

Abstract
The identification of the chemokine receptors as receptors for HIV-1 has boosted interest in these molecules, raising expectations for the development of new strategies to prevent HIV-1 infection. The discovery that chemokines block HIV-1 replication has focused attention on identifying their mechanism of action. Previous studies concluded that this inhibitory effect may be mediated by steric hindrance or by receptor down-regulation. We have identified a CCR5 receptor-specific mAb that neither competes with the chemokine for binding nor triggers signaling, as measured by Ca2+ influx or chemotaxis. The antibody neither triggers receptor down-regulation nor interferes with the R5 JRFL viral strain gp120 binding to CCR5, but blocks HIV-1 replication in both in vitro assays using peripheral blood mononuclear cells as HIV-1 targets, as well as in vivo using human peripheral blood mononuclear cell-reconstituted SCID (severe combined immunodeficient) mice. Our evidence shows that the anti-CCR5 mAb efficiently prevents HIV-1 infection by inducing receptor dimerization. Chemokine receptor dimerization also is induced by chemokines and is required for their anti-HIV-1 activity. In addition to providing a molecular mechanism through which chemokines block HIV-1 infection, these results illustrate the prospects for developing new tools that possess HIV-1 suppressor activity, but lack the undesired inflammatory side effects of the chemokines.
Keywords: AIDS, chemokine, chemokine receptor
The chemokines are a structurally related, proinflammatory cytokine family that control activation and chemotaxis in specific types of leukocytes (1). This protein family is defined by shared structural and functional characteristics, as well as by the chromosomal location of the corresponding genes. Four subfamilies have been described, classified by the position of the first two canonical cysteine residues; these are the CC (or α) and CXC (β) chemokines, the C (γ) chemokines, characterized by a single cysteine residue, and the CX3C (δ) family, represented by a unique membrane-bound chemokine (2, 3). They mediate their biological effects via interactions with a family of seven-transmembrane glycoprotein receptors coupled to a G protein signaling pathway. These receptors consist of a single polypeptide chain with an extracellular amino-terminal domain and three extracellular loops (ECL1–3) that participate in receptor-ligand interactions, as well as a cytoplasmic carboxyl-terminal domain and three intracellular loops that cooperate to bind and activate G proteins (4) and other signaling molecules.
Chemokine receptors are also coreceptors for HIV-1 (5, 6). Recent results show that, in contrast to CD4-independent HIV-1 entry, there is no example of HIV-1 infection that is independent of the chemokine receptor (5). The probable role of CD4 is to concentrate virus on the cell surface and to facilitate interaction with the chemokine receptor. A major breakthrough in this area was the demonstration that the dichotomy in HIV-1 viral tropism is related to the use of the CXCR4 or CCR5 chemokine receptors (7–10). This resulted in a new nomenclature for HIV-1 strains, R5 and X4, depending on the chemokine receptor used for viral entry.
An important advance in understanding the role of chemokine receptors as HIV-1 receptors was the identification of a CCR5 polymorphism that renders homozygous individuals highly resistant to viral infection (11, 12). This polymorphism is a 32-bp deletion (ccr5-Δ32) that results in the production of a truncated molecule not expressed on the cell surface; ccr5-Δ32 homozygous individuals are thus resistant to infection by M-tropic HIV-1 viral strains (11, 12). A recently described CCR5 promoter mutation may explain the delay in HIV-1 disease progression (13). An additional polymorphism associated with delayed disease progression is a single, conservative amino acid substitution (Val-64 to Ile) in the first transmembrane domain of the CCR2b receptor (14, 15). A means by which this mutation prevents disease progression recently has been proposed; the ability of CCR2V64I to form heterodimers with the CCR5 and the CXCR4 receptor may reduce receptor expression levels, impairing the ability of both R5 and X4 HIV-1 strains to infect target cells (16). Despite these advances, the mechanism through which chemokines prevent HIV-1 infection remains essentially unknown.
We generated mAbs specific for the human CCR5 receptor by immunizing mice with synthetic peptides corresponding to various extracellular domains of this receptor (18). CCR5–02, a CCR5 N-terminal domain-specific mAb with a potent suppressive effect on HIV-1 replication, triggers CCR5 receptor dimerization, as does the natural ligand RANTES (regulated upon activation, normal T cell expressed and secreted). This mAb does not trigger Ca2+ flux or chemotactic responses, does not affect RANTES-induced responses, binds to the receptor simultaneously with RANTES, and does not affect the binding of R5 JRFL viral strain gp120; however, the antibody is perfectly competent in blocking HIV-1 infection in the absence of chemokine receptor signaling or internalization. We therefore postulate that chemokine receptor dimerization may be the mechanism through which the chemokines impede HIV-1 infection.
