

Abstract
In this paper, we investigate the coevolution of diploid sexual reproduction and cell senescence (i.e., cell aging). We use probability analysis, computer simulation, and exact numerical computation to analyze the impacts of deleterious recessive mutations on sexual and asexual reproduction. The main conclusion is that, without cell senescence, the evolutionary advantages of sexual reproduction cannot be realized in unicellular organisms that reproduce both sexually and asexually. Also, cell senescence is found to be useful in the maintenance of sexual reproduction. This result suggests that diploid sexual reproduction was unlikely to establish itself as a widespread reproduction mechanism without the complementary process of cell senescence.
Evolutionary theories of sex (1–7) and senescence (8–15) were developed independently. However, it has been suggested that sex and cell senescence may have profound relations in their early origins (16). Among the earliest life forms to evolve cell senescence might be protists like paramecia. Paramecia reproduce both sexually and asexually, i.e., by conjugation or fission. If a single paramecium produced by recent conjugation is placed in an ideal environment, it will begin to reproduce asexually by simple fission. However, the rate of cell division will slow continually, and after a number of generations (e.g., about 200 for Paramecium tetraurelia, about 300 for Paramecium biaurelia, and about 350 for Paramecium primaurelia), the clonal offspring will stop dividing and die (17). However, if at any point during these generations two of the offspring conjugate, their senescence clocks are reset, and their offspring are granted a renewed clonal life span. Those who fail to conjugate continue to senesce and die inevitably (16).
To analyze the coevolution of cell senescence and diploid sexual reproduction, we consider two species of unicellular organism, which have the fundamental genetic properties of unicellular eukaryotes such as paramecia. They have diploid genomes (a pair of homologous chromosomes) in which a number of genetic loci may undergo deleterious mutations. Two kinds of alleles may occur at each locus: a wild-type allele or a mutated allele. When a deleterious mutation occurs at a certain locus in one of the homologous chromosomes, a mutated allele is induced. If a cell is homozygous for mutated alleles at m loci, the relative fitness is f = (1 − s)m, where s is the selection coefficient. (In a finite population, the probability of survival of an individual during selection is proportional to its relative fitness. For an infinite population, the exact mathematical formulation of our model is given later in Theoretical Analysis; see Eq. 7.) These cells may reproduce both sexually and asexually, i.e., conjugation and fission, respectively. In each generation, a fixed small proportion of individuals is selected randomly to take part in sexual reproduction. Each individual has a parameter called age to count how many generations of successive asexual reproduction the individual has undertaken since the last sexual reproduction. If two individuals undertake sexual reproduction, the clocks of their offspring are reset. The two species are subject to the same rate of deleterious mutation. The only difference between these two species is that one employs cell senescence (if an individual reaches its age limit, it will be eliminated by cell senescence) while the other does not.
Consider an individual that has a pair of homologous chromosomes. It carries n1 and n2 mutated alleles on each chromosome, respectively, and is homozygous of mutated alleles at m loci. Hence, the fitness of this individual is f = (1 − s)m. The expected number of its asexual offspring that have the same fitness in the next generation (we will refer to this kind of offspring as “same-fitness-asexual-offspring” in this paper) is given by
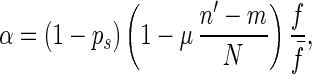 |
1 |
where n′ = (n1 + n2)/2, ps is the probability for an individual to take part in sexual reproduction, f̄ is the average fitness of the population, μ is the rate of mutation (i.e., the probability that a mutation occurs per diploid genome per generation), and N is the number of genetic loci of the single-celled organism. We let
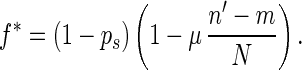 |
2 |
Because 0 ≤ n′ − m ≤ N/2, we have
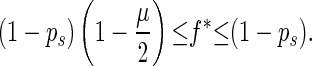 |
3 |
If f̄ > 1 − ps, α is less than 1 and is equal to the probability that this individual has same-fitness-asexual-offspring in the next generation. With the accumulation of deleterious mutations, f̄ decreases. If f̄ becomes lower than fl∗, where fl∗ = ( 1 − ps)(1 − μ/2), then for high-fitness individuals (i.e., f > f̄/fl∗), α > 1. This means that the number of same-fitness-asexual-offspring of high-fitness individuals will increase in the subsequent generations. The increase of high-fitness individuals will lead f̄ to move up. This negative feedback mechanism keeps f̄ above fl∗. Because μ and ps are typically small, fl∗ is high. Hence a significant fraction of the individuals in the population will maintain a high fitness. Eq. 1 also implies that high-fitness individuals have a better chance to keep propagating through same-fitness-asexual-offspring in a larger number of generations. In this process, mutations accumulate but they are not eliminated by selection. So high-fitness individuals will eventually lead to individuals with a mutated allele at almost all loci but with very few loci that have two mutated alleles. Such individuals will be called high-fitness-highly-mutated (HFHM) individuals. In the end, the population will be dominated by HFHM individuals. With the growth of HFHM individuals, the average number of mutations increases; these mutations impact sexual and asexual reproduction in different ways. For example, if an individual carrying n1 and n2 mutations of its two homologous chromosomes is homozygous for mutated alleles at m loci, the expected fitness of its offspring in fission is
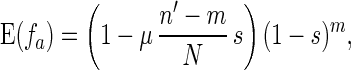 |
4 |
where n′ = (n1 + n2)/2.
