


Abstract
We have identified two novel proteins that interact specifically with the C-terminal repression domain of Interferon Regulatory Factor-2 (IRF-2). These proteins, which we term IRF-2 binding proteins 1 and 2 (IRF-2BP1 and IRF-2BP2, the latter having two splicing isoforms, A and B), are nuclear proteins, and have the properties of IRF-2-dependent transcriptional co-repressors that can inhibit both enhancer-activated and basal transcription in a manner that is not dependent upon histone deacetylation. IRF-2BP1 and IRF-2BP2A/B contain an N-terminal zinc finger and a C-terminal RING finger domain of the C3HC4 subclass, but show no homology to other known transcriptional regulators; they therefore define a new family of co- repressor proteins. An alternatively spliced form of IRF-2 that lacks two amino acids (valines 177 and 178) in the central portion of the protein (IRF-2[S]) cannot bind to these co-repressors and cannot mediate repression despite having the same C- terminal repression domain as IRF-2, suggesting that the relative conformation of the DNA binding domain and the C-terminal region of IRF-2 is crucial for transcriptional repression.
INTRODUCTION
Interferon Regulatory Factor-2 (IRF-2) is a member of a family of proteins (the IRFs) that play a major role in the transcriptional regulation of type I interferon (IFN) genes in response to viral infection and genes that are regulated in response to type I and type II IFNs [reviewed in (1)]. IRF-2 was originally described as a protein that bound to the IFN-β promoter and antagonised the effect of the transcriptional activator, IRF-1 (2). Further studies have implicated IRF-2 as a negative regulator of many IFN-responsive genes that contain IRF binding sites in their promoters [for example, 2′-5′-oligoadenylate synthetase, iNOS, MHC class I; reviewed in (1)]. Consistent with this, mice lacking IRF-2 demonstrate dramatic over-expression of genes induced by type I IFN, and develop an inflammatory skin disease in response to antigenic stimulation, indicating that IRF-2 plays an essential role in modulating the response to IFN (3). As many of these genes play a role in the negative regulation of the cell cycle and/or apoptosis, IRF-2 is also a putative oncogene (4–6).
In addition to its ability to inhibit expression of some genes, IRF-2 has been shown to be a transcriptional activator of others. This was first shown for the cell-cycle regulated transcriptional activation of the histone H4 gene (7,8) and has since been demonstrated for the gp91 phox (9), EBV EBNA-1 (10), vascular cell adhesion molecule-1 (11) and MHC class II transactivator (CIITA) genes (12,13). A requirement for IRF-2 in CIITA transactivation is supported by the finding that mutations in the IRF-2 DNA binding domain (DBD) are found in a pancreatic tumour cell line and in fresh pancreatic tumour explants and are associated with loss of CIITA transcription (14).
Analysis of the domain structure of IRF-2 shows that the N-terminal 113 amino acids encompass the highly conserved IRF DBD containing a characteristic motif consisting of five tryptophan residues, which forms a ‘winged helix’ structure (15,16). The transcriptional activation domain (AD) has been shown to reside between amino acids 160 and 220 (11,17). On promoters that are not activated by IRF-2 the transactivation domain is dominantly inhibited by the C-terminus (17), and the C-terminus of IRF-2 can also repress transcription when fused to a heterologous DBD (17,18), indicating that this region of IRF-2 contains an autonomously acting repression domain.
The mechanisms of repression and transactivation by IRF-2 are poorly characterised, but it has been shown that IRF-2 can interact with the histone acetylase factors, GCN5, PCAF and p300/CBP (19), and with TFIIB (20), all of which are known to play important roles in transcriptional stimulation. Furthermore, a cDNA for the bromodomain-containing protein, Celtix-1, has been isolated from a yeast two-hybrid screen using IRF-2 as bait, and it has been suggested that Celtix-1 may play a role in transactivation by IRF-2 (21). It is also possible that IRF-2 activates transcription by recruiting IRF-1 to some promoters (12). To date, no factors capable of interacting with the C-terminal repression domain have been identified. In this manuscript we describe the cloning and characterisation of two novel nuclear proteins (which we call IRF-2 binding proteins 1 and 2; IRF-2BP1 and IRF-2BP2) that bind to the C-terminal repression domain and have the properties of IRF-2-dependent transcriptional co- repressors that can inhibit both enhancer-activated and basal transcription.
MATERIALS AND METHODS
Plasmids
Schematics of the reporter gene and effector plasmids used in this report are shown in Figure 1. Plasmids with the firefly luciferase gene under the control of the Herpes Simplex Virus thymidine kinase promoter [ptkΔ(–105)lucter], the minimal TATA box only [ptkΔ(–39)lucter] or an IRF-dependent promoter [p[(AAGTGA)4]5tkΔ(–39)lucter] have been described previously (22,23). The GAL4-responsive minimal promoter reporter construct, p(GALUAS)5tkΔ(–39)lucter, was constructed by inserting the filled-in HindIII–XbaI fragment containing the GALUAS pentamer from pG5E4CAT (24) into BamHI-linearised and filled-in ptkΔ(–39)lucter. pSV40 (GALUAS)5tkΔ(–39)lucter contains the SV40 enhancer fragment (SV40 co-ordinates 272 to 100 from πSVHNE) (25) inserted upstream of the GALUAS in p(GALUAS)5tkΔ (–39)lucter. In pSV40(lacP/O)tkΔ(–39)lucter the GALUAS pentamer was replaced by the 70 bp BamHI–BglII polylinker fragment from πVX (26) to create a GAL4-unresponsive SV40 enhancer-driven luciferase reporter. All luciferase reporter plasmids were co-transfected with pJATlac, a plasmid in which the Escherichia coli β-galactosidase gene is under the control of the constitutively active rat β-actin promoter (27). In p(GALUAS)5tkΔ(–39)β-globin, the luciferase gene of p(GALUAS)5tkΔ(–39)lucter is replaced by a genomic fragment of human β-globin (co-ordinates –5 to +2160 relative to the β-globin cap site). The plasmid πSVHSα118 (28), which directs the constitutive expression of human α-globin, was used as a co-transfection control in β-globin experiments.
Figure 1.

Reporter and effector gene constructs. (A) Reporter constructs used in this report. (B) GAL.IRF-2 fusion effector constructs used in this report. The GAL4 DBD is fused in-frame to the indicated fragments of IRF-2 (white boxes). The hatched area corresponds to the repression domain defined in this study. (C) Alignment of the extreme C-termini of human IRF-2 (amino acids 284–349) with the equivalent amino acids from sheep, mouse and chicken. Amino acids highlighted in black represent residues conserved with human IRF-2, and grey highlighting indicates functional conservation.
Expression in mammalian cells was achieved using pEF.plink2 (29). pEF.IRF-1 and pEF.IRF-2 contain full-length cDNAs for IRF-1 and IRF-2, respectively, cloned between the NcoI and XbaI sites of pEF.plink2. pEF.IRF-2BP1, pEF.IRF-2BP2A and pEF.IRF-2BP2B contain full-length open reading frames from pGAD10 clones isolated from the yeast two-hybrid screen described below; each of these clones is N-terminally tagged with the SV5 epitope tag. pEF.GAL147 contains the DBD (amino acids 1–147) of the yeast GAL4 transcription factor cloned between the NcoI and EcoRI sites of pEF.plink2. To construct pEF.GALIRF-1(105–325), pEF.GALIRF-2(88–349), pEF.GALIRF-2(88–280), pEF.GALIRF-2(224–349), pEF.GALIRF-2(269–349), pEF.GALIRF-2(292–349), pEF.GALIRF-2(332–349), pEF.GALIRF-2(292–340) and pEF.GALIRF-2(292–330), the indicated fragments of IRF-1 or IRF-2 were generated from precursor plasmids and fused in-frame to the GAL4 DBD of pEF.GAL147 using standard procedures; all end-points and fusion junctions were verified by dideoxy sequencing. A plasmid related to pEF.GALIRF-2(88–349) but expressing the equivalent IRF-2 fragment from the IRF-2[S] isoform [pEF.GALIRF-2[S](88–347)] was constructed from a full-length IRF-2[S] cDNA isolated from Jurkat cells by RT–PCR. pEF.GALIRF-2BP1, pEF.GALIRF-2BP2A, pEF.GALIRF-2BP2B and pEF.GALIRF-2BP2(456–587) were constructed by inserting cDNA fragments from pGAD10 clones in-frame with and downstream of amino acid 147 of the GAL4 DBD of pEF.GAL147.