Material and Methods
Cell Culture and Transfection.
Human peripheral blood mononuclear cells (PBMCs) from healthy donors were purified on Ficoll-Paque. HEK-293 cells (ATCC TIB202; American Type Culture Collection) were transfected with CCR5 constructs by calcium phosphate precipitation. Transfected cells were selected in G-418 (Calbiochem) and analyzed by flow cytometry for receptor expression using anti-CCR5 antibodies. The CCR5-transfected MT2 cell line was donated by J. Alcamí (Hospital 12 Octubre, Madrid).
Flow Cytometry Analysis.
Untransfected or CCR5-transfected HEK-293 cells were centrifuged (250 × g, 10 min, room temperature), plated in V-bottom 96-well plates (2.5 × 105 cells/well) and incubated with or without RANTES (10 nM, 60 min, 4°C) (17). After washing in PBS with 2% BSA and 2% FCS and centrifugation (250 × g, 5 min, 4°C), cells were incubated with 50 μl/well biotin-labeled CCR5–02 mAb (5 μg/ml, 60 min, 4°C) and washed as above. Fluorescein isothiocyanate-labeled streptavidin (Southern Biotechnology Associates) was added and incubated (30 min, 4°C), and the plates were washed twice. Cell-bound fluorescence was determined in a Profile XL flow cytometer at 525 nm (Coulter).
Binding of R5 JRFL HIV-1 Strain gp120 to CCR5.
CCR5–02 mAb blocking of R5 JRFL viral strain gp120 binding to CCR5 was tested as described (18, 19). 125I-labeled JRFL gp120 was allowed to bind to CCR5-transiently transfected HEK-293 cells in Hepes binding buffer (50 nM Hepes, pH 7.4/5 nM MgCl2) with 0.5% BSA, alone or with 2D7 mAb (20), CCR5–02, or mIg. The labeled agonist was recovered on 25 mm GF/C glass fiber filters presoaked in 0.2% polyethyleneimine and counted in a γ counter. Untransfected cells were used as a nonspecific binding control.
Receptor Crosslinking, Immunoprecipitation, SDS/PAGE, and Western Blot Analyses.
Serum-starved, CCR5-transfected HEK-293 cells (20 × 106) were stimulated for 1 min at 37°C with RANTES (10 nM), CCR5–02 mAb (5 μg/ml), or an isotype-matched control mAb (5 μg/ml). The reaction was terminated by addition 1 ml of cold PBS and centrifugation (250 × g, 10 min). Receptor crosslinking, lysis, immunoprecipitation, and Western blot were as described (21), using 5 μg/ml of CCR5–03 mAb for immunoprecipitations and CCR5–01 to develop the blot.
Calcium Determination.
Changes in intracellular calcium concentration were monitored by using the fluorescent probe Fluo-3,AM (Molecular Probes). Calcium mobilization in response to RANTES (10 nM; Peprotech, Rocky Hill, NJ), CCR5–02 (5 μg/ml in PBS), or isotype-matched control mAb (5 μg/ml in PBS) was determined (37°C, 525 nm) in an EPICS XL flow cytometer (Coulter), as described (22).
Chemotaxis.
CCR5-transfected HEK-293 cell migration was studied in a 96-well microchamber (NeuroProbe, Gaithersburg, MD). RANTES and mAb (CCR5–02 and CXCR4–01) were added to the lower well in RPMI containing 0.25% BSA; the cells, untreated or preincubated with mAb CCR5–02 or isotype-matched control mAb CXCR4–01 (10 μg/ml, 60 min, 37°C), were added to the upper well. Polyvinylpyrrolidone-free, 10 μm-pore filters (NeuroProbe), precoated with type VI collagen (Sigma; 2 h, 37°C) were used. The chamber was incubated (5 h, 37°C, 5% CO2), filters were removed, and cells were wiped off the upper filter surface. Filters were fixed and stained (0.5% crystal violet, 20% methanol). Migration was quantified by densitometry and expressed as a migration index.
