

Abstract
We determined the genomic sequence at the site of insertion in 2,266 unselected P element insertion events. Estimating physical properties of the genomic DNA at these insertion sites—such as base composition, bendability, A-philicity, protein-induced deformability, and B-DNA twist—revealed that they differ significantly from average chromosomal DNA. By examining potential hydrogen bonding sites in the major groove, we identified a 14-bp palindromic pattern centered on the 8-bp target site duplication that is generated by P element insertion. Our results suggest that the P-element transposition mechanism has a two-fold dyad symmetry and recognizes a structural feature at insertion sites, rather than a specific sequence motif.
Transposable elements exist in the genomes of many organisms and have become important tools in genome research. By generating a simple, reproducible lesion on insertion that can be detected easily, transposable elements provide a powerful means of correlating genetic and molecular information (1, 2). In Drosophila melanogaster, the P transposable element has been particularly useful because strains whose genomes are free of this element exist (3), its movement can be controlled by limiting the availability of its transposase (4, 5), and modified elements can be constructed in vitro and reintroduced into the genome (6). It has long been appreciated that P elements insert nonrandomly (7); however, the factors that influence this specificity are not well understood. There is an apparent preference for chromosomal sites that are likely to be accessible in chromatin; euchromatic sites are favored over heterochromatic sites (8), interbands appear to be favored over bands (9), and there is a marked tendency to integrate at the 5′-end of genes (10). Local sequence composition at the site of insertion also appears to play a role. O'Hare and Rubin (7) examined the sequences flanking 18 P element insertions and noted that the 8-bp target sequence that is duplicated on insertion is GC rich.
Attempts to study the insertion site preferences of P elements have been hindered by the fact that the available collections of insertions were biased in that the insertions in these collections had been selected based on their phenotype. As part of its effort to understand gene function, the Berkeley Drosophila Genome Project (BDGP) is carrying out an insertional mutagenesis project (10) that utilizes an engineered P transposable element, the EP element (11, 12). In these experiments, no selection, other than the ability of the EP element to express the dominant eye color marker it carries, was applied. In this report, we have used this first large collection of unselected insertion events to examine what features of the genomic sequence at the site of insertion are correlated with P element insertion.
DNA secondary structure depends at least in part on the sequence of nucleotides. There are a number of methods for measuring DNA physical properties from di- or trinucleotides based on calculating stacking energy (13), propeller twist (14), nucleosome positioning (15), bendability (16), A-philicity (17), protein-induced deformability (18), duplex stability (19, 20), DNA denaturation (21), DNA bending stiffness (22), B-DNA twist (23), protein-DNA twist (18), or stabilizing energy of Z-DNA (24). Stacking energy, propeller twist, nucleosome positioning, and bendability have been applied to the analysis of specific DNA sequences (25–27), and we have used bendability, A-philicity, protein-induced deformability, and B-DNA twist here to compare sequences at the sites of P element insertion to unselected chromosomal DNA. We show that all four of these measures of DNA structure deviate significantly from random at P element insertion sites. Our results argue that the donor DNA and transposase complex performing P element integration may recognize a structural feature of the target DNA rather than a specific sequence of nucleotides.
Many protein-DNA interactions occur by hydrogen-bonding of amino acid side chains to sites in the DNA's major groove (see, for example, ref. 28). Fig. 1 shows the potential hydrogen bonding sites by protein to DNA base pairs. There are six potential hydrogen-bonding sites found in the major groove as described by Seeman et al. (29). We developed a new tool to visualize potential hydrogen-bonding patterns in DNA, which we call HbondView. Using this tool to examine P element insertion sites, we show that the 8-bp target site duplication created by P element insertion (7) is contained within a 14-bp palindromic pattern. This result suggests that the complex of P transposase and donor DNA that mediates P element integration may be two-fold symmetrical.
Figure 1.

Diagram showing the potential hydrogen bonding sites presented in the major groove of DNA by G-C and A-T base pairs. Adapted from figures and descriptions in work by Seeman et al. (29).
