constitutive actin 6 promoter, by prestalk, or by prespore promoters were mixed with wild-type cells before development.
Cell-Type-Specific Marker Studies Using Prestalk and Prespore Specific Promoters.
Wild-type and mutant strains were transformed with plasmids (V/PsA-β-gal and V/63-β-gal) that contain the lacZ gene driven by cell-type-specific promoters, psA or ecmA. The cells were plated for development as described above. At 16, 20, and 24 hr, structures on the filter were fixed and stained for β-gal according to the methods of ref. 23. Images were taken with a Hamamatsu Orca CCD camera attached to a Zeiss Axioplan-2 microscope. The images were captured by using the ip lab program (Scanalytics, Fairfax, VA). Images were then transferred to Adobe photoshop 3.0. For measurement of slug compartments, cells containing the V/PsA-β-gal or V/63-β-gal constructs were allowed to develop on filters until the slug stage and stained for β-gal. Images of the slugs were taken with a Pixera camera (Los Gatos, CA) attached to a dissecting microscope. These were cut out of photos, and then the entire slug image and the prespore compartments were weighed. The results were recorded as a percentage of slug area.
Confocal Studies.
A wild-type and a mutant strain were transformed with the plasmids V/PsA-I-s65t or V/63-I-s65t, which contain the GFP gene driven by cell-type-specific promoters. Twenty-five microliters of a suspension of 1 × 107 cells/ml in SorC were placed in the center of 35-mm Petri dishes with 2% Bactoagar in distilled H2O and allowed to dry in a laminar flow hood. When the slug stage of development was reached (16–18 hr), the GFP-labeled cells were visualized by confocal microscopy on the inverted dish. Optical sections were taken at 10 μm or 20 μm intervals at ×10 or ×20. GFP images were stored in the green channel and overlaid on the phase photos by using the LSM program (Zeiss, Oberkochen, Germany).
Results
Selection for Mutants That Preferentially Form Spores.
If a mutant exists in the population that does not form stalks, but rather forms exclusively spores, its failure to contribute to the stalk population should result in a slow increase in the proportion of the mutant in the population. The rate of increase should be 1.0/0.8 (100% spore-forming efficiency of the mutant/80% spore-forming efficiency of the wild type = 1.2/generation), if the mutation is cell autonomous and fully penetrant. If the mutant actively causes the wild-type cells in the chimera to become stalk (or in some other way to fail to become spores), the rate of increase in the population could be more rapid. A REMI mutagenized population was plated on SM/5 plates with a lawn of Klebsiella aerogenes and allowed to grow and develop (24). Spores were then recovered, and contaminating nonspore cells were killed by incubating for 15 min in 0.3% Triton-X-100 in SorensenC buffer (see Methods). Approximately 106 spores of the heterogeneous population were replated, and the selection process was repeated to enrich for cheater mutants, which were detected as those with an abnormal morphology (see below) and which increased in frequency with developmental cycle. Note that potential cheater mutants are competing against other mutants and that a cheater mutant with normal morphology would go undetected by this method. There is an obligatory growth phase in each cycle during which each cell divides six to eight times before the bacterial lawn is consumed and a new developmental cycle begins. Because spores from each cycle had been frozen, it was possible to find the increase in frequency of the mutant as a function of selection cycle. At the beginning of the selection, no mutants with the aberrant phenotype described below were found among 593 clones analyzed, which means that the frequency of the mutant in the selected mutant population was less than 0.2%. At the end of 20 cycles, slightly less than 3% of the clones were mutant. The original population had about 14,000 independent mutants. We could have selected a mutant that grows faster with this procedure, but that was not the case with chtA, as measured by the expansion rate of the clones (data not shown). This method would not detect minor differences in growth rate.
We asked whether the mutant that was eventually recovered and characterized would increase in the population under the conditions originally used for selection. In a reconstruction experiment that started with one mutant among 1,000 isogenic wild-type cells, the mutant had an advantage and the frequency of increase was rapid, perhaps because 1/1,000 is a higher starting number than in the original selection. In the first through third cycles, only one or no clones with chtA phenotype were observed among several hundred plaques. In the fourth cycle, the frequency was 1.7%, in the fifth, 2.7%, in the sixth, 5.5%, in the seventh, 8.1% and finally in the eighth cycle, 39%.
Developmental Phenotype of chtA Null Strains.