pHON1 and pHON3 allow the expression of GAL4 DBD and AD fusions, respectively, in yeast, and are derived from pGBT9 and pGAD424 (30) by replacing the part-length ADH promoter with the full-length ADH promoter from pAS2-1 (31); these vectors also contain additional cloning sites in their polylinkers. To construct pHON1.IRF-2(1–349), pHON1.IRF-2(1–224), pHON1.IRF-2(1–163), pHON1.IRF-2(172–349), pHON1.IRF-2(224–349), pHON1.IRF-2(269–349), pHON1. IRF-2(292–349) and pHON1.IRF-2(332–349), and pHON1. IRF-2[S](1–347), IRF-2 or IRF-2[S] fragments were, respectively, generated from precursor plasmids and fused in-frame to the GAL4 DBD of pHON1. pHON1.IRF-2BP1, pHON1.IRF-2BP2A, pHON1.IRF-2BP2B and pHON1.IRF-2BP2(456–587) were constructed by transferring the EcoRI cDNA fragments from the pGAD10 clones into the EcoRI site of pHON1. To construct pHON3.IRF-1(1–325), pHON3.IRF-2(292–349), pHON3.IRF-9, fragments of IRF-1, IRF-2 or IRF-9 were generated from precursor plasmids and fused in-frame to the GAL4 AD of pHON3. Plasmids for the expression of fusions with either DBD (pHON1) or AD (pHON3 or pGAD424) and p65, Jak1, STAT1α and PKR (Table 1) were constructed in this laboratory using standard procedures (details available on request).
Table 1. Interactions between IRF-2BPs and cellular proteins.
| DBD | AD Vector | IRF-2BP1 | IRF-2BP2A | IRF-2BP2B | IRF-2 (292–349) | IRF-1 | p65 |
|---|---|---|---|---|---|---|---|
| Vector | – | – | – | – | – | – | – |
| IRF-2 (292–349) | – | ++++ | +++ | +++ | – | – | – |
| IRF-9 | + | + | + | + | ND | + | ND |
| Jak-1 | – | – | – | – | – | ND | ND |
| STAT1α | – | – | – | – | – | ND | ND |
| PKR | – | – | – | – | – | ND | ND |
| IRF-2BP1 | – | – | – | – | ++++ | – | – |
| IRF-2BP2A | – | – | – | – | +++ | – | – |
| IRF-2BP2B | – | – | – | – | ++++ | – | – |
| IRF-2 (1–349) | – | ++ | + | ++ | – | – | ND |
| IRF-2 (1–224) | – | – | – | – | – | – | ND |
| IRF-2 (1–163) | – | – | – | – | – | – | ND |
| IRF-2 (172–349) | – | + | + | ++ | – | – | ND |
| IRF-2 (224–349) | – | ++++ | +++ | ++++ | – | – | ND |
| IRF-2 (269–349) | – | +++ | ++ | ++ | – | – | ND |
| IRF-2 (332–349) | – | + | – | – | – | – | ND |
| IRF-2[S] (1–347) | – | – | – | – | – | – | ND |
PJ69-4a-derivative yeast strains transformed with the indicated ‘bait’ proteins as GAL4 DBD fusions were mated with PJ69-4α-derivatives transformed with the indicated ‘prey’ proteins as GAL4 AD fusions (see Materials and Methods). Successful matings were selected on SD-Leu-Trp and diploid colonies were streaked in triplicate onto SD-Leu-Trp, SD-Leu-Trp-His and SD-Leu-Trp-Ade and their growth was monitored. Absence of interaction resulted in no growth after 6 days on SD-Leu-Trp-His and SD-Leu-Trp-Ade (indicated by ‘–’); positive interactions were graded according to the following scale: + = growth on SD-leu-trp-his, but no growth on SD-leu-trp-ade after 6 days; ++ = abundant growth on SD-leu-trp-his and limited growth on SD-leu-trp-ade after 4 days; +++ = abundant growth on SD-leu-trp-his and on SD-leu-trp-ade after 4 days; ++++ = abundant growth on SD-leu-trp-his and on SD-leu-trp-ade after 2 days; ND = interaction not determined. All diploids showed equivalent growth on SD-Leu-Trp indicating that growth rates are unaffected by competition for yeast transcription factors (‘squelching’). The relative expression levels of the GAL4-IRF-2 fusions were determined by western blotting and found to be comparable (data not shown). Attempts to quantitate the strengths of interaction using the GAL7-lacZ reporter gene in the PJ69 yeast strains (32) were unsuccessful due to high background β-galactosidase activity.
Yeast two-hybrid library screen
Saccharomyces cerevisiae PJ69-4a (MATa, ura3-52, his3-200, ade2-101, trp 1-901, leu2-3, 112, gal4Δ, gal80Δ, GAL2-ADE2, LYS2::Gal1-HIS3 met2::GAL7-lacZ) (32) was transformed (33) with the IRF-2 bait plasmid pHON1.IRF-2[292–349]. This strain was transformed with a human placental cDNA library in the GAL4 AD vector pGAD10 (Clontech catalogue number HL4003AB) and selected on SD medium plus 2 mM 3-aminotriazole but lacking tryptophan, leucine and histidine. From a total of 107 yeast colonies screened (equivalent to a 3–4-fold representation of the library), 209 colonies were selected, and of these, 43 also grew on SD medium lacking adenine. Plasmid DNA was recovered from each of these colonies by marker rescue in E.coli KC8 (hsdR, leuB600, trpC9830, pyrF::Tn5, hisB463, lacΔX74, strA, galU, K), transformed into PJ69-4α (32) and the individual transformants were mated with PJ69-4a carrying either pHON1 or pHON1.IRF-2[292–349] and the diploids tested for the ability to rescue growth on SD lacking adenine. DNA from 36 primary yeast colonies rescued adenine auxotrophy in a bait-dependent manner. The cDNA inserts were found to fall into four classes (see Fig. 3A). To confirm the interactions, the cDNA insert from a representative of each class was fused in-frame to the DBD of GAL4 (in pHON1) and tested for the ability to rescue adenine auxotrophy in combination with the C-terminal repression domain of IRF-2 fused in-frame to the AD of the yeast GAL4 protein (pHON3.IRF-2[292–349]). The cDNA inserts from the pGAD10 clones were sequenced using a combination of manual and automated (Oswel and Lark Technologies) dideoxy sequencing.
Figure 3.