Now consider the offspring from sexual reproduction. Two chromosomes of this offspring are taken from two randomly paired individuals. Suppose the two chromosomes carry n1 and n2 mutations, respectively; then the expected fitness of this offspring is
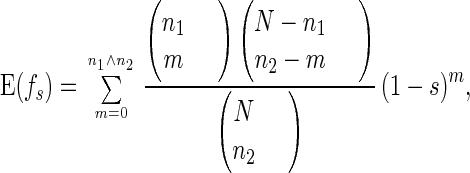 |
5 |
where n1 ∧ n2 is the minimum of n1 and n2.
Recall we have already argued that average fitness f̄ will remain high (above fl∗). Hence, there will always be high-fitness individuals (i.e., those with small m) going through fission. Fig. 1 compares the decrease of average fitness in Eq. 4 and Eq. 5. In this figure, we assume n1 = n2 = n. Note that because of genetic crossover, the two homologous chromosomes are likely to carry similar numbers of mutated alleles. We see that the expected fitness of an offspring from sexual reproduction decreases rapidly with the accumulation of deleterious mutations, whereas the expected fitness of an offspring from asexual reproduction decreases very slowly. So with the increase of accumulated mutations, the expected fitness of the offspring from sexual reproduction decreases rapidly. The genotype produced by sexual reproduction will be eliminated through selection so that evolutionary advantages of sexual reproduction cannot be realized.¶
Figure 1.

Expected fitness of offspring (N = 3,000, s = 0.015, μ = 0.03) from sexual reproduction (solid line) and asexual reproduction. (Because f̄ ≥ fl∗ = 0.96, for vast majority of the individuals, m is small. The dashed lines stand for the cases where m = 0, 1, 2, and 3.)
In contrast, in the species employing cell senescence, for the offspring of an individual to propagate, it must take part in sexual reproduction before reaching the clonal life span limit. The pathway of accumulating mutations through same-fitness-asexual-offspring of high-fitness individuals is interrupted by cell senescence. As a result, the average number of accumulated mutations can be kept at a low level, and sexual reproduction has a substantial chance to produce high-fitness offspring.
Results from Simulation
To demonstrate the effects of cell senescence, we performed a computer simulation using an individual-based model. Thegenome has 3,000 genetic loci. The population is 10,000 for each of the two species in the first generation. The ages of these individuals are set to be one. If an individual belongs to the species employing cell senescence, its age is checked at the beginning of every generation. If its age is more than 200 (the limit of clonal life span), it is eliminated from the population. In each generation, 2.5% of the individuals are randomly selected to take part in sexual reproduction. The first step is genetic crossover: paternal and maternal chromosomes in these selected individuals exchange part (in this simulation 30%) of their alleles randomly. Then, randomly selected pairs of individuals are allowed to conjugate (exchange one of their chromosomes). At the same time, their ages are reset to one. After that, each of them undergoes cell division to form two identical cells barring mutations. The other 97.5% individuals reproduce asexually. In asexual reproduction, one cell divides into two identical cells barring mutations. Recent experiments suggested the rate of deleterious mutations in Caenorhabditis elegans and Drosophila are from 0.005 to 1 per diploid genome per generation (18). Here we set this parameter to be 0.03. After reproduction, the population is doubled. The individuals in this population are then subjected to selection. The probability for an individual to survive through the selection is determined by two factors: p = k · f, where f is relative fitness and k is an environmental factor (the population is limited by resources in the environment):
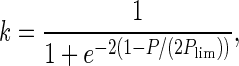 |
6 |
where P is the current population size and Plim is the limit of population size given the resources of the environment (represented in Fig. 2). In this simulation, Plim is 12,500.