On bacterial lawns and Bactoagar plates, the initial steps in development, including precursor cell differentiation and sorting to the proper compartment, appear normal (see below). After the tipped-mound stage, the mutant forms slugs, and these are longer than wild-type slugs, as shown in Fig. 1 D and E. When growing and developing at high density on lawns of bacteria or developing on nonnutrient agar, the elongated structures of the mutant extended above the substratum until their tips made contact with the cover of the Petri dish, and the front of the slug-like structure crawled onto it. A long strand of material was left between the lid and agar surface, as shown in Fig. 1 B and C. This could be mistaken for a stalk but is actually a collapsed slime sheath that contains few cells (data not shown). The slime sheath is a casing of extracellular matrix protein and cellulose that surrounds slugs and is left behind as they migrate (25, 26). These structures have been noted previously and called aerial slugs (27). Under some conditions of development, on Millipore filters, for example, the chtA strains, arrest development as a large mound of cells and have trouble initiating a tip unless a few percent of wild-type cells are included in a chimera.
Figure 1.

The phenotype of DH1 (wild type) and chtA: a normal fruiting body is shown in A. The extended vertical slug-like structure of chtA is shown in B and also as a drawing in C. Relative slug sizes of DH1 and the mutant are shown in D and E. Bar = 0.4 mm in A and D and 0.45 mm in B and E. Arrows indicate stalk in A or slime sheath in B and C.
Expression of Cell-Type-Specific Genes.
To determine whether the mutant differentiates normally into prestalk cells, the chtA and parental cells were transformed with constructs that contain the ecmAO promoter region fused to the β-gal reporter gene. The ecmA gene is used as a marker for prestalk cells because it encodes an extracellular matrix-like protein that is synthesized only in prestalk cells (28–30). During the early stages of development, up to the tipped-mound stage, the chtA cells correctly express the lacZ gene from the full ecmAO promoter. As in wild-type cells, the chtA mutant's prestalk cell differentiation is first noticed around the periphery of the mound and then at the tip as the prestalk cells sort to the apical region (data not shown) (31). Although chtA cells can differentiate into prestalk cells, they do not form mature stalk.
To ask whether prespore differentiation is normal in the cheater mutant, the prespore specific psA promoter fused to the lacZ reporter gene was introduced into parental and chtA cells (32). The patterns of gene expression and cell compartmentalization up to the tipped aggregate stage were indistinguishable in the mutant and the wild type (data not shown). Thus, aggregation and the early differentiation of the prestalk and prespore lineages are not grossly affected by the mutation in the chtA gene, which appears to act later in development.
Using a construct with the ecmAO promoter fused to GFP to monitor prestalk cells in the slug, we observed that the slugs of mutant and wild type are marked with GFP in the anterior prestalk region, but there is no sharp boundary of expression (Fig. 2 C and D). When the psA promoter fused to GFP was used, the boundary between prestalk and prespore zone is sharp (Fig. 2 A and B). The prespore region in the chtA slugs is increased. In 13 wild-type slugs, the prespore zone composed 85.7 +/− 3.3% of the total slug area, whereas in 13 chtA slugs, the prespore zone comprised 94.3 +/− 2.5% of the total area.
Figure 2.
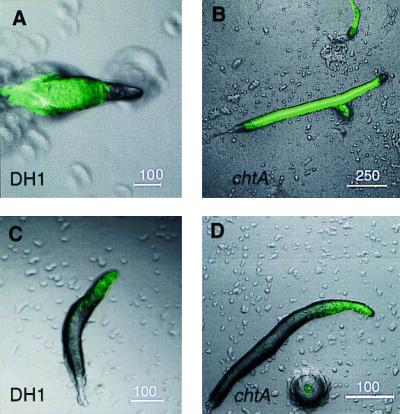
Mutant slugs have an increased prespore zone. Expression of prespore and prestalk markers in wild-type (DH1) and mutant slugs: A and B: psA-GFP. C and D: ecmA-GFP. Note the larger prespore region in B and differences in scale bars.
The chtA Mutant Adopts the Prespore Cell Fate in Chimeras.
To ask whether the mutant is preferentially incorporated into spores, wild-type and mutant amoebae were mixed and deposited on a solid substratum for development. The chtA cells were marked with β-gal or GFP driven from the ubiquitously expressed actin 6 or actin 15 promoters (see Methods). We discovered that chtA cells marked with β-gal sort to the prespore compartment of the slug and fruiting body stages, with occasional labeling in the basal disc cells (Fig. 3 A and B).