The repression domain of IRF-2 interacts with two novel factors, IRF-2BP1 and IRF-2BP2. (A) Organisation of clones isolated for IRF-2BP1, IRF-2BP2A and IRF-2BP2B. The yeast two-hybrid cDNA library screen generated five independent isolates of a 2579 bp insert that encodes IRF-2BP1. This sequence derives from an uncharacterised gene on chromosome 19 (GenBank accession number AC008623, nucleotides 134839–137352). The first in-frame ATG is located 348 bp from the 5′ end of the insert; this was determined to be the initiating methionine, as an in-frame stop codon is present 600 bp upstream from this in the genomic sequence, and no EST clones have been identified containing upstream sequences. The ATG is followed by an uninterrupted open reading frame of 584 amino acids followed by a 3′UTR, polyadenylation signal (AATAAA) and a poly(A) tail. A single isolate of the insert shown for IRF-2BP2A was obtained. The insert comprises ∼2500 bp derived from an uncharacterised gene on chromosome 1 (GenBank accession numbers AL161640, nucleotides 28362–30847, and AL160408 nucleotides 4723–2239). Due to the high G-C content of the 5′ end of this insert (and that for IRF-2BP2B; see below) we were unable to obtain accurate sequence data at the 5′ end and we note that the available sequences of both genomic clones and the ESTs stop in the same place. We have tentatively assigned the initiator ATG for IRF-2BP2A on the basis that: (i) the sequence is identical to IRF-2BP2B and very similar to IRF-2BP1 in this region; (ii) the encoded protein initiating from this ATG is highly homologous to proteins found in Drosophila and C.elegans proteins (see text and B); (iii) the assigned initiator ATG conforms to the consensus Kozak sequence; (iv) such G-C-rich sequences are frequently found in 5′UTRs. The ATG is followed by an open reading frame of 587 amino acids, which is terminated by a 3′ TAG stop codon; this cDNA clone does not have a poly(A) tail. Two independent isolates of the 2600 bp insert encoding IRF-2BP2B were obtained. IRF-2BP2A and IRF-2BP2B differ in the size of the single intron, and thus represent alternative splice forms of a single gene. IRF-2BP2B lacks 48bp, representing 16 amino acids, that are present in IRF-2BP2A. The presumptive splice acceptor site is the same in both inserts (AL161640 nucleotide 30026) and conforms to the consensus of 10 pyrimidines followed by NCAG. The presumptive donor sites are AGGT at nucleotide 29433 for IRF-2BP2A and AGGC at nucleotide 29385 for IRF-2BP2B. We also recovered 28 independent isolates of the indicated insert of 775 bp that encodes a 3′ region common to both IRF-2BP2A and IRF-2BP2B. (B) The primary amino acid sequences of IRF-2BP1 and IRF-2BP2 are aligned with each other using the BLAST pairwise programme and common residues are highlighted in black (functionally conserved residues are highlighted in grey). The asterisk-underlined sequence shown in IRF-2BP2A is not present in IRF-2BP2B. The conserved zinc finger and C3HC4 RING domains at the N- and C-terminus, respectively, are boxed and the cysteines and histidines are indicated. Homologies with the hypothetical human C14orf4 (polyQ) protein, and the C.elegans MO4G12 and the D.melanogaster CG11138 and CG1855 gene products in these regions are indicated.
Immunoprecipitation and western blotting
Cell lysates were prepared in RIPA buffer (∼5 × 106 cells in 500 µl), and 100 µl aliquots were pre-cleared using pre-immune serum and protein A–Sepharose (Pharmacia) using standard procedures. One microlitre of anti-IRF-2 (34) was added to the supernatant and incubated at 4°C for 1 h, and then specific complexes were collected on protein A–Sepharose beads by centrifugation. The beads were washed four times in 1 ml RIPA buffer, then resuspended in 20 µl 2× Laemmli’s buffer. Samples were heated to 95°C for 5 min, fractionated by SDS–PAGE, blotted onto 0.45 µ PVDF, probed with a monoclonal antibody raised against the SV5 PK epitope tag and visualised using enhanced chemiluminescence according to the manufacturers’ recommendations (Amersham). GAL4 DBD fusions were detected in extracts from yeast or transiently transfected 293 cells using a monoclonal antibody purchased from Clontech (catalogue number 5399-1) under the manufacturer’s recommended conditions.
Transfection and analysis of gene expression
HeLaE cells (E.Laufer, ICRF) were cultured and transfected as described previously (35). For all transfections except that shown in Figure 5C (see figure legend), plasmids were mixed in a relative mass ratio of three parts reporter construct:three parts transfection control construct:four parts effector. Luciferase and β-galactosidase levels were determined as described previously (22). Cellular RNA was prepared from transiently transfected cells and analysed by RNase protection using human α-globin- and β-globin-specific probes (28). Where indicated, cells were treated with 200 nM trichostatin A (Sigma) in DMSO or 0.01% v/v DMSO as a control for 26 h prior to harvesting.
Figure 5.

IRF-2BP1, IRF-2BP2A and IRF-2BP2B can act as transcriptional co-repressors. (A) pSV40(GALUAS)5tkΔ(–39)lucter or (B) p(GALUAS)5tkΔ(–39)lucter were co-transfected into HeLa cells with either pEF.GAL147, pEF.GAL.IRF-2BP1, pEF.GAL.IRF-2BP2A, pEF.GAL.IRF-2BP2B or pEF.GAL.IRF-2BP2[456–587]), and the β-galactosidase expression vector, pJATlac. (C) Four micrograms of pSV40(GALUAS)5tkΔ(–39)lucter were co-transfected using calcium phosphate co-precipitation into HeLa cells with 4 µg of pEF.GAL.IRF-2[88–349], 4 µg of the β-galactosidase expression vector, pJATlac and increasing amounts of either full-length IRF-2BP1 (pEF.IRF-2BP1) or IRF-2BP2A (pEF.IRF-2BP2A) as indicated (the total amount of plasmid DNA being made up to a total of 30 µg with the ‘empty’ effector vector, pEFplink2). Reporter activity was determined, and corrected for squelching by dividing the results by the values obtained for an identical experiment using pSV40(lacP/O)tkΔ(–39)lucter as the reporter. (D) The IRF-dependent reporter, p[(AAGTGA)4]5tkΔ(–39)lucter was co-transfected into HeLa cells with either pEF.plink2 (lane 1), pEF.IRF-1 (lane 2), pEF.IRF-1 plus pEF.IRF-2 (lane 3), pEF.IRF-1 plus pEF.IRF-2BP1 (lane 4), pEF.IRF-1 plus pEF.IRF-2BP2A (lane 5) or pEF.IRF-1 plus pEF.IRF-2.BP2B (lane 6), and the β-galactosidase expression vector, pJATlac. For each of (A) to (D), luciferase and β-galactosidase activities were determined from cellular extracts and relative expression values calculated accordingly (expressed relative to the level of the vector-only sample = 1.0). Values shown represent data from three independent experiments and averages with error bars are shown.
RESULTS
The IRF-2 C-terminal domain can repress both activated and basal transcription
Although it has been shown previously that C-terminal fragments of IRF-2 can inhibit adjacent activators when fused to a heterologous DBD, the precise requirements for repression appear to vary depending upon the transcriptional activator used (17,18). To clarify this situation we initially examined the ability of GAL4-IRF-2 fusions to repress transcription from a promoter containing a synthetic multimer of the DNA binding site for GAL4 inserted between the TATA box of HSV-1 and the SV40 enhancer [pSV40(GALUAS)5tkΔ(–39)lucter]. Figure 2A shows that a fusion with IRF-2 amino acids 88–349 [GAL.IRF- 2(88–349)] represses SV40-driven transcription in contrast to the properties of the GAL4 DBD alone (compare lanes 1 and 3); as expected, a fusion between GAL4 and IRF-1 [GAL.IRF-1(105–325)] strongly activated transcription (lane 2). These effects were dependent upon GAL.IRF-2(88–349) recruitment to the promoter since repression was not seen when the multimeric GAL UAS was replaced with a random sequence of DNA of similar length [pSV40(lacP/O)tkΔ(–39)lucter; data not shown].
Figure 2.

IRF-2 contains a C-terminal domain that can repress both activated and basal transcription. (A) pSV40(GALUAS)5tkΔ(–39)lucter was co-transfected into HeLa cells with a mammalian expression plasmid driving over-expression of the GAL4 DBD (pEF.GAL147–GAL DBD) or a fusion between the GAL4 DBD and the putative effector domains of IRF-1 (pEF.GAL.IRF-1[105–325]) or fragments of IRF-2 (amino acids as indicated), and the β-galactosidase expression vector, pJATlac. (B) p(GALUAS)5tkΔ(–39)lucter was co-transfected into HeLa cells with the indicated effector plasmid and pJATlac. For both (A) and (B), luciferase and β-galactosidase activities were determined from cellular extracts and relative expression values calculated accordingly (expressed relative to the level of the vector-only sample = 1.0). Values shown represent data from between three and eight independent experiments and averages with error bars are shown. (C) Expression levels of GAL4 DBD fusions were determined by western blotting (see Materials and Methods). (D) p(GALUAS)5tkΔ(–39)β-globin] was co-transfected into HeLa cells with either pEF.GAL147 (GAL DBD), pEF.GAL.IRF-2[88–349]) (GAL.IRF-2) or pEF.GAL.IRF-1[105–325] (GAL.IRF-1), and πSVHSα118 as a co-transfection control. RNA was prepared from transfections and mapped by RNase protection with α- or β-globin-specific probes.