Figure 2.

The population limit imposed by environment.
The second factor f is the relative fitness. If a cell is homozygous for mutated alleles at n loci, relative fitness is f = (1 − s)n, where s is selection coefficient. Most direct information of the effects of deleterious mutations in eukaryotes came from experiments in Drosophila, which indicate that most deleterious mutations have relatively small homozygous effects on fitness (19–21). Here we set s to be 0.015. We have also performed computer simulations in which s was set to be 0.01, 0.05, 0.1, and 0.2. The results are similar to those reported below.
In the simulation, the species employing cell senescence accumulated by far fewer recessive deleterious mutations (Fig. 3A) and the average clonal age stays low (Fig. 3C). This means that the population is composed of “young” individuals, i.e., their clonal ancestors are offspring from sexual reproduction within last tens of generations. In the species without cell senescence, a large number of deleterious mutations accumulated (Fig. 3A), and the average clonal age of individuals in this species continues to increase (Fig. 3C). This result clearly confirms that almost all of the offspring from sexual reproduction in late generations were eliminated through selection.
Figure 3.

The results of the individual-based model (ps = 0.025, μ = 0.03, s = 0.015, N = 3,000, AL = 200). The dashed line stands for the species without cell senescence; the solid line stands for the species that employs cell senescence. The two species were in separate environment. (A) Average number of deleterious mutations per diploid genome. (B) Mean population fitness. (C) Average clonal age. The average clonal age of individuals in the species employing cell senescence fluctuates around 50.
We have also performed computer simulations in which the mutated (deleterious) allele is not completely recessive; the dominance degree of the mutated allele is 0.01 and 0.1. The results still support our conclusion.
Theoretical Analysis
To give a more rigorous analysis of this model, we analyze the evolution of the two species at the large population limit. For the species without senescence, let Pgn,n1,n2,n be the proportion of individuals in generation gn with n1, n2 mutated alleles in the two homologous chromosomes respectively and with n loci at which there are two mutated alleles. Then we have
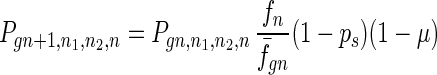 |
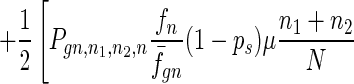 |
7 |
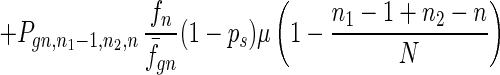 |
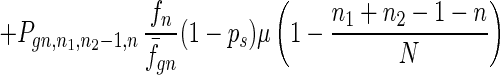 |
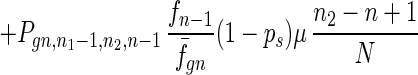 |
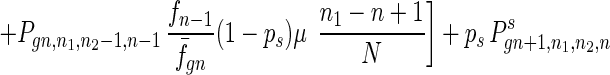 |
where fn = (1 − s)n, f̄gn = ∑n1,n2,n Pgn,n1,n2,nfn, and Pgn+1,n1,n2,ns is the contribution of sexual reproduction as given below.
The sexual reproduction has two stages: meiosis‖ and gamete production.
