

Abstract
Copper (I) promoted [3+2] Huisgen cycloaddition of azides with terminal alkynes was used to prepare triazole-containing macrocycles based on the Grb2 SH2 domain-binding motif, “Pmp-Ac6 c-Asn”, where Pmp and Ac6 c stand for 4-phosphonomethylphenylalanine and 1-aminocyclohexanecarboxylic acid, respectively. When cycloaddition reactions were conducted at 1 mM substrate concentrations cyclization of monomeric units occurred. At 2 mM substrate concentrations the predominant products were macrocyclic dimers. In Grb2 SH2 domain-binding assays the monomeric (S)-Pmp-containing macrocycle exhibited a Kd value of 0.23 μM, while the corresponding dimeric macrocycle was found to have greater than 50-fold higher affinity. The open-chain dimer was also found to have affinity equal to the dimeric macrocycle. This work represents the first application of “click chemistry” to the synthesis of SH2 domain-binding inhibitors and indicates its potential utility.
The growth factor receptor bound protein 2 (Grb2) is an SH2 domain-containing signal transducer that represents an attractive therapeutic target.1 For Grb2 SH2 domains, where o pen-chain “pTyr-Xxx-Asn” sequences are preferentially recognized in type-I β-turn conformations, induction of turn geometries through macrocyclization using ring-closing olefin metathesis2 (RCM) has resulted in a number of potent binding inhibitors.3 Recent reports have highlighted the growing utility in drug discovery of copper (I) promoted [3+2] Huisgen cycloadditions of azides with terminal alkynes, which is one example of “click chemistry.”4 This led us to examine the potential usefulness of this approach to prepare Grb2 SH2 domain-binding antagonists, including macrocyclic inhibitors. To date few reports of ring closure using azide-alkyne click chemistry have appeared for peptides or peptide mimetics.5 Therefore, the current study was undertaken to examine click chemistry in the synthesis of Grb2 SH2 domain-binding inhibitors.
Macrocyclization using ring-closing [1,3] dipolar cycloaddition was undertaken using the protected monomeric sequences 8 that contain the tripeptide motif, “Pmp-Ac6 c-Asn-amide” (Scheme 1). (Here Pmp represents the phosphatase-stable pTyr mimetic 4-phosphonomethylphenylalanine6 and Ac6 c stands for 1-aminocyclohexanecarboxylic acid.7) Preparation of 8 began with the known protected amine 1, which was obtained by Mitsunobu coupling of phthalimide with commercially-available 4-pentyn-1-ol. 8 Direct hydrazinolysis of 1 to yield 1-amino-4-pentyne had previously been reported to fail or to require tedious ion exchange chromatographic workup if an alternate reductive phthalimide cleavage procedure was used.9 However, we were able to employ 1 as a source of 1-amino-4-pentyne by reacting this material with hydrazine in aqueous EtOH (reflux, 2 h),10 then cooling the reaction mixture to room temperature, filtering and concentrating the volatile product. Without further purification the crude amine was coupled directly with N-Boc L-Asn using active ester protocols [1-hydroxybenzotriazole (HOBT) and 1-(3-dimethylaminopropyl)-3-ethyl carbodiimide hydrochloride (EDC)]. This provided the N-Boc Asn[1-(5-pentynyl)] amide 2 in 48% yield from 1. Conversion of 2 to the N-Fmoc protected dipeptide 4 required initial TFA-mediated N-Boc deprotection followed by neutralization and chromatographic purification. Active ester coupling of the resulting free amine with N-Fmoc Ac6 c provided 4 in 84% yield from 2. Removal of N-Fmoc protection followed by
Scheme 1.

(i) a) N2H4•H2O, EtOH, H2O, reflux, 2 h; b) Boc-Asn-OH, EDC, HOBt, DMF, rt, 12 h, (48% yield for two steps); (ii) a) TFA, CH2Cl2, rt, 1 h (95% yield); (iii) a) Fmoc-1-amino-cyclohexanecarboxylic acid, EDC, HOBt, DMF, rt, 12 h (84% yield); (iv) a) piperidine, CH3CN, rt, 2 h, b) EDCI•HCl, HOBt, DMF, rt, 12 h (39% yield for two steps); (v) piperidine, CH3CN, rt, 2 h (37% yield for 7a; 38% yield for 7b); (vi) a) (BrCH2CO)2O DMF, rt, 1h; b) NaN3, DMF, rt, 24 h (two steps, 81% yield for 8a, 62% yield for 8b); (vii) TFA-HS(CH2)2SH-H2O, rt, 1 h (69% yield); (viii) CuI, L-ascorbate, DIPEA, CH3CN/tBuOH/H2O, rt; (ix) TFA-HS(CH2)2SH-H2O, rt, 1 h.