HIV-1 Neutralization Assays: p24 Determination.
Human PBMCs were phytohemagglutinin-activated (10 μg/ml, 48 h, 37°C, 5% CO2), washed, preincubated for 1 h with mAb, then infected (2 ng p24/106 cells per assay, 37°C, 2 h) with HIV-1 strains NL4–3 (X4), BaL (R5), SF2 (dual-tropic), or primary R5 isolates. Cells were washed extensively in PBS and cultured in complete RPMI 1640 containing rhIL-2 (10 ng/ml), alone, or with CCR5–02 or isotype-matched control mAb. Every 2 days, half the culture supernatant was replaced with fresh medium containing IL-2 and antibodies at their initial concentrations (23). Cell-free supernatants were tested for HIV-1 p24 antigen content at days 4–8 postinfection by using a commercial ELISA kit (Coulter). SF2 infection experiments also were performed in CCR5-transfected MT2 cells.
SCID (Severe Combined Immunodeficient) Mouse Reconstitution and Viral Challenge.
CB.17 SCID/SCID mice were bred and maintained under specific pathogen-free conditions in the Centro Nacional de Biotecnología animal facilities. Eight- to 10-week-old nonleaky phenotype mice were reconstituted by i.p. injection of 20 × 106 freshly isolated normal human PBMCs. To confirm reconstitution, serum was tested 2 weeks later in ELISA for human Ig; only human Ig-positive mice were used for HIV-1 infection studies. Four hours before viral challenge and on the next 2 days, mice were injected i.p. with purified CCR5–02 mAb or an isotype-matched mAb (200 μg/mouse) in PBS. Mice were infected 2 weeks after PBMC reconstitution by i.p. injection of 0.5 ml of diluted cell-free HIV-1 BaL stocks containing 100 TCID50. Viral replication was assessed 2 weeks after challenge by measuring plasma HIV RNA copy number in individual mice by using the Amplicor HIV-1 Monitor Assay (Roche Molecular Systems, Branchburg, NJ). Simultaneously, mice were killed by cervical dislocation and a peritoneal cell suspension was obtained by washing with ice-cold PBS. Cells (1 × 106) were incubated with 1 × 106 phytohemagglutinin-activated PBMCs from HIV-1-seronegative donors, in RPMI 1640 with 10% heat-inactivated FCS and recombinant human IL-2 (10 ng/ml). Cocultures were monitored in ELISA for HIV-1 p24 core antigen in supernatant and were considered positive when p24 was >30 pg/ml (24).
CCR5 Receptor Down-Regulation.
CCR5 internalization was analyzed by using CCR5-transfected HEK-293 cells stimulated at different times with 10 nM RANTES, 10 μg/ml of CCR5–02 mAb, or 10 μg/ml isotype-matched control mAb (CXCR4–01). After washing, CCR5 levels were detected by cytofluorometry using an anti-CCR5 mAb (CCR5–03), as above.
Results
The Human CCR5 Receptor-Specific CCR5–02 mAb Interferes Neither with RANTES Binding Nor with Its Function.
We produced a set of mAbs against the CCR5 extracellular domains (17); one of these, mAb CCR5–02, which recognizes the CCR5(15–29) amino terminal domain peptide, is further characterized here. Antibody specificity was analyzed by cytofluorometry in HEK-293 cells, mock-transfected, or transfected with the human CCR5 gene. Whereas CCR5–02 mAb does not bind to mock-transfected HEK-293 cells (17), specific binding is observed when CCR5-transfected cells are used (Fig. 1A). CCR5–02 mAb recognition of CCR5 was unaffected in the presence of RANTES previously bound to the receptor (Fig. 1A), showing that CCR5–02 and RANTES do not compete for binding to this receptor.
Figure 1.

The human CCR5-specific CCR5–02 mAb interferes neither with RANTES binding nor function. (A) CCR5-transfected HEK-293 cells were incubated alone or with 10 nM RANTES (30 min, 4°C). After washing, cells were incubated with biotin-labeled CCR5–02 mAbs, as described. An isotype-matched mAb was used as control. (B) RANTES-induced Ca2+ mobilization in CCR5-transfected HEK-293 cells was unaffected by pretreatment with 10 μg/ml CCR5–02 mAb or isotype-matched control mAb (30 min, 37°C). As a control, Ca2+ mobilization by CCR5–02 mAb (10 μg/ml) is shown. (C) RANTES-induced CCR5-transfected HEK-293 cell migration was unaffected by pretreatment with CCR5–02 mAbs (10 μg/ml, 30 min 37°C) or isotype-matched control mAbs. The figure depicts one of five experiments performed, with SD indicated.