Figure 3.
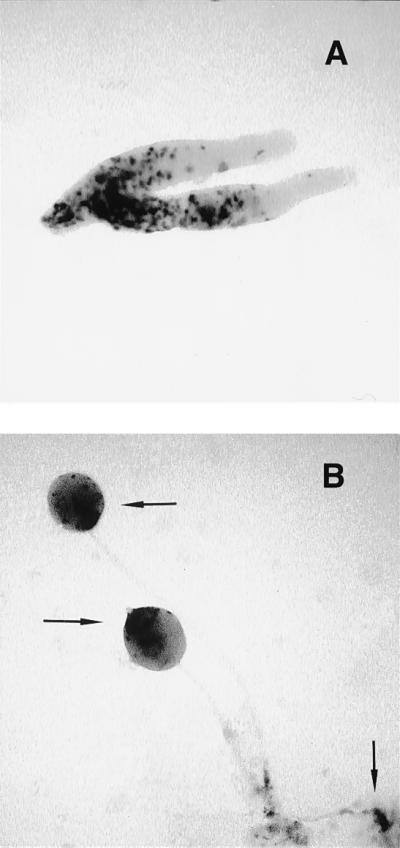
ChtA cells form spores in chimeras with the wild type. Mutant cells labeled with an actin promoter expressing β-gal were mixed 1:10 with unlabeled wild type (DH1). A shows the presence of the mutant cells in the prespore zone of slugs, and B shows the movement to the spore mass several hours later. The top two arrows point to mutant cells in the spore mass and the lower arrow to basal disc cells, which are sometimes labeled.
If the mechanism by which the mutant increases in the population is based on a simple inability to form stalk, without an effect on the wild-type population, the rate of increase in a chimeric population should be about 1.2-fold per developmental cycle. If there is active suppression of spore differentiation in wild-type cells, perhaps because a natural suppression pathway has been overexpressed in the mutant, a more rapid increase of the mutant's frequency in the population could occur. To ask which mechanism applied, we performed several chimeric studies with chtA mutants and an isogenic parental strain. The two cell types were mixed in the ratios shown in Table 1. An aliquot of each mixture was plated clonally before development to confirm the ratios of mutant to wild type. Then the mixture of cells was allowed to develop into fruiting bodies, and the resulting detergent-resistant spores were dispersed at low density on lawns of bacteria so they could form individual plaques. The ratio of mutant to wild type was determined in the spore population from fruiting bodies. The mutant and wild-type spores are both completely resistant to the detergent treatment. As the results in Table 1 show, there was a dramatic increase in the mutant's frequency, even over one cycle. Starting with low numbers (4.8% or 1.9%) of the chtA cells, there is a 5-fold or 6.5-fold increase in the proportion of chtA spores in the resulting spore mass. At higher inputs of mutant cells, there was approximately a 2-fold increase in spores with the chtA mutant phenotype. The increase was more rapid than found in the original selection, which took a number of cycles to go from undetectable to about 3% of the population. In the original selection, the population was genetically heterogeneous, there were obligatory growth cycles, and growth and development was on nutrient agar plates with bacterial lawns. In the experiments shown in Table 1, cells were carefully washed, and relatively high numbers of cells were deposited on Millipore filters and sampled immediately after sori had formed, with no bacteria and no growth phase.
Table 1.
The ChtA cells increase in the population over one cycle of development
| Input vegetative cells
|
Spores after 48 hr
|
||
|---|---|---|---|
| % chtA vs. wild type
|
% chtA vs. wild type
|
||
| Expt. 1 | Expt. 2 | Expt. 1 | Expt. 2 |
| 4.8 | 1.9 | 23.9 (5×) | 12.5 (6.5×) |
| 24.5 | 20.2 | 46.9 (1.9×) | 49.6 (2.5×) |
| 41.7 | 43.4 | 85.2 (2×) | 73.0 (1.7×) |
Chimeras were made with AX3 and chtA. After fruiting bodies formed, the spores were plated clonally and examined for wild-type or chtA phenotypes. Between 200 and 300 clones were examined in each case.