coupling with 4-[bis(tert-butyl)phosphonomethyl]-N-Fmoc-D,L-phenylalanine ([N-Fmoc-D,L-(OBut)2Pmp], 5)11 gave the tripeptide 6 as an inseparable mixture of (R/S) epimers at the Pmp α-carbon. Following N-Fmoc removal, the epimeric free amines could be separated by flash chromatography to yield the (S)-Pmp- and (R)-Pmp-containing diastereomers 7a and 7b, respectively. Assignment of relative configurations was done by comparison with an authentic sample of (S)-Pmp-containing tripeptide (co rresponding to the faster eluting material) prepared using enantiomerically pure N-Fmoc-L-(OBut)2 Pmp.12 Treatment of 7a and 7b with bromoacetic anhydride, followed after 1 h by the addition of sodium azide, yielded the cycloaddition precursors 8a and 8b in 81% yield and 62% yield, respectively.
Initial attempts at [1,3] Huisgen cycloaddition using CuI and L-ascorbate13 proceeded poorly and yielded mainly unreacted starting material. However, addition of N,N-diisopropylethylamine (DIEA) significantly facilitated the reaction, apparently by promoting formation of the copper-(I)-alkyne complex.14 When the cycloaddition reaction was conducted using a substrate concentration of 2 mM, the expected monomeric cyclization product 10 was obtained only as a minor component. The major product under these conditions proved to be the dimeric macrocycle 11 (26% yield for 11a, 56% yield for 11b). The dimerization reaction was highly concentration dependent, since repeating the procedure at 1 mM
substrate concentration reversed the product ratios and provided monomeric 10 (29% yield for 10a and 19% yield for 10b) as the predominant product with dimeric 11 appearing as a minor component. Similar peptide cyclodimerization by copper-catalyzed azide-alkyne cycloaddition has recently been reported.5a Cleavage of the phosphonate tert-butyl esters (TFA-ethanedithiol-H2O) and purification by HPLC gave the final monomeric (12) and dimeric (13) products, respectively.
For comparison purposes the (S)-Pmp-containing open-chain monomer (9a) (Scheme 1) and dimer (18) (Scheme 2) were prepared. In the latter case, starting from the (S)-Pmp-containing tripeptide 7a, cycloaddition precursors 15 and 16 were synthesized bearing C-terminal alkyne and N-terminal azide functionality, respectively. Performing [3+2] cycloaddition of 15 and 16 using CuI and L-ascorbate in the presence of DIEA gave the protected product 17, which was converted to 18.
Scheme 2.

(i) Pd/C, H2, EtOAc; (ii) a) (BrCH2CO)2O DMF, rt, 1 h; b) NaN3, DMF, rt, 24 h (93% yield from 7a); (iii) 1-acetylimidazole, DMF, 24 h (quantitative); (iv) CuI, L-ascorbate, DIPEA, CH3CN/tBuOH/H2O, rt (73% yield); (v) TFA-HS(CH2)2SH-H2O, rt, 1 h (76% yield).
Evaluation of Grb2 SH2 domain-binding affinities was conducted using surface plasmon resonance (SPR) as previously described.15A key feature of this technique was its use of Biacore S51 instrumentation, which allowed direct measurement of synthetic peptide binding to surface-bound Grb2 SH2 domain protein. In similar studies conducted on the reference macrocycle 19 (Figure 1) using a Biacore 3000 instrument, close agreement was observed between the SPR binding affinity (Kd = 0.9 nM) and affinity determined by ELISA methods (IC50 = 1.4 nM).3d
Figure 1.