Materials and Methods
Determination of the P Element Insertion Site Sequences.
The 2,266 EP insertion lines are described in ref. 12. Flanking DNA sequences were determined by sequencing inverse PCR products as described in detail at http://www.fruitfly.org/p_disrupt/inverse_pcr.html. In brief, DNA was prepared from 30 adult flies, digested with either Sau3A or HinPI, and then ligated under dilute conditions to favor intermolecular ligation. PCR was performed for 35 cycles using primers specific for appropriate P element sequences. Generally strong and unique products resulted that could be directly sequenced without extensive purification.
To identify the EP insertion sites on genomic DNA, we used blastn to align the assembled EP flanking sequences against ≈25 Mb of available genomic DNA sequence at the time the analysis was done. Only those matches with >95% identity for >95% of the length of EP flanking sequences were used. EP flanking sequences hitting multiple genomic clones, implying insertion into repetitive DNA, were excluded.
HbondView.
In this visualization method, a set of aligned nucleotide sequences is represented by their potential hydrogen-bonding donor and acceptor sites, using the conversion matrix derived in ref. 29 and shown in Table 1. Each of the six potential hydrogen-bonding positions in the major grove at each base pair is represented by a color: donor (red), acceptor (blue), and non-hydrogen-bonding (gray). In the current analysis, multiple insertion site sequences were aligned at the first nucleotide (base 0 in Fig. 3) of the 8-bp target site duplication. The final color at each position is determined by the percentages of hydrogen-bond donors, hydrogen-bond acceptors, and non-hydrogen-bonding sites at that position.
Table 1.
Conversion matrix for nucleotide sequence
| Potential hydrogen bonding position | w1 | w2 | w3 | w3′ | w2′ | w1′ |
|---|---|---|---|---|---|---|
| A/T | a | n | d | n | a | n |
| T/A | n | a | n | d | n | a |
| G/C | a | n | a | n | d | n |
| C/G | n | d | n | a | n | a |
Each base pair can be converted to a representation of six potential hydrogen bonding positions. a, a potential hydrogen bond acceptor site; d, a potential hydrogen bond donor site; n, no hydrogen bonding.
Figure 3.

Potential hydrogen bonding pattern at P transposable element insertion sites. Nucleotide positions are numbered with position 0 corresponding to the first nucleotide of the 8-bp target site duplication. At each nucleotide position, the six potential sites of hydrogen bonding in the major groove (see Table 1) are color coded as follows: red represents a potential acceptor site, blue represents a potential donor site, and gray indicates a site that can be neither a donor nor an acceptor. In the pattern shown the colors have been averaged over 1,185 aligned sequences so that the color at each site reflects how often that site is a hydrogen bond donor, acceptor, or non-hydrogen-bonding site in this sequence set. A 14-bp palindromic pattern centered on the 8-bp target site duplication is apparent. Above and below this 14-bp region, red predominates at the outer sites (w1 and w1′), and purple predominates at the other positions. This is the expected pattern for random sequences because, regardless of the DNA sequence, there is no potential hydrogen-bonding donor at site w1 and w1′. Close inspection of the pattern uncovered several significant potential hydrogen-bonding donor and acceptor sites. Donor sites predominate at six positions: the w2 position of bases 2 and 7, w3 of base −3, w3′ of base 10, and w2′ of bases 0 and 5. Acceptor sites predominate at 10 positions: the w1 position of bases 0, 4, and 5, w3 of bases 0 and 5, w3′ of bases 2 and 7, and w1′ of bases 2, 3, and 7.
We developed the following scoring function to measure how well a given sequence matches an n-bp pattern:
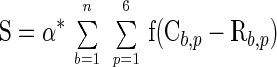 |
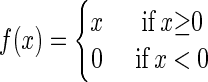 |
where α is a constant, C is the pattern value of a given color at position p of base b, and R is the expected value of same color of position p of base b if there is no pattern. We do not impose a penalty if the color at a position did not match the pattern.