We asked what happened to the wild-type cells that would account for the increase in the frequency of mutant spores in the mosaic fruiting body. If the mutant cells force the wild-type cells in the chimera to enter the stalk cell pathway or otherwise fail to enter the spore pathway, which leads to death, in a chimera with chtA cells, more wild-type cells should end up as prestalk than would normally occur. This is the case. When 80% wild-type cells and 20% wild-type cells marked with β-gal expressed from an ecmAO (prestalk) promoter are mixed, about 4% (20% of 20%) of the labeled cells should end up in the prestalk region of the slug. This results in light staining, as shown in Fig. 4A. When 80% unlabeled chtA cells are mixed with the 20% ecmAO-β-gal-labeled wild-type cells, many more should turn into prestalk cells, making a darker prestalk zone, if the model of forced conversion of wild-type cells to prestalk and stalk cells is correct. The slugs shown in Fig. 4B are representative of many hundreds and indicate that chtA cells cause the wild-type cells to enter the prestalk zone and to express the ecmAO prestalk promoter. When the chtA cells were labeled with the ecmAO-β-gal construct, no expression occurs in chimeras with wild type, although expression is detected in a prestalk zone when the mutant develops alone (data not shown). Thus mutant cells do not enter the prestalk pathway when wild type is present, but wild-type cells preferentially enter the prestalk pathway when mutant is present.
Figure 4.

ChtA drives wild-type cells into the prestalk region of the sorocarp: A shows the results of mixing 20% wild-type cells marked with β-gal driven by the ecmAO prestalk specific promoter and 80% unlabeled wild-type cells (AX3). In B, 20% AX3 with an ecmAO driven β-gal was mixed with 80% unlabeled chtA cells. C, E, and G show a mixture of 20% wild type labeled with the prespore psA promoter driving β-gal and 80% unlabeled wild type (AX3). D, F, and H show what happens when the unlabeled wild-type cells are replaced with the chtA mutant.
Are wild-type cells that have begun to differentiate in the prespore pathway influenced by the mutant? If a prespore promoter fused to β-gal is used to follow wild-type cells that have become prespore cells, β-gal is expressed in the rear of the slug and then in the spore mass, as expected (Fig. 4 C, E, and G). If the mutant is the major partner in the chimera, the wild-type cells differentiate into prespore promoter in the rear of the slug (Fig. 4D), but with time these cells appear in the front of the slug, in the prestalk zone (Fig. 4F). The fate of the cells can be followed because β-gal is relatively stable. The labeled wild-type cells go up not to the spore mass but to the upper cup, on top of the spore mass (Fig. 4H). The upper cup is normally populated by a category of prestalk cell, but we cannot yet say that these cells have converted to prestalk cells—only that they have moved to a prestalk cell region and do not form spores.
The chtA Gene Codes for a Protein with an F-box and WD40 Repeats.
The blasticidin resistance plasmid inserts 26 nucleotides upstream from an apparent TAA termination codon. The full sequence is presented in Fig. 5. The chtA gene encodes a protein of 1,247 amino acids and is produced from an mRNA of about 4.4 kb. The total coding sequence is 3,741 nucleotides, and we can account for 229 bases as 5′ untranslated region. There is one intron of 141 nucleotides that is inserted between nucleotides 239 and 380 of the genomic sequence. Within the sequence is a domain that codes for an F-box (33). Fig. 5B compares the Dictyostelium F-box to those in humans, Saccharomyces cerevisiae, and Caenorhabditis elegans. The carboxy terminus of the ChtA protein codes for five WD40 repeats (indicated by light gray in Fig. 5) (13, 34).
Figure 5.

The chtA gene codes for a protein with F-box and WD40 repeats: the diagram (A Upper) shows the relative positions of the F-box and the WD40 repeats (gray). GenBank accession nos. are AF151111 and AF151112 for chtA. The insertion site of pBSR3 in chtA is shown. The position of the primer used for 5′ rapid amplification of cDNA ends experiments is indicated by an arrow. The sequence is shown with the F-box (dark gray) and WD40 repeats in lighter gray and underlined. B shows the homology with several other F-box sequences: humans (skp2, accession no. AAC50242) (51), C. elegans (sel10, accession no. AF020788) (14), and S. cerevisiae (met30, accession no. P39014) (52, 53).