RANTES induces Ca2+ mobilization and cell migration in CCR5-transfected HEK-293 cells. To further characterize the biological activity of the CCR5–02 mAb, we analyzed its effect on RANTES-induced Ca2+ influx. At all concentrations tested, CCR5–02 was unable to promote Ca2+ mobilization or to affect the RANTES-induced response in Fluo-3,AM-loaded CCR5-transfected HEK-293 cells (Fig. 1B). Similar results were obtained when cell transmigration was analyzed; CCR5–02 promoted no effect and did not affect RANTES-induced CCR5-transfected HEK-293 migration (Fig. 1C). We thus conclude that CCR5–02 has no biological activity and does not block the RANTES/CCR5 interaction, nor does it prevent RANTES activation.
mAb CCR5–02 Neither Induces CCR5 Down-Regulation nor Blocks JRFL gp120-CCR5 Interaction.
We showed that RANTES promotes CCR5 tyrosine phosphorylation (17), which is simultaneous with Janus kinase activation, phosphorylation, and association to the receptor immediately after chemokine binding. These events are required for G protein activation and subsequent chemokine sensitization (22), as well as for chemokine receptor polarization (25) and internalization (26).
Although the CCR5–02 mAb triggers no biological responses, we tested whether it induces a signal leading to CCR5 down-regulation. When RANTES-stimulated CCR5-transfected HEK-293 cells are analyzed by CCR5 membrane staining, a clear decrease is observed in membrane expression (Fig. 2), whereas these levels are unaltered by stimulation with CCR5–02 or isotype-matched control mAb (Fig. 2). We next analyzed whether CCR5–02 competes with HIV-1 gp120 binding to the CCR5 receptor, by testing the CCR5 binding of R5 HIV-1 strain JRFL gp120 in the presence of soluble CD4. CCR5–02 mAb did not interfere with JRFL gp120 binding, whereas neutralizing antibodies did, as for example the anti-CCR5 2D7 mAb (20) (Fig. 3), showing that the CCR5–02 mAb does not compete with R5 HIV-1 gp120 binding.
Figure 2.
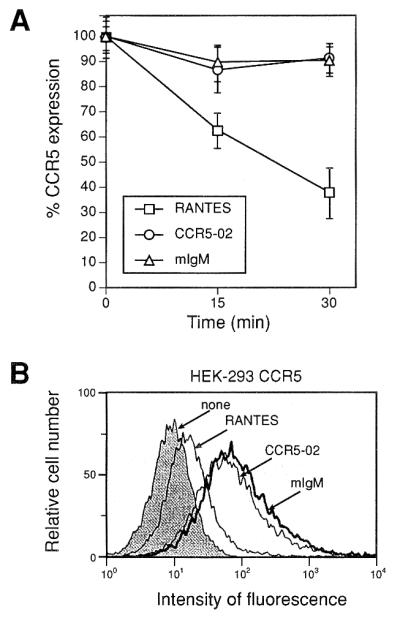
mAb CCR5–02 does not promote CCR5 down-regulation from the surface of stably transfected HEK-293 cells. (A) Serum-starved, CCR5-transfected HEK-293 cells were incubated as indicated with RANTES (10 nM), CCR5–02 mAbs (10 μg/ml), or isotype-matched control mAbs (10 μg/ml) at 37°C. Surface CCR5 was detected by FACS analysis using biotin-labeled CCR5–03 mAbs, followed by streptavidin-phycoerythrin; an isotype-matched mAb was used as a staining control. Results are expressed as the percentage of maximum binding in the absence of chemokine, with SD indicated. (B) A representative flow cytometry figure is shown for data from A, corresponding to RANTES or CCR5–02 mAb treatment (15 min, 37°C).
Figure 3.