Removal of the C-terminal 69 amino acids [GAL.IRF-2(88–280)] led to a complete loss of repression (Fig. 2A, lane 4), indicating, as expected, that residues in this region contribute to negative regulation. Constructs containing as few as 58 amino acids from the extreme C-terminus [GAL.IRF-2(292–349)] are capable of repressing transcription to the same extent as the full-length protein, whilst a fusion containing the C-terminal 18 amino acids [GAL.IRF-2(332–349)] only showed ∼30% of this repression (Fig. 2A, lanes 5–8). Removal of the nine C-terminal-most amino acids from the GAL.IRF-2(292–349) construct [GAL.IRF-2(292–340)] had no effect, whilst removal of the 19 C-terminal-most amino acids [GAL.IRF-2(292–330)] completely eliminates repression (Fig. 2A, lanes 9 and 10). Thus, amino acids 292–340 of IRF-2 contain an independently acting repression domain, which is strongly conserved between species (see Fig. 1C).
We also investigated whether GAL4-IRF-2 fusions could repress transcription from a minimal promoter (i.e. a promoter that lacks identifiable upstream activator sequences). Figure 2B shows that GAL.IRF-2(88–349) is capable of repressing transcription from a promoter containing a synthetic multimer of the DNA binding site for GAL4 placed upstream of the TATA box of HSV-1 [p(GALUAS)5tkΔ (–39)lucter]; the repressive effects of other GAL4.IRF-2 fusions on this promoter were very similar to those seen on SV40-driven transcription, although repression by GAL.IRF-2(332–349) was slightly more efficient on the minimal promoter. The ability of IRF-2 to mediate repression of a minimal promoter has not been reported previously.
To confirm that the effects of GAL-IRF fusions are operating at the level of transcription, we analysed RNA from transient transfections of the fusions and a reporter gene consisting of a human β-globin gene under the control of a minimal promoter [p(GALUAS)5tkΔ(–39)β-globin]. Figure 2D shows that transcription from this promoter is markedly repressed by GAL.IRF-2(88–349); as expected, transcription is strongly stimulated by expression of GAL.IRF-1(105–325).
Isolation of cDNA clones encoding proteins that interact with the C-terminal repression domain of IRF-2
We next sought to identify interacting partners for the C-terminus of IRF-2 using the yeast two-hybrid system. Thirty-six colonies containing verifiable bait-dependent partners were isolated from a total of 107 yeast colonies of a human placental cDNA library screened with a fusion containing IRF-2 amino acids 292–349 (see Materials and Methods). The cDNA inserts in these colonies were mapped and sequenced and shown to encode two novel related products [referred to hereafter as IRF-2-binding proteins 1 and 2; IRF-2BP1 (GenBank accession number AY278022) and IRF-2BP2], with two distinct splicing isoforms of IRF-2BP2 being isolated [IRF-2BP2A (GenBank accession number AY278023) and IRF-2BP2B (GenBank accession umber AY278024) that lacks 16 amino acids present in IRF-2BP2A; see Fig. 3A].
The translated ORFs of IRF-2BP1 and IRF-2BP2A encode proteins of 584 and 587 amino acids, respectively, with IRF-2BP2B encoding 571 amino acids. IRF-2BP1 and IRF-2BP2A/B are related proteins with two highly conserved regions (Fig. 3B); at the N-terminus there is a 64 amino acid domain which contains a single C4 zinc finger motif, and at the C-terminus the final 82 amino acids encompass a C3HC4 or RING finger. The IRF-2BPs are also related to a third human cDNA that encodes a hypothetical protein of unknown function called C14orf4 (36), and to the Caenorhabditis elegans MO4G12 and the Drosophila melanogaster CG11138 and CG1855 gene products, also of unknown function. The C-terminal RING finger of IRF-2BP2 is sufficient for the interaction with IRF-2 since we had isolated an insert encoding amino acids 456–587 of IRF-2BP2A/B as a primary interacting clone in our library screen.
The specificity of interaction of IRF-2BP1, IRF-2BP2A and IRF-2BP2B with other members of the IRF family was investigated using the yeast two-hybrid assay. Whilst all three proteins could interact with GAL.IRF-2 fusions containing a variety of C-terminal IRF-2 fragments (see below), they were not capable of interacting with fusions with the C-terminal effector domains of IRF-1 or IRF-9 (Table 1). Furthermore, interactions could not be observed with non-IRF preys (Jak-1, STAT1, PKR or the p65 subunit of NF-κB; Table 1). IRF-2BP1, IRF-2BP2A and IRF-2BP2B failed to interact with themselves or each other indicating that they may not be able to form dimers.
The IRF-2 domain dependence for IRF-2BP binding was investigated in more detail. None of IRF-2BP1, IRF-2BP2A and IRF-2BP2B interacted with IRF-2 fragments lacking the C-terminal region (Table 1). These data show that the C-terminal 58 amino acids of IRF-2 are necessary and sufficient for interaction with the IRF-2BPs. Although all of the IRF-2BP fusions interacted with the IRF-2 repression domain encoding amino acids 292–349, IRF-2BP1 was capable of interacting with fusions containing just the C-terminal 18 amino acids of IRF-2 (amino acids 332–349), albeit weakly (Table 1).
The fact that multiple copies of cDNAs encoding two highly related IRF-2-interacting partners were the only factors isolated from a library screen of ∼4-fold transcriptome equivalents strongly suggests that the IRF-2BPs are physiologically important partners for IRF-2. To verify interaction with IRF-2 in vivo we constructed vectors that permitted mammalian expression of epitope-tagged forms of IRF-2BP1, IRF-2BP2A or IRF-2BP2B, and transiently transfected them into HeLa cells. Whole cell extracts were prepared from these cells and immunoprecipitated with a rabbit polyclonal antibody raised against IRF-2 (34) and the immunoprecipitates were fractionated on an SDS–polyacrylamide gel and analysed by western blotting using a monoclonal antibody that recognised the epitope tag. Figure 4A shows that each of the IRF-2BPs is co-immunoprecipitated, indicating a strong interaction with IRF-2. To determine the sub-cellular localisation of the IRF-2BPs, transiently transfected cells were harvested and both cytoplasmic and nuclear extracts prepared and analysed by western blotting. Figure 4B shows that, as expected for factors that interact with IRF-2 and potentially regulate transcription, each of IRF-2BP1, IRF-2BP2A and IRF-2BP2B is localised to the nucleus.
Figure 4.

IRF-2BP1, IRF-2BP2A and IRF-2BP2B interact with IRF-2 in vivo and are nuclear proteins. (A) HeLa cells were transiently transfected with pEF.plink2, pEF.IRF-2BP1, pEF.IRF-2BP2A or pEF.IRF-2BP2B and whole cell lysates prepared. An aliquot of each lysate was fractionated on a SDS–PAGE gel and analysed by western blotting using a monoclonal antibody against the SV5 epitope tag (left hand panel). To determine association with cellular IRF-2, whole cell extracts were immunoprecipitated with rabbit polyclonal anti-human IRF-2 serum and the immunoprecipitates were analysed by western blotting with SV5 epitope tag antibody (right hand panel). (B) HeLa cells were transiently transfected with pEF.plink2, pEF.IRF-2BP1, pEF.IRF-2BP2A or pEF.IRF-2BP2B and extracts from cytoplasmic (C) and nuclear (N) subcellular fractions were prepared. An aliquot of each extract was fractionated on an SDS–PAGE gel and analysed by western blotting using a monoclonal antibody against the SV5 epitope tag.
IRF-2BP1 and IRF-2BP2 are novel co-repressor proteins
To investigate the function of IRF-2BP1, IRF-2BP2A and IRF-2BP2B we initially examined their effect on transcription when tethered to promoters through the DBD of GAL4. IRF-2BP1, IRF-2BP2A and IRF-2BP2B are all capable of specifically repressing transcription on both SV40-driven (Fig. 5A) and minimal (Fig. 5B) promoters. The magnitude of repression is similar to that seen with a GAL4 fusion to a fragment of IRF-2 spanning amino acids 88–349. Interestingly, although a fusion with just the RING domain of IRF-2BP2 (amino acids 456–587) is unable to repress transcription from the SV40-driven promoter (Fig. 5A), it retains the ability to repress transcription from a minimal promoter (Fig. 5B). The repression effects were dependent upon recruitment of GAL4 fusion proteins to the promoter since repression was not seen when the multimeric GAL UAS was replaced with a random sequence of DNA [pSV40(lacP/O)tkΔ(–39)lucter], nor with reporters under the control of other promoters lacking a GAL UAS [ptkΔ(–39)lucter; ptkΔ(–105)lucter; data not shown].