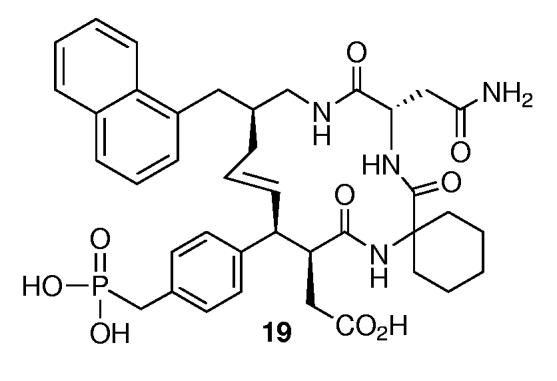
Structure of reference macrocycle examined by both SPR
As shown in Table 1, the (R)-Pmp-containing monomeric and dimeric macrocycles (12b and 13b, respectively) exhibited binding constants above 1 μM. However, the monomeric (S)-Pmp-containing macrocycle (12a)16 bound with su b-micromolar affinity. Of particular note were the affinities of the dimeric (S)-Pmp-containing peptides, which provided low nanomolar Kd values for both the macrocyclic (13a)17 and open-chain (18)18 forms. These high affinities were somewhat unexpected based on the low affinity of the open-chain precursor 9a (Kd > 1 μM).
Table 1.
Grb2 SH2 domain-binding affinities of synthetic peptides.
| Compds | Binding Affinity Kd, μMa |
|---|---|
| 9a | >1 |
| 12a | 0.23 |
| 12b | >1 |
| 13a | Kd1= 0.0018; Kd2= 0.0040 |
| 13b | >l |
| 18 | Kd1= 0.0011; Kd2 = 0.087 |
Values were determined as described in reference 15.
The binding kinetics of 13a and 18 were complex and consistent with multiple binding components. The quality of the Biacore data was very high and this was not felt to an artifact of the data. Multiple binding components have been reported previously for Grb2 SH2 domain-binding macro cycles, however the physical interpretation is not clear.19 Fitting data to a two-component system (Figure 2) provided the Kd1 and Kd2 values shown in Table 1.
Figure 2.
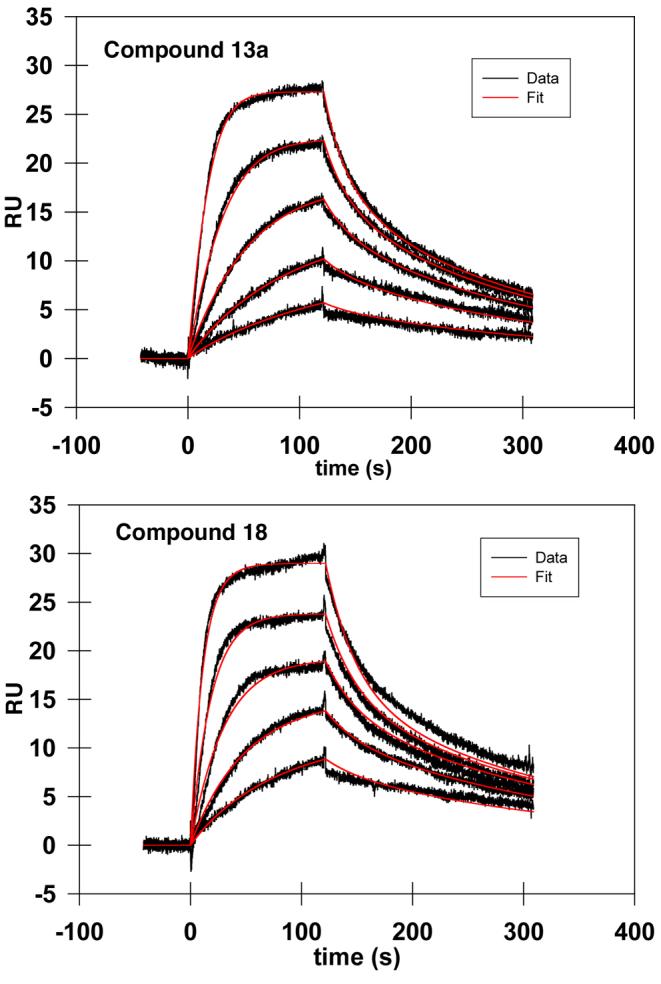
Representative SPR data for 13a and 18 (panel B) showing response in RU units versus time. Traces in black are for two-fold dilutions of inhibitor from 62 nM to 0.98 nM. Curve fitting for two-component models are shown in red.
reported previously for a peptide containing two pTyr mimetic residues.20 However, in this latter case both residues were situated in the proximal pTyr and pTyr+1 positions with the pTyr+1 residue participating in a non-canonical interaction with the Grb2 Arg142.21 Molecular modeling indicates that this type of interaction is not possible for dimeric peptides 13a and 18 (Figure 3).
Figure 3.