Calculation of the Probability of Obtaining a Peak in a GC Content Distribution.
To calculate GC content over window size w for s aligned sequences, we took (s × w) nucleotides and counted the number of Gs and Cs. This process can be considered analogous to flipping a coin (s × w) times and counting how many times heads is obtained, in which case the probability of s DNA sequences giving a peak value v (0 < v < 1) with average GC content p (0 < p < 1) in genome DNA is the same as the probability of obtaining v heads, after flipping a coin (s × w) times, assuming a probability p of obtaining heads and a probability q = 1 − p of obtaining tails on each flip. Applying the binomial probability formula:
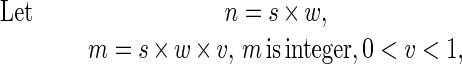 |
then the probability
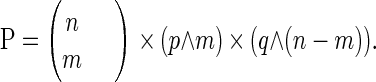 |
Results
Determining the Insertion Site Sequences for a Large Collection of Unselected P Element Insertions.
We determined the insertion site sequences for 2,266 independent insertions of the EP element that were present in the lines described by Rørth et al. (12) by using a method based on inverse PCR (see Materials and Methods). For most lines, we were able to obtain sequences corresponding to both the 5′ and 3′ junctions between the inserted element and genomic DNA. Because P element insertion generates an 8-bp target site duplication, it was possible to assemble these sequences to reconstruct a contiguous sequence of genomic DNA spanning the insertion site. In this way, we were able to obtain insertion site sequences that averaged 400 bp for 1,577 (69.6%) of the EP insertions. For 611 (27%) of the insertions, we successfully obtained sequence only across the junction between the element and the genome on one end of the element; these sequences had an average length of about 200 base pairs. For 78 (3.4%) of the EP lines, the 8-bp direct target site duplications did not agree in sequence. Such events can result from either a deletion being associated with the P element insertion event or from a mistracking of samples. These lines were excluded from further analysis. For 2,241 (98.9%) EP lines, we obtained enough DNA sequence (>25 bp) to allow us to compare the sequence with genomic DNA (see Materials and Methods); the insertion site sequences obtained for these 2,241 lines averaged 311 bp. We were able to use available genomic DNA sequences to extend the insertion site sequences of 637 lines, giving us a total of 587 insertions for which we had at least 250 bp of sequence on either side of the insertion site. To prevent the introduction of bias into our analysis of insertion sites, we grouped these sequences if the distance between insertion sites was less than 250 bp and only included one sequence from each group in the data set we used in subsequent analyses. This data set contained 467 sequences, which were aligned at the first nucleotide of 8-bp direct repeat (position 0 in Fig. 2).
Figure 2.

Profiles of GC content and the four indicated DNA physical measures in 500-bp segments of DNA derived from 467 independent, aligned P insertion sites. Position 0 corresponds to the first nucleotide of the 8-bp target site duplication that is shaded gray. In the profile of GC content, the heavy line represents actual EP insertion sites whereas the light line represents the same number of randomly picked 500-bp genomic sequences. Profiles of A-philicity, protein-induced deformability, B-DNA twist, and bendability are shown.
P element insertions occur nonrandomly and some sites are known to be highly preferred. We compared the sequences of the insertion sites to determine how many different sites were represented in our collection. We considered two EP insertions to be at the same site if their 8-bp target site duplications overlapped. We found 2,045 distinct insertion sites, with 118 sites having been hit more than once. The most preferred site in our data set was located at polytene band 12C on the X chromosome and was hit by 40 independent insertions.
Table 2 shows a summary of 8-bp target site duplications from 1,469 different P insertion sites. For this analysis, we only used data from insertions for which we could independently determine the 8-bp target site duplication sequence from each end of the element. Although there are base preferences at each position, these are not strong enough to generate a clear consensus sequence.
Table 2.