To test whether IRF-2BP1/2 retain the ability to repress transcription when recruited to DNA indirectly through association with IRF-2, we examined whether they could enhance the degree of repression exerted by GAL.IRF-2 fusions. Figure 5C shows that repression by GAL.IRF-2 could be significantly enhanced by co-expression of IRF-2BP1 or IRF-2BP2A (IRF-2BP2B was not tested in this experiment). We conclude from this that IRF-2BP1 and IRF-2BP2A can repress transcription by their recruitment to promoters by IRF-2.
In order to test whether the IRF-2BPs affect transcriptional regulation through IRFs bound to their cognate DNA binding sites we investigated the behaviour of the IRF-dependent promoter, [p[(AAGTGA)4]5tkΔ(–39)lucter], in transient transfection experiments. We have shown previously that the activity of this reporter in HeLa cells represents a balance of the opposing activities of IRF-1 and IRF-2 (23). Figure 5D (lane 1) shows that in the absence of co-transfected IRF expression plasmids this reporter is only weakly active; this observation was expected since HeLa cells contain abundant IRF-2 but no detectable IRF-1 (34). The activity is strongly stimulated by IRF-1 expression (Fig. 5D, lane 2), and this stimulation is antagonised by co-expressing IRF-2 (Fig. 5D, lane 3). Co-transfection of expressing plasmids for IRF-2BP1 (Fig. 5D, lane 4), and IRF-2BP2A (Fig. 5D, lane 5), reduced IRF-1-dependent activation to 20–30%, whilst IRF-2BP2B (Fig. 5D, lane 6), reduced activation to ∼60%. Since the IRF-2BPs were unable to influence the transcriptional activity of IRF-1 when examined as a GAL4 fusion (the function of which is independent of IRF-2; data not shown), and were unable to interact with IRF-1 in the yeast two-hybrid assay (Table 1), the effects of IRF-2BP1, IRF-2BP2A and IRF-2BP2B are likely acting through endogenous IRF-2, and thus they repress transcription in an IRF-2-dependent manner.
Repression by IRF-2 and the IRF-2BP co-repressors is still seen in the presence of a histone deacetylase inhibitor
Many transcriptional repressors exert their effect either directly or indirectly through the deacetylation of histones in the vicinity of their binding site [reviewed in (37)]. To test whether IRF-2 and its associated co-repressors work in this manner we treated cells with the histone deacetylase (HDAC) inhibitor trichostatin A. Figure 6 shows that repression of SV40-driven transcription by the GAL.IRF-2(88–349) fusion is unaffected by trichostatin A suggesting that it is HDAC-independent. Although transcriptional repression by GAL4 fusions with IRF-2BP1, IRF-2BP2A or IRF-2BP2B was seen in the presence and absence of trichostatin A, repression by IRF-2BP1 and IRF-2BP2A is partially relieved by the addition of trichostatin A. This effect was not seen with IRF-2BP2B, which retained full repression in the presence of trichostatin A. These results suggest that the IRF-2BPs must exert a significant part of their repressive activity through a mechanism(s) that is independent of histone deacetylation.
Figure 6.
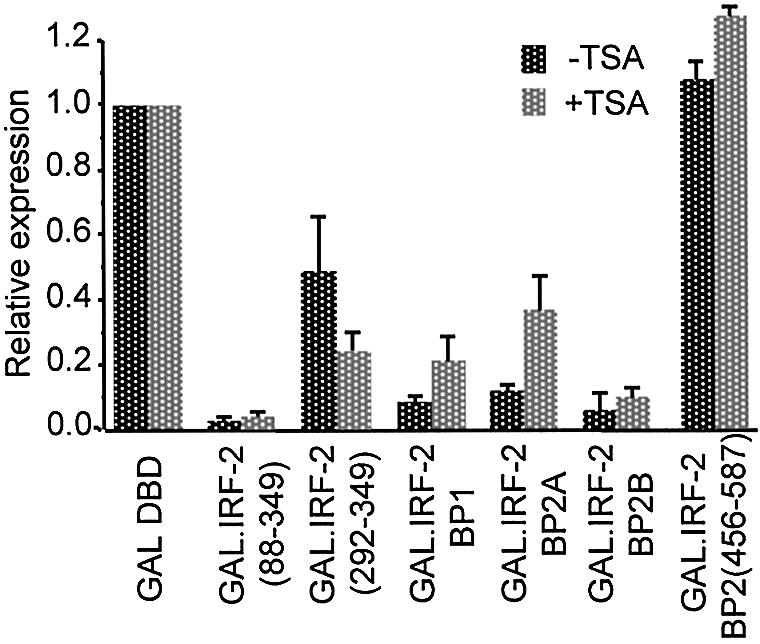
IRF-2BP1- and IRF-2BP2-mediated repression does not require histone deacetylase activity. pSV40(GALUAS)5tkΔ(–39)lucter was co- transfected into HeLa cells with either pEF.GAL147, pEF.GAL.IRF-2[88–349], pEF.GAL.IRF-2(292–349), pEF.GAL.IRF-2BP1, pEF.GAL.IRF-2BP2A, pEF.GAL.IRF-2BP2B or pEF.GAL.IRF-2BP2[456–587]), and the β-galactosidase expression vector, pJATlac. Cells were incubated with the histone deacetylase inhibitor trichostatin A (200 nM) where indicated, for 26 h prior to harvesting. Control cells not treated with trichostatin A were treated with 0.01% v/v DMSO. Luciferase and β-galactosidase activities were determined from cellular extracts and relative expression values calculated accordingly (expressed relative to the level of the vector-only sample = 1.0). In the presence of trichostatin A both luciferase and β-galactosidase activities were increased relative to the DMSO control. This global effect has been observed by others (57). Therefore, repression is expressed relative to the GAL4 DBD alone in each case. Values shown represent data from three independent experiments.
An isoform of IRF-2, IRF-2[S], which lacks two amino acids, cannot interact with the IRF-2BPs nor repress transcription
During the course of our investigations we identified a form of IRF-2 that lacks valine 177 and valine 178; this alternative form of IRF-2 lacks 6 bp that derive from the exon 5/exon 6 junction and has recently been described by others (38), and according to their convention we refer to this alternative form as IRF-2[S]. It was reported that IRF-2[S] is a less efficient transcriptional activator of the CIITA promoter than IRF-2 (38). In order to test whether the repressive activities of IRF-2[S] and IRF-2 are also different we constructed a GAL.IRF-2[S] fusion containing the equivalent of amino acids 88–349 [GAL.IRF-2[S](88–347)] and compared its ability to repress transcription with that seen with GAL.IRF-2(88–349). Figure 7A and B show that the IRF-2[S] fusion is unable to repress transcription from either the enhancer-activated or basal reporter genes. This result is surprising since both forms of IRF-2 contain an identical C-terminal repression domain (292–349) that is sufficient for repression when fused to GAL4 and sufficient for strong interaction with the IRF-2BPs.
Figure 7.

An isoform of IRF-2, IRF-2[S], which lacks two amino acids, cannot repress transcription. (A) pSV40(GALUAS)5tkΔ(–39)lucter or (B) p(GALUAS)5tkΔ(–39)lucter was co-transfected into HeLa cells with either pEF.GAL147, pEF.GAL.IRF-1[105–325], pEF.GAL.IRF-2[88–349] or pEF.GAL.IRF-2(S)[88–347], and the β-galactosidase expression vector, pJATlac. Luciferase and β-galactosidase activities were determined from cellular extracts and relative expression values calculated accordingly (expressed relative to the vector only sample = 1.0). Values shown represent data from three separate experiments and averages with error bars are shown. (C) p(GALUAS)5tkΔ(–39)β-globin was co-transfected into HeLa cells with either pEF.GAL147, pEF.GAL.IRF-2[88–349] or pEF.GAL.IRF-2(S)[88–347], and πSVHSα118 as a co-transfection control. RNA was prepared from transfections and mapped by RNase protection with α- or β-globin-specific probes.