Recapitulation studies, in which the wild-type gene was again destroyed by homologous recombination, gave rise to strains with the same phenotype as chtA. In addition to AX3, a V12 (HH309) axenic strain was disrupted by homologous recombination, and this produced a strain with the same phenotype as the mutant (data not shown).
The chtA transcript is developmentally regulated in the wild type. None can be found during vegetative growth, and it appears by about 4 hr of development, as shown in Fig. 6. Little or no transcript could be detected in Northern blots of RNA from the mutant.
Figure 6.

The mRNA of chtA is developmentally regulated. A band of 4.4–4.5 kb was detected in wild-type cells after 4 hr of starvation (A Upper). Ribosomal RNA loading standards are shown below.
Discussion
The selection used here derives from a consideration of the problems involved in the evolution of organisms in which not all participants are genetically identical. We postulate that mutants that are successful at increasing their frequency in chimeras are affected in mechanisms that are fundamental to cell–cell communication and signaling. The REMI mutagenesis procedure results in the insertion of a plasmid within a gene and in most cases causes a null. This allows us to impose genetic diversity on a previously homogeneous population of amoebae, but it does not provide a full range of mutagenic possibilities, as would occur in wild strains. The other limitation of the selection is that the cheater must present an abnormal phenotype when it is not part of a chimera. In this selection, the only mutant recovered was chtA, but this does not mean that other mutants, including mutants with normal phenotype, could not exist, nor are we suggesting that this particular mutant would be selected in the wild.
The ctrA mutant expressed both prestalk and prespore markers: several models could account for the accumulation of the mutant cells in the spore masses of fruiting bodies. The chtA mutation could promote more efficient entry into the aggregate in the earlier part of development. Time-lapse recordings made of a chimera of 90% wild-type cells and 10% GFP-labeled chtA cells showed that both mutant and wild-type cells enter the aggregate with equal efficiency (data not shown). Another possibility is that the mutant ignores signals to become stalk and is passive in its cheating. If so, it would increase slowly in the population, at a maximum rate of 1.2-fold per developmental cycle. The reconstruction experiment shown in Table 1 confirmed that the mutant increases more rapidly, especially at lower inputs. Even with an input of 1/1,000, there is a rapid increase over eight generations. One way to explain these results is to postulate that the mutant affects the wild-type cells, preventing them from turning into detergent-resistant spores or causing them to turn into inviable stalk cells. The failure of cells missing a crucial F-box component of the SCF complex to degrade key regulatory proteins could lead to diversion to the spore pathway and increased production of stalk cell inducers by an as yet poorly understood cell-type homeostasis mechanism (35, 36).
An alternative explanation is that chtA cells divide more than twice when the wild type does not. The literature on cell division during Dictyostelium development is controversial. Most studies do not distinguish between initiation of DNA synthesis and the cytokinesis of the amoebae, which are often binucleate. Thus, some evidence exists for division of prespore cells (37, 38). Other studies using different conditions show that DNA synthesis during development is primarily mitochondrial (39). If chtA were involved in the cell cycle, one might expect it to be expressed in growing cells, but it appears only after development begins, as shown in Fig. 6. If the mutant were increasing from 5% of the population to about 25% by cell division, without affecting the wild-type cells, the final fruiting structures should not suffer from a dearth of wild-type cells and should be normal. However, we consistently observe that when there is a 5% input of chtA cells in chimeras with 95% wild type, the resulting spores contain about 25% chtA mutants (see Table 1), and the resulting sori are smaller than controls. Despite these observations and the results shown in Fig. 4, we cannot exclude a contribution to the increase in chtA cells by division, although we consider it unlikely that starving cells would divide more than twice to give the results presented.
F-box proteins mediate irreversible transitions of the cell cycle, cell-fate decisions, and other events (40), by serving as adapters between protein targets and the proteolytic machinery (11, 12, 41). These proteins contain two notable domains, an F-box domain, such as those shown in Fig. 5, and protein–protein interacting domains such as WD40 repeats. The F-box domain binds to a protein called Skp1. A Dictyostelium Skp1 is known (42). Skp1, in turn, is connected by cullins (43) to a ubiquitinating enzyme of the E2 class. The WD40 repeats are the domain of the protein that binds the target of the ubiquitination machinery (11, 12). Targets must be phosphorylated. There is considerable divergence within the WD40 repeats, perhaps accounting for the diversity of phosphorylated proteins that can be bound and degraded. The composite of Skp1, cullin, and F-box protein is known as an SCF complex. The target proteins that bind to this potential SCFchtA complex are not yet known.