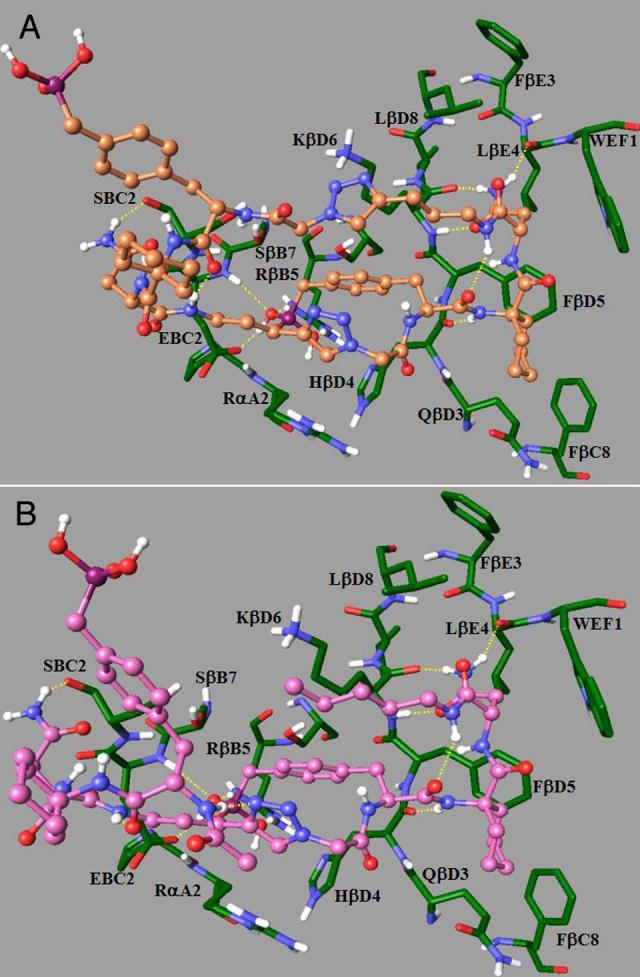
Potential Grb2 SH2 domain binding interactions of synthetic ligands as determined by molecular modeling. Panel A: dimeric macrocycle 13a; Panel B: open-chain dimer 18.
Peptides based on immunoreceptor tyrosine activation motifs (ITAMs) also present tandem pTyr-containing motifs that bind with high affinity to targets such as the ZAP-70 tyrosine kinase that contain multiple SH2 domains. X-ray analysis has confirmed that this binding involves simultaneous interaction of each pTyr-containing motif with a separate SH2 domain.19 To our knowledge, peptides 13a and 18 are among the first examples of dimeric pTyr-mimetic-containing peptides that show high affinity against a single SH2 domain construct. The structural basis for the more than two orders of magnitude affinity enhancement in going from the monomer 9a to the dimers 13a and 18 is not apparent from docking studies with the isolated Grb2 SH2 domain.
Reported herein is the first application of [3+2] Huisgen cycloaddition click chemistry for the synthesis of SH2 domain-binding peptides. By appending azide/alkyne functionality to the N- and C-termini, respectively, macrocyclization could be effected in monomeric or dimeric fashion depending on substrate concentration. Open-chain dimeric analogues could also be prepared by reacting monomers that contained a single azide/alkyne group each. Although the open chain (S)-Pmp-containing monomer exhibited very low binding affinity, dimeric analogues based on the same motif bound with very high affinity and displayed complex kinetics. These results support the potential utility of click chemistries for the preparation of novel SH2 domain-binding antagonists.