To confirm that the effects of GAL.IRF fusions are operating at the level of transcription, we analysed RNA from transient transfections of the fusions and a reporter gene consisting of a human β-globin gene under the control of a minimal promoter. Figure 7C shows that transcription from this promoter is markedly repressed by GAL.IRF-2(88–349), whereas the IRF-2[S] counterpart, GAL.IRF-2[S](88–347)], is without effect.
One explanation for the difference in the negative regulatory properties of IRF-2 and IRF-2[S] may be that the C-terminal repression domains are in different conformations. Structural predictions for IRF-2 suggest that the loss of V177 and V178 disrupts the secondary structure in this region of IRF-2[S], and generates a protein which has an alternative β-sheet structure (38). Whilst this putative structural change does not affect the ability of IRF-2[S] to bind to DNA, it may influence the conformation of other domains and this has been suggested to account for the difference in transactivation properties of IRF-2 and IRF-2[S] on the CIITA promoter (38). To test whether the C-terminal repression domains of IRF-2 and IRF-2[S] differ in their ability to recruit the IRF-2BPs, we constructed a GAL.IRF-2[S] fusion that could be used in the yeast two-hybrid assay and investigated the interaction between this and the IRF-2BPs. Unlike the equivalent GAL.IRF-2 fusion, GAL.IRF-2[S] was incapable of interacting with any of IRF-2BP1, IRF-2BP2A or IRF-2BP2B (Table 1), demonstrating that the C-terminal repression/IRF-2BP-interacting domain is non-functional in the context of the IRF-2[S] protein. We also noted that the interaction between the IRF-2BPs and a GAL4 fusion with full-length IRF-2 (GAL.IRF-2[1–349]) is weaker than that with GAL.IRF-2(292–349), consistent with IRF-2 N-terminal sequences having an effect on interactions with the C-terminus (Table 1).
DISCUSSION
We have identified two novel nuclear factors, IRF-2BP1 and IRF-2BP2 (comprising two closely related splicing isoforms, A and B), which interact with the C-terminal 58 amino acids of IRF-2. This region of IRF-2 is a fully independent transcriptional repression domain and IRF-2BP1, IRF-2BP2A and IRF-2BP2B have the properties of co-repressor molecules that act by recruitment to DNA through this domain. Although this domain is intact in the IRF-2[S] isoform, the loss of valines 177 and 178 prevents interaction with IRF-2BP1, IRF-2BP2A and IRF-2BP2B leading to loss of transcriptional repression. These data independently verify the importance of IRF-2BP1, IRF-2BP2A and IRF-2BP2B as transcriptional co-repressors. An understanding of the biological functions of IRF-2[S] awaits further work.
IRF-2-BP1 and IRF-2BP2A/B have an N-terminal C4 zinc finger and a C-terminal C3HC4 RING finger, and these features are conserved between the IRF-2BPs and the uncharacterised human C14orf4 gene product (36). This sequence is sufficiently related to the IRF-2BPs that it is possible that the C14orf4 gene product also interacts with IRF-2, but we have not tested this. The IRF-2BPs are also related to the C.elegans MO4G12 and the D.melanogaster CG11138 and CG1855 gene products. In none of these cases has the function been ascertained; since IRF homologues do not occur in C.elegans or D.melanogaster the IRF-2BPs may be members of a wider group of proteins that interact with a range of transcription factors.
The C3HC4 RING is a feature found in diverse families of proteins (including some co-repressors) (39–41) and serves as a domain for protein–protein interaction [reviewed in (42)]. A major function of the RING finger in some of these proteins is to serve as a bridge between E2 and E3 ubiquitin ligases [reviewed in (43)], and in the case of NOT4 this activity is essential for repression by the CCR4–NOT complex (44). Furthermore, members of the protein inhibitor of activated STATs (PIAS) family have recently been shown to act as SUMO E3 ligases; the transfer of SUMO to a number of transcriptional activators has been shown to alter their nuclear distribution and correlates with a down-regulation in their activity (45–51). Although we have yet to determine whether IRF-2BP1, IRF-2BP2A and IRF-2BP2B have ubiquitin or SUMO ligase activity it should be stressed that they have no homology with known ligases.
The RING finger motif of IRF-2BP2A/B (amino acids 456–587) is sufficient to interact with the C-terminus of IRF-2, but is not sufficient to repress enhancer-driven transcription, implying that some other region of IRF-2BP2A/B (and presumably IRF-2BP1) is responsible for repression. Additional regions of IRF-2BP1 and IRF-2BP2 have features in common with other repressors or co-repressors. Immediately after the N-terminal conserved C4 zinc finger is a segment rich in alanine, glutamine and proline residues (also present in the C14orf4 gene product where the extended alanine and glutamine stretch is inserted). There are also several clusters of prolines, another feature that is commonly found in or near to repression domains, as evidenced in p53 (52), Groucho (53) and HNF4 (54).
Since C4 type zinc fingers are typically found in DBDs of proteins, it is possible that the N-terminal zinc finger of IRF-2BP1/2 can bind to DNA. If this is the case, then its role may be to stabilise the interaction of the IRF-2–IRF-2BP1/2 complex with the target promoter. It is also possible that this domain could mediate an interaction with another protein, either a transcription factor bound to the promoter adjacent to IRF-2 or a component of the basal transcriptional machinery.
Studies on co-repressors indicate that they often either possess or recruit HDAC activities to the target promoter and thereby modify local nucleosome structure to make the promoter less accessible to the transcriptional machinery [reviewed in (37)]. Our experiments indicate that repression by GAL.IRF-2 fusions containing amino acids 88–349 of IRF-2 and by GAL.IRF-2BP2B is unaffected by an inhibitor of HDAC activity, and that repression by GAL.IRF-2BP1 and GAL.IRF-2BP2A is only partially sensitive to this inhibitor. These experiments suggest that repression by IRF-2 and its associated co-repressors does not depend upon histone deacetylation. This is consistent with observations on several other repressors (55–57). It is possible that the low level of HDAC-dependency seen with IRF-2BP1 and IRF-2BP2A reflects an activity that is required for repression when recruited to DNA through some transcription factor other than IRF-2.
Since the activity of the IRF-2BPs is largely independent of HDAC activity, repression must be achieved by other means. Several examples have been described in which repressors or co-repressors act by preventing the formation of a stable initiation complex. For example, the N-terminal repression domain of N-CoR has been shown to prevent the interaction between TFIIB and TAFII32 that is required for transcriptional activation (58). We note that IRF-2BP1 can bind (albeit weakly) to a sub-region of the C-terminal repression domain of IRF-2 (amino acids 332–349), and that this IRF-2 sub-domain is sufficient to mediate repression of a minimal promoter, consistent with a repressor that is functioning by interference with the basal transcriptional machinery. We also note that at very high levels of expression, IRF-2BP1 exerts a limited degree of interference with promoters to which it should not bind (data not shown); this behaviour might be explained by sequestering some essential component of the basal transcriptional apparatus (commonly referred to as squelching). It is interesting to note that IRF-2 has been reported to interact with TFIIB (20); perhaps the IRF-2BPs prevent an interaction between IRF-2-bound TFIIB and another transcriptional co-factor. The IRF-2BPs might also act by preventing the recruitment or activity of the histone acetylases GCN5, PCAF and p300 to promoters by IRF-2 (19,59).