The process of meiosis includes DNA replication, in which mutations may occur; crossover, in which we assume alleles are randomly redistributed on the two homologous chromosomes; and two cell divisions. After cell division I, the distribution function is
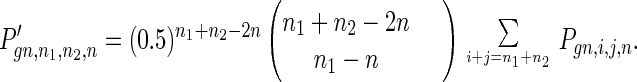 |
8 |
Gamete production: the distribution of deleterious alleles in the haploid gamete cells is
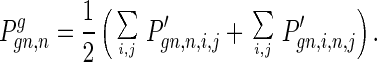 |
9 |
The coupling of two gametes to form a new individual is shown by
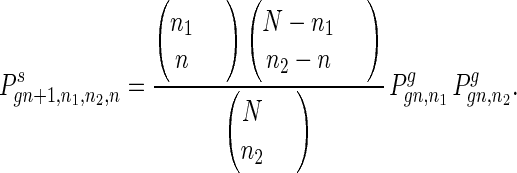 |
10 |
For an individual in the species that employs cell senescence, when its age a reaches a certain limit the individual will be eliminated because of senescence. The distribution function is
 |
11 |
where AL is the age limit, cgn+1 is a normalizing factor that has to be calculated in each generation to compensate for the population eliminated by senescence, and 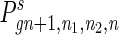 can be calculated in the same way as that described in Eqs. 7–10, except that the distribution function should also depend on a and Eq. 8 should sum over a also. With Eqs. 7–11, we can calculate the distribution function of a, n1, n2, and n in each generation. Unlike the computer simulation described above, this method is deterministic and gives exact results. If the stochastic simulation were carried out with a large population size, the result would approximate that of the deterministic method. However, the memory requirement of the deterministic method is of order O(N3) in Eq. 7 and O(AL · N3) in Eq. 11; hence a numerical solution is only applicable to small genomes. We will perform the computation for a genome of 100 loci.
can be calculated in the same way as that described in Eqs. 7–10, except that the distribution function should also depend on a and Eq. 8 should sum over a also. With Eqs. 7–11, we can calculate the distribution function of a, n1, n2, and n in each generation. Unlike the computer simulation described above, this method is deterministic and gives exact results. If the stochastic simulation were carried out with a large population size, the result would approximate that of the deterministic method. However, the memory requirement of the deterministic method is of order O(N3) in Eq. 7 and O(AL · N3) in Eq. 11; hence a numerical solution is only applicable to small genomes. We will perform the computation for a genome of 100 loci.
Exact Results for Small Genome
Given the initial distributions P0,0,0,0 = 1 for the species without senescence and P0,0,0,0,0 = 1 for the species that employs senescence, we compute the distribution function for 30,000 generations by using Eqs. 7–11 iteratively. The average number of deleterious alleles and average fitness during evolution are shown in Fig. 4. We can see that genetic equilibrium has been reached rapidly. We also find a rapid accumulation of recessive deleterious mutations in the species without senescence. In this computation, ps = 0.025, μ = 0.05, N = 100. According to footnote ¶, the average fitness f̄ should converge to an equilibrium value approximately equal to fl∗ = 0.95. This prediction is confirmed by the result shown in Fig. 4B.
Figure 4.

The results of the deterministic model (ps = 0.025, μ = 0.05, s = 0.05, N = 100, AL = 50). The dashed lines stand for the species without cell senescence; solid lines stand for the species that employs cell senescence. (A) Average number of deleterious mutations per diploid genome. (B) Mean population fitness. (C) Mean fitness of the offspring from sexual reproduction. (D) Percentage that offspring from sexual reproduction that have above-average fitness.
After the genetic equilibrium is reached, the species that employs senescence has far fewer deleterious mutations (Fig. 4A). The equilibrium distribution for the total number of mutations (n1 + n2) and the number of loci with both alleles mutated (n) are presented in Fig. 5 A and B, respectively for the two species. It is clear in Fig. 5B that the population (without senescence) contains HFHM individuals exclusively. For such individuals, the number of mutated alleles on the two homologous chromosomes is very close to N.
Figure 5.

Population distribution on number of mutated alleles (n1 + n2) and number of loci (n) at which two mutated alleles occur. (A) Species that employs cell senescence. (B) Species without cell senescence.
The distributions at genetic equilibrium of the two species are very different (Fig. 4 and Fig. 5). For the species without cell senescence, the population is mainly composed of HFHM individuals. If two HFHM individuals undertake sexual reproduction, their offspring will have a very low expected fitness according to Eq. 5. This leads to a very low mean fitness of offspring from sexual reproduction (Fig. 4C; it will converge to a value near zero for large genome), and there are no above-average-fitness offspring from sexual reproduction even for a very large population (Fig. 4D). So the genotype created by sexual reproduction will be eliminated through selection. Thus sexual reproduction cannot realize whatever evolutionary benefits it has. In the species that employs cell senescence, there is no highly mutated individual; the number of mutated alleles is kept at a low level (Fig. 4A) so that sexual reproduction may have a substantial chance to produce offspring that have above-average fitness (Fig. 4C). Sexual reproduction can be maintained by the benefits of itself.