Acknowledgments
The authors wish to thank Drs. James A. Kelley and Christopher Lai of the Laboratory of Medicinal Chemistry, NCI, for mass spectral data. This research was supported in part by the Intramural Research Program of the NIH, Center for Cancer Research, NCI-Frederick. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
References and notes
- 1(a).Feller SM, Lewitzky M. Curr. Pharm. Design. 2006;12:529. doi: 10.2174/138161206775474369. [DOI] [PubMed] [Google Scholar]
- 1(b).Dharmawardana PG, Peruzzi B, Giubellino A, Burke TR, Jr., Bottaro DP. Anti-Cancer Drugs. 2006;17:13. doi: 10.1097/01.cad.0000185180.72604.ac. [DOI] [PubMed] [Google Scholar]
- 2.Miller SJ, Blackwell HE, Grubbs RH. J. Am. Chem. Soc. 1996;118:9606. [Google Scholar]
- 3(a).Gao Y, Wei C-Q, Burke TR. Org. Lett. 2001;3:1617. doi: 10.1021/ol0157609. [DOI] [PubMed] [Google Scholar]
- 3(b).Wei C-Q, Gao Y, Lee K, Guo R, Li B, Zhang M, Yang D, Burke TR., Jr. J. Med. Chem. 2003;46:244. doi: 10.1021/jm0203635. [DOI] [PubMed] [Google Scholar]
- 3(c).Dekker FJ, de Mol NJ, Fischer MJE, Kemmink J, Liskamp RMJ. Org. Biomol. Chem. 2003;1:3297. doi: 10.1039/b306681a. [DOI] [PubMed] [Google Scholar]
- 3(d).Shi Z-D, Wei C-Q, Lee K, Liu H, Zhang M, Araki T, Roberts LR, Worthy KM, Fisher RJ, Neel BG, Kelley JA, Yang D, Burke TR., Jr. J. Med. Chem. 2004;47:2166. doi: 10.1021/jm030510e. [DOI] [PubMed] [Google Scholar]
- 3(e).Shi Z-D, Lee K, Wei C-Q, Roberts LR, Worthy KM, Fisher RJ, Burke TR., Jr. J. Med. Chem. 2004;47:788. doi: 10.1021/jm030440b. [DOI] [PubMed] [Google Scholar]
- 3(f).Oishi S, Karki RG, Kang S-U, Wang X, Worthy KM, Bindu LK, Nicklaus MC, Fisher RJ, Burke TR., Jr. J. Med. Chem. 2005;48:764. doi: 10.1021/jm0492709. [DOI] [PubMed] [Google Scholar]
- 3(g).Shi Z-D, Karki RG, Worthy KM, Bindu LK, Fisher RJ, Burke TR., Jr. Chem. Biodiversity. 2005;2:447. doi: 10.1002/cbdv.200590024. [DOI] [PubMed] [Google Scholar]
- 3(h).Burke TR., Jr. Int. J. Pept. Res. Therap. 2006;12:33. doi: 10.1007/s10989-006-9014-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kolb HC, Sharpless KB. Drug Discovery Today. 2003;8:1128. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 5(a).Punna S, Kuzelka J, Wang Q, Finn MG. Angew. Chem. Int. Ed. 2005;44:2215. doi: 10.1002/anie.200461656. [DOI] [PubMed] [Google Scholar]
- 5(b).Billing JF, Nilsson UJ. J. Org. Chem. 2005;70:4847. doi: 10.1021/jo050585l. [DOI] [PubMed] [Google Scholar]
- 5(c).Angell Y, Burgess K. J. Org. Chem. 2005;70:9595. doi: 10.1021/jo0516180. [DOI] [PubMed] [Google Scholar]
- 5(d).Bock VD, Perciaccante R, Jansen TP, Hiemstra H, van Maarseveen JH. Org. Lett. 2006;5:919. doi: 10.1021/ol053095o. [DOI] [PubMed] [Google Scholar]
- 6.Marseigne I, Roques BP. J. Org. Chem. 1988;53:3621. [Google Scholar]
- 7.Garcia-Echeverria C, Gay B, Rahuel J, Furet P. Bioorg. Med. Chem. Lett. 1999;9:2915. doi: 10.1016/s0960-894x(99)00501-6. [DOI] [PubMed] [Google Scholar]
- 8.Tiecco M, Testaferri L, Temperini A, Bagnoli L, Marini F, Santi C, Terlizzi R. Eur. J. Org. Chem. 2004:3447. doi: 10.1039/b712861d. [DOI] [PubMed] [Google Scholar]