In a previous analysis of the repressor functions of IRF-2, Senger et al. were unable to identify a single domain that was capable of repressing a range of different GAL4-transactivator fusion proteins tethered to the DNA through a common site (18). Instead, the magnitude of repression gradually diminished as more of IRF-2 was removed, with some activators requiring larger fragments of IRF-2 than others. The authors proposed that basic amino acids in the IRF-2 repression domain build a positively charged microenvironment that repels activator-recruited incoming CBP, or destabilises the CBP-activator interactions (18). Such a model is intrinsically devoid of requirement for co-repressors. These results might also be explained if IRF-2 makes specific direct or indirect contacts with several individual components of the transcription machinery (and with chromatin and chromatin-modifying components) as seen for conventional transcriptional repressors. The identification of the IRF-2BPs as IRF-2-dependent co-repressors is consistent with this hypothesis.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Philip James for yeast strains PJ69-4a and PJ69-4α, Dave Slack, Jo Brown and Peter King for reagents and valuable discussions and Melanie Lee, Kathy Weston, John McCauley, Paula Barnard and Emma Poole for comments on the manuscript. We thank the Cancer Prevention Research Trust for support for this work, the Medical Research Council of Great Britain for a postgraduate student award for K.S.C., and The Wellcome Trust for a University Award to S.G.
REFERENCES
- 1.Taniguchi T., Ogasawara,K., Takaoka,A. and Tanaka,N. (2001) IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol., 19, 623–655. [DOI] [PubMed] [Google Scholar]
- 2.Harada H., Fujita,T., Miyamoto,M., Kimura,Y., Maruyama,M., Furia,A., Miyata,T. and Taniguchi,T. (1989) Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell, 58, 729–739. [DOI] [PubMed] [Google Scholar]
- 3.Hida S., Ogasawara,K., Sato,K., Abe,M., Takayanagi,H., Yokochi,T., Sato,T., Hirose,S., Shirai,T., Taki,S. and Taniguchi,T. (2000) CD8(+) T cell-mediated skin disease in mice lacking IRF-2, the transcriptional attenuator of interferon-alpha/beta signaling. Immunity, 13, 643–655. [DOI] [PubMed] [Google Scholar]
- 4.Harada H., Kitagawa,M., Tanaka,N., Yamamoto,H., Harada,K., Ishihara,M. and Taniguchi,T. (1993) Anti-oncogenic and oncogenic potentials of interferon regulatory factors-1 and -2. Science, 259, 971–974. [DOI] [PubMed] [Google Scholar]
- 5.Nguyen H., Mustafa,A., Hiscott,J. and Lin,R. (1995) Transcription factor IRF-2 exerts its oncogenic phenotype through the DNA binding/transcription repression domain. Oncogene, 11, 537–544. [PubMed] [Google Scholar]
- 6.Futaki M., Inokuchi,K., Hanawa,H., Tanosaki,S., Dan,K. and Nomura,T. (1996) Possible transforming activity of interferon regulatory factor 2 in tumorigenicity assay of NIH3T3 cells transfected with DNA from chronic myelogenous leukemia patients. Leuk. Res., 20, 601–605. [DOI] [PubMed] [Google Scholar]
- 7.Vaughan P.S., Aziz,F., van Wijnen,A.J., Wu,S., Harada,H., Taniguchi,T., Soprano,K.J., Stein,J.L. and Stein,G.S. (1995) Activation of a cell-cycle-regulated histone gene by the oncogenic transcription factor IRF-2. Nature, 377, 362–365. [DOI] [PubMed] [Google Scholar]
- 8.Vaughan P.S., van der Meijden,C.M.J., Aziz,F., Harada,H., Taniguchi,T., van Wijnen,A.J., Stein,J.L. and Stein,G.S. (1998) Cell cycle regulation of histone H4 gene transcription requires the oncogenic factor IRF-2. J. Biol. Chem., 273, 194–199. [DOI] [PubMed] [Google Scholar]
- 9.Luo W. and Skalnik,D.G. (1996) Interferon regulatory factor-2 directs transcription from the gp91 phox promoter. J. Biol. Chem., 271, 23445–23451. [DOI] [PubMed] [Google Scholar]
- 10.Schaefer B.C., Paulson,E., Strominger,J.L. and Speck,S.H. (1997) Constitutive activation of Epstein–Barr virus (EBV) nuclear antigen 1 gene transcription by IRF1 and IRF2 during restricted EBV latency. Mol. Cell. Biol., 17, 873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jesse T.L., LaChance,R., Iademarco,M.F. and Dean,D.C. (1998) Interferon regulatory factor-2 is a transcriptional activator in muscle where it regulates expression of vascular cell adhesion molecule-1. J. Cell Biol., 140, 1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xi H., Eason,D.D., Ghosh,D., Dovhey,S., Wright,K.L. and Blanck,G. (1999) Co-occupancy of the interferon regulatory element of the class II transactivator (CIITA) type IV promoter by interferon regulatory factors 1 and 2. Oncogene, 18, 5889–5903. [DOI] [PubMed] [Google Scholar]
- 13.Xi H., Goodwin,B., Shepherd,A.T. and Blanck,G. (2001) Impaired class II transactivator expression in mice lacking interferon regulatory factor-2. Oncogene, 20, 4219–4227. [DOI] [PubMed] [Google Scholar]
- 14.Xi H. and Blanck,G. (2000) Interferon regulatory factor-2 point mutations in human pancreatic tumors. Int. J. Cancer, 87, 803–808. [PubMed] [Google Scholar]
- 15.Escalante C.R., Yie,J., Thanos,D. and Aggarwal,A.K. (1998) Structure of IRF-1 with bound DNA reveals determinants of interferon regulation. Nature, 391, 103–106. [DOI] [PubMed] [Google Scholar]
- 16.Uegaki K., Shirakawa,M., Harada,H., Taniguchi,T. and Kyogoku,Y. (1995) Secondary structure and folding topology of the DNA binding domain of interferon regulatory factor 2, as revealed by NMR spectroscopy. FEBS Lett., 359, 184–188. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto H., Lamphier,M.S., Fujita,T., Taniguchi,T. and Harada,H. (1994) The oncogenic transcription factor IRF-2 possesses a transcriptional repression and a latent activation domain. Oncogene, 9, 1423–1428. [PubMed] [Google Scholar]
- 18.Senger K., Merika,M., Agalioti,T., Yie,J., Escalante,R.C., Chen,G., Aggarwal,K.A. and Thanos,D. (2000) Gene repression by coactivator repulsion. Mol. Cell, 6, 931–937. [DOI] [PubMed] [Google Scholar]
- 19.Masumi A., Wang,I.M., Lefebvre,B., Yang,X.J., Nakatani,Y. and Ozato,K. (1999) The histone acetylase PCAF is a phorbol-ester-inducible coactivator of the IRF family that confers enhanced interferon responsiveness. Mol. Cell. Biol., 19, 1810–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang I.M., Blanco,J.C., Tsai,S.Y., Tsai,M.J. and Ozato,K. (1996) Interferon regulatory factors and TFIIB cooperatively regulate interferon-responsive promoter activity in vivo and in vitro. Mol. Cell. Biol., 16, 6313–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Staal A., Enserink,J.M., Stein,J.L., Stein,G.S. and van Wijnen,A.J. (2000) Molecular characterization of celtix-1, a bromodomain protein interacting with the transcription factor interferon regulatory factor 2. J. Cell Physiol., 185, 269–279. [DOI] [PubMed] [Google Scholar]
- 22.King P. and Goodbourn,S. (1994) The β-interferon promoter responds to priming through multiple independent regulatory elements. J. Biol. Chem., 269, 30609–30615. [PubMed] [Google Scholar]
- 23.Whiteside S.T., King,P. and Goodbourn,S. (1994) A truncated form of the IRF-2 transcription factor has the properties of a postinduction repressor of interferon-β gene expression. J. Biol. Chem., 269, 27059–27065. [PubMed] [Google Scholar]
- 24.Flint K.J. and Jones,N.C. (1991) Differential regulation of three members of the ATF/CREB family of DNA binding proteins. Oncogene, 6, 2019–2026. [PubMed] [Google Scholar]
- 25.Treisman R. and Maniatis,T. (1985) Simian virus 40 enhancer increases number of RNA polymerase II molecules on linked DNA. Nature, 315, 72–75. [DOI] [PubMed] [Google Scholar]