There is one other known F-box/WD40 protein in Dictyostelium.
The other gene codes for MEK kinase (MEKKα) (44). Deletion of this gene leads to an expansion of the rear subcompartment (pstO) region of the prestalk zone and a reduction in the prespore compartment. The mosaic fruiting body of a mixture of a MEKKα mutant and wild type shows that the mutant adopts the stalk cell fate.
The library of mutants known in Dictyostelium allows us to ask whether any other mutants have a phenotype similar to that of the chtA mutant and which could act in the same pathway. At the slug stage, the prespore compartment in the chtA mutant slug increased, as shown in Fig. 2B, indicating a flaw in proportionality regulation. This could occur because of a failure to convert prespore cells to prestalk cells, which occurs during normal development, or it could result from defects at the time of the initial differentiation of prespore and prestalk cells. Several mutants with reduced prestalk regions have been recovered in the past, before the time when their genes could be isolated (35, 36). A mutant described by Newell and Ross, NP429, of the slugger E complementation group, may be similar to chtA because it makes the same extended aerial slugs (27). More recently, the characteristic short prestalk region of the chtA mutant has been observed in cells that lack the serpentine receptor cAR4 (45). This receptor is present only in prestalk cells and negatively regulates the Dictyostelium Gsk3 homologue, and thus this system may represent a version of the wingless signaling pathway that is used to create and regulate a normal pattern of prestalk and prespore cells (46, 47). Gsk3 is required for spore cell differentiation. Gsk3-null cells develop into a mound containing stalk cells but devoid of spore cells (46). The substrates of Gsk3 are not characterized yet, but by analogy with Wnt systems, it is possible that an F-box WD40 domain protein targets a β-catenin homologue for degradation.
We postulate that ChtA controls the concentration of a key regulatory protein(s) by regulating degradation via the SCF complex. High levels of this protein would cause the cells to initiate the spore differentiation pathway. Removing the protein would be necessary to initiate the prestalk to stalk differentiation pathway during culmination. We speculate that cells with high amounts of the protein target(s) of the F-box protein (the chtA mutant) produce a signal that forces cells with lower amounts of the protein target (the wild type) to convert to prestalk cells. We do not yet know the nature of such a signal. These ideas are consistent with the observation that chtA-null cells in chimeras with wild type turn into spores and wild type becomes stalk cells. Signals from the stalk cells (wild type) in the chimera may rescue the sporulation defect of the mutant that occurs when it develops alone. A signal from prestalk cells is necessary for final differentiation of the spore cells (48–50).
Acknowledgments
We thank Harry MacWilliams of the University of Munich for generously supplying the plasmids used in this work. We also thank Margaret Nelson and Jeffrey Williams (University of Dundee) for their collegial sharing of a strain and Jan Kitajewski for many helpful discussions. Jakob Franke, Grant Otto, and Mary Wu were of great assistance. We thank Theresa Swayne for help with the confocal microscopy. Supported by grant GM33136–17 from The National Institutes of Health to R.H.K. and grant IBN-9727184 from the National Science Foundation to H.L.E.
Abbreviations
- REMI
restriction enzyme-mediated integration
- β-gal
β-galactosidase
- GFP
green fluorescent protein
Footnotes
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AF151111 and AF151112).
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.050005097.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.050005097
References
- 1.Bonner J T. The Evolution of Complexity by Means of Natural Selection. Princeton, NJ: Princeton Univ. Press; 1988. [Google Scholar]
- 2.Gadagkar R, Bonner J T. J Biosci. 1994;19:219–245. [Google Scholar]
- 3.Bonner J T. Am Nat. 1982;119:530–552. [Google Scholar]
- 4.Buss L W. Proc Natl Acad Sci USA. 1982;79:5337–5341. doi: 10.1073/pnas.79.17.5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellison A M, Buss L W. Am J Bot. 1983;70:298–302. [Google Scholar]
- 6.Armstrong D P. J Theor Biol. 1984;109:271–283. [Google Scholar]
- 7.Francis D, Eisenberg R. Mol Ecol. 1993;2:385–392. doi: 10.1111/j.1365-294x.1993.tb00031.x. [DOI] [PubMed] [Google Scholar]
- 8.Filosa, M. F. (1962) Am. Nat.XCVI (no. 887), 79–92.