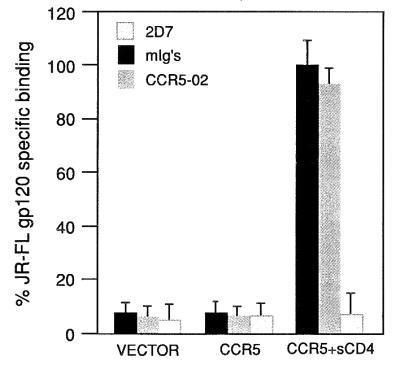
The CCR5–02 mAb does not interfere with JRFL gp120 binding to the CCR5 receptor. Iodinated 125I-labeled JRFL gp120, produced from recombinant vaccinia virus, was bound to HEK-293 cells transfected with plasmids expressing the indicated receptors. Shown are representative raw values of binding in the presence or absence of the indicated antibodies. Background binding to pcDNA3-transfected cells has been subtracted.
mAb CCR5–02 Triggers Receptor Dimerization and Blocks HIV-1 Infection by R5 Strains in Vitro and in Vivo.
One of the earliest chemokine-activated signals is the induction of chemokine receptor dimerization, which may occur in the absence of additional biochemical signals (21). We previously showed that MCP-1, RANTES, and SDF-1α induce homodimerization of their respective receptors (17, 21, 27). As CCR5–02 displays no agonist or antagonist activity, and induces neither steric hindrance nor receptor down-regulation, we tested whether it triggers CCR5 receptor homodimerization. After RANTES and CCR5–02 stimulation and DSS crosslinking, we detected a high molecular mass receptor species (75 kDa) corresponding to the expected molecular mass of two CCR5 molecules, as assessed by immunoprecipitation and Western blot with anti-CCR5 antibodies (Fig. 4); only the monomer form appeared when cells were stimulated with an isotype-matched control mAb (CXCR4–01). We thus conclude that both RANTES and CCR5–02 can trigger CCR5 receptor dimerization.
Figure 4.

The CCR5–02 mAb triggers receptor dimerization. CCR5-transfected HEK-293 cells were stimulated with 10 nM RANTES, 10 μg/ml CCR5–02 mAb, or 10 μg/ml isotype-matched control mAb (60 sec, 37°C), and crosslinked by using 1 mM disuccinimidyl suberate. Cell lysates were immunoprecipitated with CCR5–03 mAb, electrophoresed, and transferred to nitrocellulose membranes. The Western blot was analyzed with CCR5–01 mAb; as a positive control, CCR5-transfected HEK-293 cell lysates were immunoblotted with CCR5–01 mAb. Arrows indicate the monomer and the dimer.
The ability of CCR5 to act as an HIV-1 receptor is widely recognized (7). We recently showed that chemokine receptor dimerization, the first event after chemokine binding, explains the delay to AIDS progression in HIV-1-infected individuals carrying the CCR2V64I mutation (16). As it promotes CCR5 dimerization, we tested whether the CCR5–02 mAb displays activity in HIV-1 infection. Potent inhibition of extracellular HIV-1 p24 antigen release was observed in the R5 BaL strain both for 10 nM RANTES and 10 μg/ml CCR5–02, whereas no effect was seen with 10 nM SDF-1α or 10 μg/ml mIgM (Fig. 5A). As a control, experiments were performed by using X4 HIV-1 strain NL4–3 as a virus source. In this case, as predicted, 10 nM SDF-1α, but neither RANTES nor CCR5–02 mAb, blocked extracellular HIV-1 p24 antigen release (Fig. 5A). To characterize inhibition of the HIV-1 R5 BaL strain, we performed a dose-response curve demonstrating an ID50 of 0.5 nM for RANTES and 1 μg/ml for the CCR5–02 mAb (Fig. 5B). In addition, CCR5–02 prevented HIV-1 p24 antigen release by PBMCs infected with two different R5 primary isolates in a dose-dependent manner, whereas no effect was seen with a control mIgM (Fig. 5C).
Figure 5.