Maintenance of Sexual Reproduction
To show the relationship between the maintenance of sexual reproduction and cell senescence in a more direct manner, it is necessary to summarize the evolutionary benefits of sexual reproduction in the model. Many mechanisms have been proposed to explain why sexual reproduction is favored by natural selection. Most of them fall into two groups, namely, that sexual reproduction brings beneficial mutations together, or that sexual reproduction purges the genome of harmful mutations (18). Both groups of theories are related to a fundamental nature of sexual reproduction: the offspring from sexual reproduction are more different from each other and more different from their parents than the offspring from asexual reproduction are, i.e., sexual reproduction increases the genetic variance in fitness. Because the exact advantages sexual reproduction confers are still not known, we simply formulate this characteristic of sexual reproduction in the model. A factor Φ is added to the function of relative fitness to reflect the variance of the offspring from sexual reproduction; thus f = cΦ · (1 − s)m. The value of Φ is calculated after sexual reproduction as Φ = (Φ1Φ2)1/2w, where Φ1, Φ2 are the corresponding factors of the parents, and w is determined according to the distribution in Table 1. Φ remains unchanged after asexual reproduction. The constant c is used to keep f ≤ 1.0 for any individual in a generation. The variable w introduces additional variance in fitness of the offspring from sexual reproduction, but it does not change the mean fitness of these offspring. Fig. 6 presents the results of an individual-based simulation using this model. The results on the accumulation of recessive deleterious mutations and its impact on sexual reproduction are seen to be the same as before (Fig. 6 A–C).
Table 1.
Probability distribution* of w
| w |
Probability |
|---|---|
| 0.975 | 0.025 |
| 0.985 | 0.135 |
| 1.0 | 0.68 |
| 1.015 | 0.135 |
| 1.025 | 0.025 |
This is a discrete approximation of the normal distribution with mean 1.0 and standard deviation 0.01.
Figure 6.

The results of the individual-based model using a modified fitness function (ps = 0.025, μ = 0.05, s = 0.05, N = 100, AL = 200). The dashed line stands for the species without cell senescence; the solid line stands for the species that employs cell senescence. (A) Average number of deleterious mutations per diploid genome. (B) Average clonal age. (The average clonal age of individuals in the species employing cell senescence fluctuates around 70.) (C) Percentage that offspring from sexual reproduction that have above-average fitness. After 10,000 generations, modifier alleles were introduced into the population. (D) Asexual reproduction modifier allele A invades the population without cell senescence. (The mutant's genotype is supposed to be AB/AB; the initial frequency of allele A is 0.01. If the genotype is AB/aB, the frequency of A will converge to 0.5. Either way the mutants will invade the whole population.) (E) The modifier allele B cannot invade the population with cell senescence (initial frequency of allele B is 0.01). (F) Frequency of modifier haplotypes AB in the population with cell senescence. (Here we suppose the genotype of mutants is AB/ab. Similar result can be produced by using other genotypes, e.g., AB/AB.)
Next we study the behavior of the population after the introduction (at generation 10,000) of dominant modifier alleles for asexual reproduction (allele A) and nonsenescence (allele B), respectively. The phenotypes of these alleles are given in Table 2. First we consider a population of individuals with genotype aB/aB (i.e., the species without cell senescence, which we discussed previously). In this population, a large number of deleterious mutations accumulate, and the offspring from sexual reproduction are of low fitness; therefore, sexual reproduction cannot realize its benefit of introducing genetic variance in fitness. Because the asexual offspring have a much higher fitness, the asexual reproduction modifier allele A will invade the population (Fig. 6D). (Here the mutant genotype can be AB/AB or AB/aB.) Thus, sexual reproduction by itself is unstable.
Table 2.