The frequency with which each nucleotide occurs at the eight positions in the direct repeats generated by 1,469 P element insertions
| A | 0.19 | 0.14 | 0.09 | 0.10 | 0.30 | 0.30 | 0.41 | 0.07 |
| T | 0.06 | 0.39 | 0.31 | 0.34 | 0.10 | 0.12 | 0.13 | 0.20 |
| G | 0.52 | 0.24 | 0.11 | 0.16 | 0.43 | 0.48 | 0.21 | 0.22 |
| C | 0.23 | 0.23 | 0.49 | 0.40 | 0.17 | 0.10 | 0.25 | 0.51 |
P Element Insertion Sites Have a High GC Content.
GC content was then calculated for a 500-bp segment of genomic DNA centered on the P element insertion site using a window size of 3 bp. The average value of all sequences over a window was assigned to the nucleotide in the middle of the window, and these data were plotted (Fig. 2, top panel). GC content shows a symmetrical pattern centered on the 8-bp target site duplication, rising from around 37% and reaching a peak of 59.2% at the 8-bp target site (Fig. 2, top panel, heavy line). We were able to observe a peak of GC content at the insertion site by using sliding window sizes ranging from 3 to 21 bp. A data set containing 467 randomly selected 500-bp genomic sequences gave a nearly flat distribution with a 42.5% average GC content (Fig. 2, top panel, light line). The chance of 467 randomly chosen 500-bp DNA sequences of 42.5% average GC content giving a peak value of 59.2% when aligned and averaged is about 10−36 (see Materials and Methods), indicating that the observed peak in GC content at the P insertion site is highly statistically significant. We also tested another data set containing 467 randomly generated sequences with same base composition as the EP insertion site data set (43.3% GC); again, no comparable peak in GC content was seen (data not shown).
Several Measures of DNA Physical Properties Show Significant Signals at the P Insertion Site.
We applied 12 different measures (13–24) of DNA physical properties to the same data sets examined above for GC content. All measures show a significant signal at the P insertion site (only results from A-philicity, protein-induced deformability, B-DNA twist, and bendability are reported here). Although these measures are obtained via different experiments, we examined the computational relationships between these measures and GC content. We calculated correlation coefficients for each pair of dinucleotide or trinucleotide measures by using a uniform distribution across dinucleotides or trinucleotides (see http://www.fruitfly.org/∼guochun/pins.html for a complete listing of measures and correlation coefficients). Most correlation coefficients between A-philicity, protein-induced deformability, B-DNA twist, bendability, and GC content are small and suggest that those measures are computational-independent and can provide independent elements of supporting evidence.
Profiles were calculated by using a window size of 3 and averaged over all sequences. As shown in Fig. 2, these profiles are each symmetrical around the site of insertion and display a significant signal at the P insertion site. Neither the data set of randomly selected genomic sequences nor of randomly generated sequences with same base composition as the test set gave any significant signal (data not shown). We also applied these same physical scales to an independent data set derived from the insertion sites of P elements selected to cause lethality (30) and obtained similar results (data not shown). Given the fact that these 500-bp sequences, as well as the sequences of the 8-bp target site duplications themselves, are highly diverse, our results strongly support the idea that P-element insertion recognizes some aspect of DNA structure rather than sequence similarity.
Analysis of the trinucleotide composition of the 8-bp target site duplication revealed that the sequences around the P insertion sites are enriched in six triplets: CAG, CTG, GAC, GCC, GGC, and GTC. The bendability and nucleosome positioning measures are based on triplet frequencies, and these six triplets are correlated with high values. Similarly, the analysis of dinucleotide frequencies revealed enrichments for the dinucleotides CC, GC, GG, and GT, consistent with the high GC content at the insertion site. CC, GG, and GT are correlated with low values of A-philicity and B-DNA twist, and CC and GG are correlated with high values of protein-induced deformability.
HbondView Identifies a 14-bp Palindromic Pattern at P Insertion Site.