- 9.Bellier B, Dugave C, Etivant F, Genet R, Gigoux V, Garbay C. Bioorg. Med. Chem. Lett. 2004;14:369. doi: 10.1016/j.bmcl.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 10.L’abbe G, Leurs S, Sannen I, Dehaen W. Tetrahedron. 1993;49:4439. [Google Scholar]
- 11.Burke TR, Jr., Russ P, Lim B. Synthesis. 1991;11:1019. [Google Scholar]
- 12.Li P, Zhang M, Peach ML, Liu H, Yang D, Roller PP. Org. Lett. 2003;5:3095. doi: 10.1021/ol035078+. [DOI] [PubMed] [Google Scholar]
- 13.Rostovtsev VV, Green LG, fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 14.Sonogashira K, Tohda Y, Hagihara N. Tetrhedron Lett. 1975;16:4467. [Google Scholar]
- 15.Oishi S, Karki RG, Shi Z-D, Worthy KM, Bindu L, Chertov O, Esposito D, Frank P, Gillette WK, Maderia MA, Hartley J, Nicklaus MC, Barchi JJ, Jr., Fisher RJ, Burke TR., Jr. Bioorg. Med. Chem. 2005;13:2431. doi: 10.1016/j.bmc.2005.01.052. [DOI] [PubMed] [Google Scholar]
- 16. Compound 12a. 1H NMR (CD3OD) δ 8.21 (s, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.76 (s, 1H), 7.68 (d, J = 4.8 Hz, 1H), 7.21 (dd, J = 8.0 & 2.4 Hz, 2H), 7.16 (d, J = 8.0 Hz, 2H), 5.18 (d, J = 16.0 Hz, 1H), 4.99 (d, J = 16.0 Hz, 1H), 4.66 (m, 1H), 4.31(t, J = 6.4 Hz, 1H), 3.33 (m, 1H), 3.05 (d, J = 21.6 Hz, 2H), 3.06-2.94 (m, 2H), 2.73 (t, J = 6.0 Hz, 2H), 2.51 (dd, J = 15.6 & 7.2 Hz, 1H), 2.39 (dd, J = 15.6 & 5.6 Hz, 1H), 2.03-1.90 (m, 3H), 1.70 (m, 2H), 1.58 (m, 1H), 1.48-1.32 (m, 4H), 1.15 (m, 1H), 0.79 (m, 1H). MALDI-MS (+Ve) m/z: 647 [MH+]
- 17. Compound 13a. 1H NMR (400 MHz, d6-DMSO) δ 8.70 (d, 1H, J = 7.8 Hz), 8.54 (s, 1H), 7.97 (d, 1H, J = 8.0 Hz), 7.45 (s, 1H), 7.42-7.39 (m, 2H), 7.22-7.16 (m, 4H), 6.96 (s, 1H), 4.95 (d, 1H, J = 16.0 Hz), 4.80 (d, 1H, J = 16.2 Hz), 4.75 (m, 1H), 4.36 (dd, 1H, J = 5.4 Hz & 13.2 Hz), 3.23-3.16 (m, 2H), 3.05 (m, 1H), 2.95 (dd, 1H, J = 6.5 Hz & 21.2 Hz), 2.76-2.66 (m, 2H), 2.58 (t, 2H, J = 7.5 Hz), 2.43 (dd, 1H, J = 4.7 Hz & 15.6 Hz), 2.02-1.14 (m, 12H). FABMS (-Ve) m/z 1291.8 [(M-H)-]
- 18. Compound 18. 1H NMR (400 MHz, CD3OD) δ 8.06 (d, 1H J = 15.2 Hz), 7.69 (s, 1H), 7.24-7.14 (m, 9H), 5.00 (dd, 2H, J = 45.6 & 16.0 Hz), 4.63 (m, 2H), 4.49 (dd, 1H, J = 6.4 & 5.2 Hz), 4.41 (t, 1H, J = 6.0 Hz), 3.21-3.02 (m, 5H), 3.05 (d, 4H, J = 21.2 Hz), 2.93 (dd, 1H, J = 14.0 & 9.2 Hz), 2.85-2.63 (m, 6H), 1.84 (s, 3H), 2.00-1.66 (m, 8H), 1.52-1.06 (m, 18H), 0.85 (t, 3H, J = 7.2 Hz). FABMS (-Ve) m/z 1254.5 [(M-H)-]
- 19.Oishi S, Shi Z-D, Worthy KM, Bindu LK, Fisher RJ, Burke TR., Jr. ChemBioChem. 2005;6:668. doi: 10.1002/cbic.200400298. [DOI] [PubMed] [Google Scholar]
- 20.Liu WQ, Vidal M, Gresh N, Rogues BP, Garbay C. J. Med. Chem. 1999;42:3737. doi: 10.1021/jm9911074. [DOI] [PubMed] [Google Scholar]
- 21.Nioche P, Liu W-Q, Broutin I, Charbonnier F, Latreille M-T, Vidal M, Roques B, Garbay C, Ducruix A. J. Mol. Biol. 2002;315:1167. doi: 10.1006/jmbi.2001.5299. [DOI] [PubMed] [Google Scholar]
- 22.Hatada MH, Lu X, Laird ER, Green J, Morgenstern JP, Lou M, Marr CS, Phillips TB, Ram MK, Theriault K, Zoller MJ, Karas JL. Nature. 1995;377:32. doi: 10.1038/377032a0. [DOI] [PubMed] [Google Scholar]