- 9.Buss L W. Proc Natl Acad Sci USA. 1999;96:8801–8803. doi: 10.1073/pnas.96.16.8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kay R R, Large S, Traynor D, Nayler O. Proc Natl Acad Sci USA. 1993;90:487–491. doi: 10.1073/pnas.90.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper J W, Elledge S J. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- 12.Skowyra D, Craig K L, Tyers M, Elledge S J, Harper J W. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- 13.Neer E J, Schmidt C J, Nambudripad R, Smith T F. Nature (London) 1994;371:297–300. doi: 10.1038/371297a0. [DOI] [PubMed] [Google Scholar]
- 14.Hubbard E J, Wu G, Kitajewski J, Greenwald I. Genes Dev. 1997;11:3182–3193. doi: 10.1101/gad.11.23.3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuspa A, Loomis W F. Proc Natl Acad Sci USA. 1992;89:8803–8807. doi: 10.1073/pnas.89.18.8803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adachi H, Hasebe T, Yoshinaga K, Ohta T, Sutoh K. Biochem Biophys Res Commun. 1994;205:1808–1814. doi: 10.1006/bbrc.1994.2880. [DOI] [PubMed] [Google Scholar]
- 17.Shaulsky G, Escalante R, Loomis W F. Proc Natl Acad Sci USA. 1996;93:15260–15265. doi: 10.1073/pnas.93.26.15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Welker D L, Hirth K P, Williams K L. Mol Cell Biol. 1985;5:273–280. doi: 10.1128/mcb.5.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu L, Hansen D, Franke J, Kessin R H, Podgorski G J. Dev Biol. 1995;171:149–158. doi: 10.1006/dbio.1995.1267. [DOI] [PubMed] [Google Scholar]
- 20.Deichsel H, Friedel S, Detterbeck A, Coyne C, Hamker U, MacWilliams H K. Dev Genes Evol. 1999;209:63–68. doi: 10.1007/s004270050228. [DOI] [PubMed] [Google Scholar]
- 21.Detterbeck S, Morandini P, Wetterauer B, Bachmair A, Fischer K, MacWilliams H K. Development (Cambridge, UK) 1994;120:2847–2855. doi: 10.1242/dev.120.10.2847. [DOI] [PubMed] [Google Scholar]
- 22.Pukatzki S, Tordilla N, Franke J, Kessin R H. J Biol Chem. 1998;273:24131–24138. doi: 10.1074/jbc.273.37.24131. [DOI] [PubMed] [Google Scholar]
- 23.Richardson D L, Loomis W F, Kimmel A R. Development (Cambridge, UK) 1994;120:2891–2900. doi: 10.1242/dev.120.10.2891. [DOI] [PubMed] [Google Scholar]
- 24.Sussman M. Methods Cell Biol. 1987;28:9–29. doi: 10.1016/s0091-679x(08)61635-0. [DOI] [PubMed] [Google Scholar]
- 25.Ti Z C, Wilkins M R, Vardy P H, Gooley A A, Williams K L. Dev Biol. 1995;168:332–341. doi: 10.1006/dbio.1995.1084. [DOI] [PubMed] [Google Scholar]
- 26.Wilkins M R, Williams K L. Experientia. 1995;51:1189–1196. doi: 10.1007/BF01944736. [DOI] [PubMed] [Google Scholar]
- 27.Newell P C, Ross F M. J Gen Microbiol. 1982;128:1639–1652. [Google Scholar]
- 28.Jermyn K A, Berks M, Kay R R, Williams J G. Development (Cambridge, UK) 1987;100:745–755. doi: 10.1242/dev.100.4.745. [DOI] [PubMed] [Google Scholar]
- 29.Ceccarelli A, McRobbie S J, Jermyn K A, Duffy K, Early A, Williams J G. Nucleic Acids Res. 1987;15:7463–7476. doi: 10.1093/nar/15.18.7463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jermyn K A, Duffy K T, Williams J G. Nature (London) 1989;340:144–146. doi: 10.1038/340144a0. [DOI] [PubMed] [Google Scholar]