The effect of modifier alleles A and B
| Modifier genotype | Phenotype trait |
|---|---|
| AA | Exclusively asexual reproduction |
| Aa | Exclusively asexual reproduction |
| aa | Facultative sexual reproduction |
| BB | Infinite clonal life span |
| Bb | Infinite clonal life span |
| bb | Finite clonal life span (cell senescence) |
In the species that employs cell senescence, the genotype is ab/ab (facultative sexual reproduction and finite clonal life span). We will see that the population of this genotype is immune from invasion by any possible modifier allele.
(i) Mutants with genotype Ab/ab or Ab/Ab (exclusively asexual reproduction and finite clonal life span) will be eliminated when they reach the clonal life span. So asexual modifier allele A cannot invade the population directly.
(ii) For mutants with genotype aB/ab or aB/aB (facultative sexual reproduction and infinite clonal life span), Fig. 6E shows that they cannot invade the population.
(iii) In rare cases, both modifier alleles A and B may occur in a mutant (genotype is AB/ab, Ab/aB, or AB/AB). As shown in Fig. 6F, such mutants still cannot invade the population of genotype ab/ab. The mutant subpopulation increases at first because the sexual reproduction produces more low-fitness offspring (impact of accumulated deleterious mutations). However, the continually arising, novel high-fitness offspring from sexual reproduction eventually dominate the population.
Thus, sexual reproduction and cell senescence can be maintained if they coevolve.
Experimental Clues.
Recent results from telomere research provide clues for a molecular mechanism that may link diploid sexual reproduction and cell senescence together in the evolution. Telomeres are protein–DNA complexes found at the termini of eukaryotic linear chromosomes. It has been postulated that telomere shortening is the “molecular clock” that leads to senescence and that the expression of telomerase overcomes telomere shortening by adding telomeric repeats to the chromosomal DNA termini (22, 23). The recent cloning of the human telomerase gene (24, 25) made it possible to test this “telomere hypothesis” experimentally. It was found that extension of replicative life span can indeed be achieved by the introduction of telomerase into human cell types normally lacking telomerase activity (26). However, recent studies of yeast telomeres have provided definitive evidence that telomeres play a critical role in meiosis (27–30). The alignment of homologous chromosomes during prophase is an essential step of meiosis. Mutations in the telomere-specific protein Taz1 lead to disruption of telemeric clustering and homolog alignment in prophase, and also to reduced recombination and defective chromosome segregation during meiosis (27, 28). A telomere protein (Ndj1p) was also found to be necessary for normal chromosome synapsis and segregation in yeast (31). Thus, telomere association is important to the proper chromosome alignment, synapsis, recombination, and segregation during sexual reproduction. These experimental results indicate that telomere may provide a common molecular basis for the coevolution of diploid sexual reproduction and cell senescence.
Acknowledgments
We thank Professors William Clark and Kenneth Lange for useful comments on a preliminary draft. This work is supported in part by National Science Foundation Grants DMS-9703918 and DBI-9904701.
Abbreviation
- HFHM
high-fitness-highly-mutated
Footnotes
These results also allow us to deduce approximately the equilibrium value of the average fitness f̄. For HFHM individuals, we have n′ − m ≅ N/2, and hence α ≅ fl∗f/f̄. If f̄ > fl∗, then α < 1. This means the number of same-fitness-asexual-offspring will decrease. Because the population is dominated by HFHM individuals, offspring from sexual reproduction cannot compensate for the loss of high-fitness individuals. So the average fitness will decrease until it reaches fl∗.
In this model, no mutation is induced in meiosis. We have tried to induce mutation in meiosis, but the results remain unchanged.