The CCR5–02 mAb interferes with R5 HIV-1 replication. (A) CCR5–02 (10 μg/ml) or an isotype-matched control mAb were tested for HIV-1 suppressive activity by using the X4 NL4–3 or the R5 BaL viral strains and activated PBMC target cells. Untreated culture supernatants were used as control (medium). As a control for suppressive activity, 10 nM of SDF-1α for NL4–3, and 10 nM RANTES or MIP-1α for BaL were used. Viral replication was monitored by quantitating gag p24 antigen levels (ng/ml) at day 7 postinfection. Data represent the mean ± SD of triplicate analyses; one representative experiment is shown of three performed (100% control for NL4–3-infected PBMCs = 13 ng/ml p24; for BaL-infected PBMCs = 9.25 ng/ml p24). (B) A dose-response curve of HIV-1 suppressive activity for RANTES and CCR5–02 mAb is shown by using the R5 BaL viral strain and activated PBMC target cells. Viral replication was monitored as in A. Data represent the mean ± SD of triplicate analyses; one representative experiment is shown of three performed. (C) A dose-response curve of HIV-1 suppressive activity for CCR5–02 mAb, using a primary R5 viral strain and activated PBMCs, and the dual-tropic SF2 viral strain and MT2-B7 cells. Viral replication was monitored as in A. (100% control for SF2-infected CCR5-transfected MT2 cells = 0.65 ng/ml p24, 4 days postinfection; for R5 primary isolate-infected PBMCs = 3.15 ng/ml p24, 7 days postinfection.) Data represent the mean ± SD of triplicate analyses; one representative experiment is shown of three performed. (D) SCID mice were reconstituted with 20 × 106 PBMCs and injected with 200 μg mAb before and after HIV-1 infection. Two weeks after infection, plasma concentrations of HIV-1 RNA were determined. Values represent RNA copies/ml of individual animals. Only mice with values above detection level (>300 copies/ml) are shown. Viral infection in mice with undetectable viral plasma levels was assessed by coculture of peritoneal cells with activated PBMCs.
As chemokine receptor heterodimerization appears to have an important role in some cases of delay to AIDS progression (16), we tested the ability of CCR5–02 mAb to block infection by the SF2 dual-tropic virus strain. Neither CCR5–02 nor RANTES blocked HIV-1 infection by SF2 (Fig. 5C), in accordance with their inability to promote heterodimerization between CCR5 and CXCR4 in the absence of SDF-1α (not shown).
We extended these in vitro results to in vivo HIV-1 infection in SCID mice reconstituted with human PBMCs. SCID mice grafted with adult human PBMCs (SCID-hu-PBMC) are sensitive to HIV-1 infection; as a consequence, they undergo loss of human CD4+ T lymphocytes, making them suitable to study the mechanisms involved in HIV-1 pathogenesis and potential therapeutic treatments (28). Reconstitution was confirmed by measuring the human Ig concentration in serum, and by human CD45 cell marker expression in cytofluometry of cells recovered from the peritoneal cavity 2 weeks after reconstitution (not shown). A large proportion (47%) of these SCID-hu-PBMC mice are susceptible to HIV-1 infection, as shown by HIV-1 p24 measurement in coculture supernatants of phytohemagglutinin-activated human PBMCs and peritoneal cells recovered from HIV-1-infected SCID-hu-PBMC mice. Two weeks after infection, the mean plasma viral RNA copy number was 10-fold lower in mice treated with CCR5–02 mAb compared with mice treated with a control antibody (3,094 ± 3,065 vs. 39,032 ± 21,497 copies/ml) (Fig. 5D). We thus conclude that the CCR5–02 mAb is a potent inhibitor of infectivity by the HIV-1 R5 BaL strain in SCID-hu-PBMC mice.
Discussion
Several models have been postulated by which chemokines inhibit HIV-1 infection (29–32). One, the so-called steric hindrance model, sustains that chemokine binding to its receptor blocks interaction of the HIV-1-env/CD4 complex with the receptor. Results of experiments using modified chemokines that are antagonists in functional assays such as chemotaxis, have been inferred to indicate that this may be a mode of blockage (31, 32). Another model suggests that chemokines induce chemokine receptor desensitization and internalization, preventing viral interaction with and infection of the target cell; evidence for this mode of action also has been reported (30).
Using HIV-1 viral strains that use the CCR5 receptor for infection, the ability of RANTES and (AOP)-RANTES to block HIV-1 infection has been demonstrated (32). Here we show the ability of CCR5–02 to prevent infection by HIV-1 R5 viral strains, both in vitro and in vivo. There are several explanations for the anti-HIV-1 activity shared by these three molecules. The first is that CCR5 ligands activate the receptor signaling pathway, as a result of which HIV-1 infection is prevented; this is nonetheless unlikely, as the CCR5–02 mAb activates no detectable signaling event. Second, that RANTES and CCR5–02 bind directly to the epitope used by HIV-1 to interact with CCR5, which is not the case, because CCR5–02 did not interfere with gp120 binding. The third possibility is that the receptor undergoes a conformational change after chemokine binding, giving rise to a conformation no longer recognized by the virus. Here we show that a conformational change, dimerization, indeed follows chemokine or antibody binding, which may in turn impede the interaction between HIV-1 gp120 and the chemokine receptor. Because RANTES, the CCR5–02 mAb, and (AOP)-RANTES trigger CCR5 receptor dimerization, we propose that chemokine receptor dimerization is the mechanism by which these molecules prevent HIV-1 gp120 interaction with the receptor. We conclude that chemokine receptor dimerization is sufficient, in the absence of any other signal, to prevent HIV-1 infection.