- 31.Williams J G, Duffy K T, Lane D P, Mcrobbie S J, Harwood A J, Traynor D, Kay R R, Jermyn K A. Cell. 1989;59:1157–1163. doi: 10.1016/0092-8674(89)90771-x. [DOI] [PubMed] [Google Scholar]
- 32.Early A E, Williams J G, Meyer H E, Por S B, Smith E, Williams K L, Gooley A A. Mol Cell Biol. 1988;8:3458–3466. doi: 10.1128/mcb.8.8.3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hofmann K, Bucher P, Falquet L, Bairoch A. Nucleic Acids Res. 1999;27:215–219. doi: 10.1093/nar/27.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith T F, Gaitatzes C, Saxena K, Neer E J. Trends Biochem Sci. 1999;24:181–185. doi: 10.1016/s0968-0004(99)01384-5. [DOI] [PubMed] [Google Scholar]
- 35.MacWilliams H, Blaschke A, Prause I. CSH Symp Quant Biol. 1985;50:779–785. doi: 10.1101/sqb.1985.050.01.096. [DOI] [PubMed] [Google Scholar]
- 36.Blaschke A, Weijer C, MacWilliams H. Differentation (Berlin) 1986;32:1–9. [Google Scholar]
- 37.Oohata A A, Nakagawa M, Tasaka M, Fujii S. Development (Cambridge, UK) 1997;124:2781–2787. doi: 10.1242/dev.124.14.2781. [DOI] [PubMed] [Google Scholar]
- 38.Bonner J T, Frascella E B. J Exp Zool. 1952;121:561–571. [Google Scholar]
- 39.Shaulsky G, Loomis W F. Proc Natl Acad Sci USA. 1995;92:5660–5663. doi: 10.1073/pnas.92.12.5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patton E E, Willems A R, Tyers M. Trends Genet. 1998;14:236–243. doi: 10.1016/s0168-9525(98)01473-5. [DOI] [PubMed] [Google Scholar]
- 41.Jiang J, Struhl G. Nature (London) 1998;391:493–496. doi: 10.1038/35154. [DOI] [PubMed] [Google Scholar]
- 42.Tengumnuay P, Morris H R, Dell A, Panico M, Paxton T, West C M. J Biol Chem. 1998;273:18242–18249. doi: 10.1074/jbc.273.29.18242. [DOI] [PubMed] [Google Scholar]
- 43.Michel J J, Xiong Y. Cell Growth Differ. 1998;9:435–449. [PubMed] [Google Scholar]
- 44.Chung C Y, Reddy T B, Zhou K, Firtel R A. Genes Dev. 1998;12:3564–3578. doi: 10.1101/gad.12.22.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ginsburg G T, Kimmel A R. Genes Dev. 1997;11:2112–2123. doi: 10.1101/gad.11.16.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harwood A J, Plyte S E, Woodgett J, Strutt H, Kay R R. Cell. 1995;80:139–148. doi: 10.1016/0092-8674(95)90458-1. [DOI] [PubMed] [Google Scholar]
- 47.Plyte S E, O'Donovan E, Woodgett J R, Harwood A J. Development (Cambridge, UK) 1999;126:325–333. doi: 10.1242/dev.126.2.325. [DOI] [PubMed] [Google Scholar]
- 48.Shaulsky G, Kuspa A, Loomis W F. Genes Dev. 1995;9:1111–1122. doi: 10.1101/gad.9.9.1111. [DOI] [PubMed] [Google Scholar]
- 49.Shaulsky G, Loomis W F. Dev Biol. 1996;174:214–220. doi: 10.1006/dbio.1996.0067. [DOI] [PubMed] [Google Scholar]
- 50.Firtel R A. Curr Opin Genet Dev. 1996;6:545–554. doi: 10.1016/s0959-437x(96)80082-7. [DOI] [PubMed] [Google Scholar]
- 51.Zhang H, Kobayashi R, Galaktionov K, Beach D. Cell. 1995;82:915–925. doi: 10.1016/0092-8674(95)90271-6. [DOI] [PubMed] [Google Scholar]
- 52.Thomas D, Kuras L, Barbey R, Cherest H, Blaiseau P L, Surdin-Kerjan Y. Mol Cell Biol. 1995;15:6526–6534. doi: 10.1128/mcb.15.12.6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaiser P, Sia R A, Bardes E G, Lew D J, Reed S I. Genes Dev. 1998;12:2587–2597. doi: 10.1101/gad.12.16.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]