Many protein-DNA interactions occur through hydrogen bonding between amino acid side-chains and sites in the major groove of the DNA double helix. Because different bases can present similar arrangements of donor and acceptor sites (29), we thought it might be more informative to examine the pattern of these sites in the major groove in our collection of P insertion sites, rather than to simply compare their nucleotide sequences. To facilitate this effort we developed a method, which we call HbondView, that converts a set of aligned nucleotide sequences into a display of potential hydrogen-bonding positions in the major groove by representing hydrogen-bond donor and acceptor sites as different colors (see Materials and Methods). We applied this method to several data sets of EP insertion sequences. In the experiment shown in Fig. 3, we studied 1,185 different 50-bp sequences, each centered on the 8-bp target-site duplication. Each row represents a base pair, and the six columns in each row represent the six potential hydrogen-bonding sites in the major groove for each base pair (see Fig. 1). Acceptor sites are represented in red and donor sites in blue. Because the pattern shown is the average of all 1,185 aligned sequences, the final color at each position indicates the tendency for that position to be a donor or an acceptor. A 14-bp palindromic pattern, composed of the 8 bp that are duplicated on insertion and 3 bp on either side, is apparent. Although this palindromic pattern is not obvious when only a single insertion site is examined, we found that as few as 50 insertion sites, when aligned and averaged, gave a clear 14-bp pattern.
We then built the 14-bp pattern using a data set of 1,284 EP insertion sites that occurred in regions that were not present in the available completed genomic sequence at the time the analysis was done. Using the scoring function described in Materials and Methods, we then calculated the scores of 254 14-mer corresponding to unrelated EP insertion sites in the completed genomic sequence. We also scanned available completed genomic sequences and scored 23 million unselected 14-mer. The average score for 14-mer from genomic sequences is 41 ± 10.5 whereas the average score for 14-mer from EP insertion sites is 61.7 ± 8. The distributions of scores are compared in Fig. 4 and are largely non-overlapping, indicating that the 14-bp pattern we derived correlated well with P insertion sites.
Figure 4.

Comparison of how well 14-mer from EP insertion sites (solid line) and 14-mer from genomic sequences (dotted line) fit the 14-bp pattern. A score for each 14-mer is calculated by using the function described in Materials and Methods. The results are then plotted as percentage of 14-mer at each score. 91.7% of 14-mer from EP insertion sites are outside the first standard deviation of the distribution of 14-mer from unselected genomic sequences, and 87.7% of 14-mer from unselected genomic sequences are outside the first standard deviations of the distribution of 14-mer from EP insertions.
Because the 14-bp pattern is a palindrome, we were interested in determining whether any palindromic sequence would be favored for P insertion, or if particular features of the 14-bp pattern we identified were responsible for the observed preference. We found that the average tendency for 14-mer from EP insertion sites to be palindromic is only 15% higher than for 14-mer chosen at random from genomic sequences. In comparison, 14-mer from EP insertion sites are about 50% better at matching the 14-bp pattern than are 14-mer from genomic sequences, implying that simply being palindromic is not the only factor in P insertion site determination.
Discussion
In this report we describe a highly efficient protocol for mapping the genomic sites of insertion of P transposable elements and its application to over 2,200 individual insertion events. This data set represents a large, unbiased collection of sequenced P insertion sites, allowing us to apply a number of statistical methods to analyze P insertion preferences.
P element insertion is nonrandom, and most insertions occur within a few hundred bases of the transcription start site of a gene. It is likely that a great deal of this preference is caused by chromatin accessibility, as these are the same chromosomal regions that must be accessed by the transcriptional control machinery. Even within these open regions of chromatin, however, P insertion does not appear to be random. In this report we present evidence that this local preference may depend more on DNA structure than on primary sequence. Similar DNA structures may be produced by DNA with different nucleotide sequences. Therefore, in addition to looking for sequence similarity, we used four existing measures of DNA physical properties, each of which shows a clear tendency for P insertion sites to differ from general chromosomal DNA.