References
- 1.Muller H L. Mutat Res. 1964;1:2–9. doi: 10.1016/0027-5107(64)90047-8. [DOI] [PubMed] [Google Scholar]
- 2.Maynard Smith J. The Evolution of Sex. Cambridge, U.K.: Cambridge Univ. Press; 1978. [Google Scholar]
- 3.Crow J F. Evolution. 1983;37:863–865. [Google Scholar]
- 4.Michod R E, Levin B R. The Evolution of Sex. Sunderland, MA: Sinauer; 1988. [Google Scholar]
- 5.Kondrashov A S. Nature (London) 1988;336:435–440. doi: 10.1038/336435a0. [DOI] [PubMed] [Google Scholar]
- 6.Charlesworth B. Genet Res. 1990;55:199–221. doi: 10.1017/s0016672300025532. [DOI] [PubMed] [Google Scholar]
- 7.Barton N H, Charlesworth B. Science. 1998;281:1986–1990. [PubMed] [Google Scholar]
- 8.Medawar P B. An Unsolved Problem in Biology. London: Lewis; 1952. [Google Scholar]
- 9.Williams G C. Evolution. 1957;11:398–411. [Google Scholar]
- 10.Edney E B, Gill R W. Nature (London) 1968;220:281–282. doi: 10.1038/220281a0. [DOI] [PubMed] [Google Scholar]
- 11.Parker G A, Maynard Smith J. Nature (London) 1978;274:849–855. doi: 10.1038/274849a0. [DOI] [PubMed] [Google Scholar]
- 12.Charlesworth B. Evolution in Age-Structured Population. Cambridge, U.K.: Cambridge Univ. Press.; 1980. [Google Scholar]
- 13.Partridge L, Barton N H. Nature (London) 1993;362:305–311. doi: 10.1038/362305a0. [DOI] [PubMed] [Google Scholar]
- 14.Charlesworth B. Curr Biol. 1996;6:20–22. doi: 10.1016/s0960-9822(02)00411-6. [DOI] [PubMed] [Google Scholar]
- 15.Burstein M. J Gerontol Nurs. 1998;24(9):16–19. doi: 10.3928/0098-9134-19980901-09. [DOI] [PubMed] [Google Scholar]
- 16.Clark W R. Sex & The Origin of Death. New York: Oxford Univ. Press; 1996. [Google Scholar]
- 17.Wichterman R. The Biology of Paramecium. New York: Plenum; 1986. [Google Scholar]
- 18.Wuethrich B. Science. 1998;281:1980–1982. doi: 10.1126/science.281.5385.1980. [DOI] [PubMed] [Google Scholar]
- 19.Charlesworth B, Charlesworth D. Genetica. 1998;102–103:3–19. [PubMed] [Google Scholar]
- 20.Simmons M J, Crow J F. Annu Rev Genet. 1977;11:49–78. doi: 10.1146/annurev.ge.11.120177.000405. [DOI] [PubMed] [Google Scholar]
- 21.Crow J F, Simmons M J. In: The Genetics and Biology of Drosophila, Ashburner M, Carson H L, Thompson J N, editors. 3C. London: Academic; 1983. pp. 1–35. [Google Scholar]
- 22.Blackburn E H. Nature (London) 1991;350:569–573. doi: 10.1038/350569a0. [DOI] [PubMed] [Google Scholar]
- 23.Greider C W. Annu Rev Biochem. 1996;65:337–365. doi: 10.1146/annurev.bi.65.070196.002005. [DOI] [PubMed] [Google Scholar]
- 24.Nakamura T M, Morin G B, Chapman K B, Weinrich S L, Andrews W H, Lingner J, Harley C B, Cech T R. Science. 1997;277:955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- 25.Meyerson M, Counter C M, Eaton E N, Ellisen L W, Steiner P, Caddle S D, Ziaugra L, Beijersbergen R L, Davidoff M J, Liu Q, et al. Cell. 1997;90:785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 26.Bodnar A G, Quellette M, Frolkis M, Holt S E, Chiu C P, Morin G B, Harley C B, Shay J W, Lischsteiner S, Wright W E. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 27.Nimmo E R, Pidoux A L, Perry P E, Allshire R C. Nature (London) 1998;392:825–828. doi: 10.1038/33941. [DOI] [PubMed] [Google Scholar]
- 28.Cooper J P, Watanabe Y, Nurse P. Nature (London) 1998;392:828–831. doi: 10.1038/33947. [DOI] [PubMed] [Google Scholar]
- 29.Rockmill B, Roeder G S. Genes Dev. 1998;12:2574–2586. doi: 10.1101/gad.12.16.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Price C M. Curr Opin Genet Dev. 1999;9:218–224. doi: 10.1016/S0959-437X(99)80032-X. [DOI] [PubMed] [Google Scholar]
- 31.Conrad M N, Dominguez A M, Dresser M E. Science. 1997;276:1252–1255. doi: 10.1126/science.276.5316.1252. [DOI] [PubMed] [Google Scholar]