- 26.Seed B. (1983) Purification of genomic sequences from bacteriophage libraries by recombination and selection in vivo. Nucleic Acids Res., 11, 2427–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masson N., Ellis,M., Goodbourn,S. and Lee,K.A.W. (1992) Cyclic-AMP response element-binding protein and the catalytic subunit of protein kinase A are present in F9 embryonal carcinoma cells but are unable to activate the somatostatin promoter. Mol. Cell. Biol., 12, 1096–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Treisman R.H. (1985) Transient accumulation of c-fos RNA following serum stimulation requires a conserved 5′ element and c-fos 3′ sequences. Cell, 42, 889–902. [DOI] [PubMed] [Google Scholar]
- 29.Marais R., Light,Y., Paterson,H.F. and Marshall,C.J. (1995) Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J., 14, 3136–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bartel P.L. and Fields,S. (1993) Analyzing protein-protein interactions using two-hybrid system. Methods Enzymol., 254, 241–263. [DOI] [PubMed] [Google Scholar]
- 31.Harper J.W., Adami,G.R., Wei,N., Keyomarsi,K. and Elledge,S.J. (1993) The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell, 75, 805–816. [DOI] [PubMed] [Google Scholar]
- 32.James P., Halladay,J. and Craig,E.A. (1996) Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics, 144, 1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gietz R.D., Schiestl,R.H., Willems,A.R. and Woods,R.A. (1995) Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast, 11, 355–360. [DOI] [PubMed] [Google Scholar]
- 34.Whiteside S.T., Visvanathan,K.V. and Goodbourn,S. (1992) Identification of novel factors that bind to the PRD1 region of the human β-interferon promoter Nucleic Acids Res., 20, 1531–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.King P. and Goodbourn,S. (1998) STAT1 is inactivated by a caspase. J. Biol. Chem., 273, 8699–8704. [DOI] [PubMed] [Google Scholar]
- 36.Rampazzo A., Pivotto,F., Occhi,G., Tiso,N., Bortoluzzi,S., Rowen,L., Hood,L., Nava,A. and Danieli,G.A. (2000) Characterization of C14orf4, a novel intronless human gene containing a polyglutamine repeat, mapped to the ARVD1 critical region. Biochem. Biophys. Res. Commun., 278, 766–774. [DOI] [PubMed] [Google Scholar]
- 37.Burke L.J. and Baniahmad,A. (2000) Co-repressors 2000. FASEB J., 14, 1876–1888. [DOI] [PubMed] [Google Scholar]
- 38.Merediz S.A., Schmidt,M., Hoppe,G.J., Alfken,J., Meraro,D., Levi,B.Z., Neubauer,A. and Wittig,B. (2000) Cloning of an interferon regulatory factor 2 isoform with different regulatory ability. Nucleic Acids Res., 28, 4219–4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Satijn D.P.E., Gunster,M.J., Van Der Vlag,J., Hamer,K.M., Schul,W., Alkema,M.J., Saurin,A.J., Freemont,P.S., Van Driel,R. and Otte,A.P. (1997) RING1 is associated with the polycomb group protein complex and acts as a transcriptional repressor. Mol. Cell. Biol., 17, 4105–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moosmann D., Georgiev,O., Le Douarin,B., Bourquin,J.-P. and Schaffner,W. (1996) Transcriptional repression by RING finger protein TIF1β that interacts with the KRAB repressor domain of KOX1. Nucleic Acids Res., 24, 4859–4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fedele M., Benvenuto,G., Pero,R., Majello,B., Battista,S., Lembo,F., Vollono,E., Day,P.M., Santoro,M., Lania,L., Bruni,C.B., Fusco,A. and Chiariotti,L. (2000) A novel member of the BTB/POZ family, PATZ, associates with the RNF4 RING finger protein and acts as a transcriptional repressor. J. Biol. Chem., 275, 7894–7901. [DOI] [PubMed] [Google Scholar]
- 42.Saurin A.J., Borden,K.L.B., Boddy,M.N. and Freemont,P.S. (1996) Does this have a familiar RING? Trends Biochem. Sci., 21, 208–214. [PubMed] [Google Scholar]
- 43.Glickman M.H. and Ciechanover,A. (2002) The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev., 82, 373–428. [DOI] [PubMed] [Google Scholar]
- 44.Albert T.K., Hanzawa,H., Legtenberg,Y.I., de Ruwe,M.J., van den Heuvel,F.A., Collart,M.A., Boelens,R. and Timmers,H.T. (2002) Identification of a ubiquitin-protein ligase subunit within the CCR4-NOT transcription repressor complex. EMBO J., 21, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jackson P.K. (2001) A new RING for SUMO: wrestling transcriptional responses into nuclear bodies with PIAS family E3 SUMO ligases. Genes Dev., 15, 3053–3058. [DOI] [PubMed] [Google Scholar]
- 46.Sachdev S., Bruhn,L., Sieber,H., Pichler,A., Melchior,F. and Grosschedl,R. (2001) PIASy, a nuclear matrix-associated SUMO E3 ligase, represses LEF1 activity by sequestration into nuclear bodies. Genes Dev., 15, 3088–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kotaja N., Karvonen,U., Janne,O.A. and Palvimo,J.J. (2002) PIAS proteins modulate transcription factors by functioning as SUMO-1 ligases. Mol. Cell. Biol., 22, 5222–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nishida T. and Yasuda,H. (2002) PIAS1 and PIASxa function as SUMO-E3 ligases toward androgen receptor and repress androgen receptor-dependent transcription. J. Biol. Chem., 277, 41311–41317. [DOI] [PubMed] [Google Scholar]
- 49.Sapetschnig A., Rischitor,G., Braun,H., Doll,A., Schergaut,M., Melchior,F. and Suske,G. (2002) Transcription factor Sp3 is silenced through SUMO modification by PIAS1. EMBO J., 21, 5206–5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmidt D. and Muller,S. (2002) Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proc. Natl Acad. Sci. USA, 99, 2872–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tussie-Luna M.I., Michel,B., Hakre,S. and Roy,A.L. (2002) The SUMO ubiquitin-protein isopeptide ligase family member Miz1/PIASxbeta/Siz2 is a transcriptional cofactor for TFII-I. J. Biol. Chem., 277, 43185–43193. [DOI] [PubMed] [Google Scholar]
- 52.Zilfou J.T., Hoffman,W.H., Sank,M., George,D.L. and Murphy,M. (2001) The corepressor mSin3a interacts with the proline-rich domain of p53 and protects p53 from proteasome-mediated degradation. Mol. Cell. Biol., 21, 3974–3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen G. and Courey,A.J. (2000) Groucho/TLE family proteins and transcriptional repression. Gene, 249, 1–16. [DOI] [PubMed] [Google Scholar]
- 54.Ilyemere V.P., Davies,N.H. and Brownlee,G.G. (1998) The activation function 2 domain of hepatic nuclear factor 4 is regulated by a short C-terminal proline-rich repressor domain. Nucleic Acids Res., 26, 2098–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lai A., Lee,J.M., Yang,W., DeCaprio,J.A., Kaelin,W.G., Seto,E. and Branton,P.E. (1999) RBP1 recruits both histone deacetylase-dependent and -independent repression activities to retinoblastoma family proteins. Mol. Cell. Biol., 19, 6632–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Polly P., Herdick,M., Moehren,U., Baniahmad,A., Heinzel,T. and Carlberg,C. (2000) VDR-Alien: a novel, DNA-selective vitamin D(3) receptor-corepressor partnership. FASEB J., 14, 1455–1463. [DOI] [PubMed] [Google Scholar]
- 57.Sun H. and Taneja,R. (2000) Stra13 expression is associated with growth arrest and represses transcription through histone deacetylase (HDAC)-dependent and HDAC-independent mechanisms. Proc. Natl Acad. Sci. USA, 97, 4058–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muscat G.E., Burke,L.J. and Downes,M. (1998) The corepressor N-CoR and its variants RIP13a and RIP13Delta1 directly interact with the basal transcription factors TFIIB, TAFII32 and TAFII70. Nucleic Acids Res., 26, 2899–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Masumi A. and Ozato,K. (2001) Coactivator p300 acetylates the interferon regulatory factor-2 in U937 cells following phorbol ester treatment. J. Biol. Chem., 276, 20973–20980. [DOI] [PubMed] [Google Scholar]