We also developed a new tool, HbondView, to visualize the hydrogen bonding potential of sites in the DNA major groove. The results of this analysis indicate that the P elements prefer a particular palindromic arrangement of hydrogen bonding sites over a 14-bp region centered on their insertion site. Individual P insertion sites are usually highly diverged from the consensus we derived, indicating that recognition of this site can only require the formation of a small subset of hydrogen bonds shown in the consensus. Our results imply that interaction of P transposase with the P insertion site is facilitated by both DNA structural features and a degenerate pattern of hydrogen bonding sites in the major groove. It is likely that other DNA binding proteins that show low sequence specificity of binding employ similar mechanisms.
HbondView is a graphical method that converts a set of aligned DNA sequences to a representation of potential hydrogen-bonding positions in the major groove. It provides a way to uncover and quantitate features of protein-binding sites that are easily ignored if only DNA sequence similarity is considered. We are currently evaluating the use of this coding strategy for other applications in bioinformatics.
Acknowledgments
The work has benefited from valuable interactions with Professor Wilma Olson. We are grateful to Professor Wilma Olson for sharing her expertise on DNA structure and to Professor Alexander Rich for his suggestion on applying Z-DNA measure on our data set. We thank ProfessorNicholas R. Cozzarelli and Professor Don Rio for their critical comments. We also thank Dr. Martin Reese for helpful discussions. This work was supported by National Institutes of Health Grant HG00750.
Footnotes
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AQ024952∼AQ025011, AQ025013∼AQ025032, AQ025035∼AQ025039, AQ025041∼AQ025041, AQ025043∼AQ025057, AQ025059∼AQ025144, AQ025146∼AQ025162, AQ025164∼AQ025175, AQ025177∼AQ025192, AQ025194∼AQ025222, AQ025224∼AQ025254, AQ025256∼AQ025261, AQ025263∼AQ025290, AQ025292∼AQ025293, AQ025295∼AQ025296, AQ025298∼AQ025353, AQ025355∼AQ025383, AQ025385∼AQ025403, AQ025405∼AQ025569, AQ025969∼AQ025973, AQ025975∼AQ025977, AQ025979∼AQ025983, AQ025985∼AQ026032, AQ026034∼AQ026035, AQ026445∼AQ026457, AQ026459∼AQ026507, AQ072890∼AQ073256, AQ073362∼AQ073820, AQ073822∼AQ074130, AQ254591∼AQ254785, AQ254789∼AQ254895).
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.050017397.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.050017397
References
- 1.Kleckner N, Roth J, Botstein D. J Mol Biol. 1975;116:125. doi: 10.1016/0022-2836(77)90123-1. -. [DOI] [PubMed] [Google Scholar]
- 2.Bingham P M, Levis R, Rubin G M. Cell. 1982;25:693–704. doi: 10.1016/0092-8674(81)90176-8. [DOI] [PubMed] [Google Scholar]
- 3.Bingham P M, Kidwell M G, Rubin G M. Cell. 1982;29:995–1004. doi: 10.1016/0092-8674(82)90463-9. [DOI] [PubMed] [Google Scholar]
- 4.Spradling A C, Rubin G M. Science. 1982;218:341–347. doi: 10.1126/science.6289435. [DOI] [PubMed] [Google Scholar]
- 5.Engels W R. Science. 1984;226:1194–1196. doi: 10.1126/science.6095450. [DOI] [PubMed] [Google Scholar]
- 6.Rubin G M, Spradling A C. Science. 1982;218:348–353. doi: 10.1126/science.6289436. [DOI] [PubMed] [Google Scholar]
- 7.O'Hare K, Rubin G M. Cell. 1983;34:25–35. doi: 10.1016/0092-8674(83)90133-2. [DOI] [PubMed] [Google Scholar]
- 8.Berg C A, Spradling A C. Genetics. 1991;127:515–524. doi: 10.1093/genetics/127.3.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Semeshin V F, Belyaeva E S, Zhimulev I F, Lis J T, Richards G, Bourouis M. Chromosoma. 1986;93:461–468. [Google Scholar]
- 10.Sprading A, Stern D, Kiss I, Rootc J, Laverty T, Rubin G M. Proc Natl Acad Sci USA. 1995;92:10824–10830. doi: 10.1073/pnas.92.24.10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rørth P. Proc Natl Acad Sci USA. 1996;93:12418–12422. doi: 10.1073/pnas.93.22.12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rørth P, Szabo K, Bailey A, Laverty T, Rehm J, Rubin G M, Weigmann K, Milan M, Benes V, Ansorge W, Cohen S M. Development (Cambridge, UK) 1998;125:1049–1057. doi: 10.1242/dev.125.6.1049. [DOI] [PubMed] [Google Scholar]
- 13.Ornstein R L, Rein R, Breen D L, MacElroy R. Biopolymers. 1978;17:2341–2360. doi: 10.1002/bip.1978.360171005. [DOI] [PubMed] [Google Scholar]
- 14.Hassan M A E, Calladine C R. J Mol Biol. 1996;259:95–103. doi: 10.1006/jmbi.1996.0304. [DOI] [PubMed] [Google Scholar]
- 15.Goodsell D S, Dickerson R E. Nucleic Acids Res. 1994;22:5497–5503. doi: 10.1093/nar/22.24.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brukner I, Sanchez R, Suck D, Pongor S. EMBO J. 1995;14:1812–1818. doi: 10.1002/j.1460-2075.1995.tb07169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ivanov V I, Minchenkova L E. Mol Biol. 1995;28:780–788. [PubMed] [Google Scholar]
- 18.Olson W K, Gorin A, Lu X J, Hock L M, Zhurkin V B. Proc Natl Acad Sci USA. 1998;95:11163–11168. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sugimoto N, Nakano S, Yoneyama M, Honda K. Nucleic Acids Res. 1996;24:4501–4505. doi: 10.1093/nar/24.22.4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breslauer K J, Frank R, Bolcker H, Marky L A. Proc Natl Acad Sci USA. 1986;83:3746–3750. doi: 10.1073/pnas.83.11.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blake R D. Encyclopedia of Molecular Biology and Molecular Medicine. New York: VCH; 1996. pp. 1–19. [Google Scholar]
- 22.Sivolob A V, Khrapunov S N. J Mol Biol. 1995;247:918–931. doi: 10.1006/jmbi.1994.0190. [DOI] [PubMed] [Google Scholar]
- 23.Gorin A, Zhurkin V B, Olson W K. J Mol Biol. 1995;247:34–48. doi: 10.1006/jmbi.1994.0120. [DOI] [PubMed] [Google Scholar]
- 24.Ho P S, Ellison M J, Quigley G J, Rich A. EMBO J. 1986;5:2737–2744. doi: 10.1002/j.1460-2075.1986.tb04558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pedersen A, Baldi P, Chauvin Y, Brunak S. J Mol Biol. 1998;281:663–673. doi: 10.1006/jmbi.1998.1972. [DOI] [PubMed] [Google Scholar]
- 26.Baldi P, Brunak S, Chauvin Y, Krogh A. J Mol Biol. 1996;263:503–510. doi: 10.1006/jmbi.1996.0592. [DOI] [PubMed] [Google Scholar]
- 27.Baldi P, Chauvin Y. ISMB. 1998;98:35–42. [PubMed] [Google Scholar]
- 28.Lesser D R, Kurpiewski M R, Waters T, Connolly B A, Jen-Jacobson L. Proc Natl Acad Sci USA. 1993;90:7548–7552. doi: 10.1073/pnas.90.16.7548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seeman N C, Rosenberg J M, Rich A. Proc Nat Acad Sci USA. 1974;73:804–808. doi: 10.1073/pnas.73.3.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spradling A C, Stern D, Beaton A, Rehm E J, Laverty T, Mozden N, Misra S, Rubin G M. Genetics. 1999;153:135–177. doi: 10.1093/genetics/153.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–404. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]