In contrast to other antibodies against the amino terminal loop, the CCR5–02 mAb specifically recognizes the 15–29 peptide and displays unique features. It is the only mAb that recognizes an epitope not primarily implicated in HIV-1 infection (18, 33), that does not interfere with chemokine or gp120 binding, yet blocks viral infection. We have obtained other anti-CCR5 mAbs, some of which do not promote receptor dimerization and are unable to block HIV-1 infection. Earlier studies indicate that the CCR5 domains involved in chemokine ligand specificity and in coreceptor usage for various HIV-1 strains are not identical (33). Experiments using receptor chimeras (33–35) and amino acid substitutions (36–40) show that env-CCR5 interactions are probably complex. Whereas the amino terminal and second extracellular loop of CCR5 have been implicated in coreceptor function, the second extracellular loop is the major determinant of ligand specificity (18). Previously described anti-ECL2-A mAbs, such as 2D7 (20), thus block viral infection, but also prevent JRFL gp120 and chemokine binding to CCR5, indicating that this effect may be related to steric hindrance. To a lesser extent, this is also the case for antibodies against the ECL2-B loop or those multidomain-specific mAbs that lack an HIV-1 blocking effect, although they interfere partially with chemokine or gp120 binding, as is observed for mAbs against the N-terminal domain (18).
Prior attempts to correlate mAb ability to block gp120 binding to CCR5 and prevention of HIV-1 infection were unsuccessful (18). Whereas N-terminal-specific mAbs block gp120-CCR5 binding very efficiently, ECL2-specific antibodies are more potent in preventing viral infection. This lack of correlation between gp120 binding and coreceptor function led to two important conclusions: (i) the amino terminal domain of CCR5 is more important for gp120 binding, whereas the extracellular loops are more important for inducing the conformational changes in env that lead to membrane fusion and viral infection, and (ii) there is differential sensitivity in gp120 binding to structurally modified CCR5 as compared with chemokine binding or signaling (19). Although we cannot rule out that other anti-CCR5 antibodies promote receptor aggregation, related or unrelated to HIV-1 neutralizing effects, our results show that the CCR5–02 mAb induces a subtle change in the CCR5 receptor, probably dimerization-triggered, sufficient to block viral infection. We showed previously that the ability of the CCR2V64I mutant, in contrast to the wild-type CCR2 receptor, to delay AIDS progression in HIV-1-infected individuals may be related to its capacity to heterodimerize with other chemokine receptors used by the HIV-1 virus to infect cells (16). Here we show that it also may operate to prevent HIV-1 infection by specifically targeting the CCR5 receptor.
The search for specific agents that impede HIV infection but do not interfere with chemokine physiology has yielded three unrelated, low molecular weight compounds that block T-tropic HIV-1 strains by preventing gp120 interaction with CXCR4 (41). This finding and the results presented here indicate that a tool such as the CCR5–02 mAb, which does not compete with RANTES binding to CCR5 and lacks any known physiological activity, but suppresses HIV-1 replication, may be useful for exploring new approaches in the prevention and treatment of AIDS infection without inducing inflammatory side effects (42). It is thus possible that chemokine receptor dimerization agents can be found that block HIV-1 entry through CXCR4 and CCR5 receptors without affecting chemokine responses.
Existing therapies for AIDS treatment are limited to inhibitors of the HIV-1 protease and reverse-transcriptase enzymes. Resistance to these drugs develops rapidly through virus mutation, limiting their efficiency. Combination therapies, in which two or more drugs targeted against one or both of these enzymes are used concurrently, appear to delay the onset of resistance. The long-term use of drug cocktails will, however, require new drugs that inhibit a variety of targets simultaneously. The most effective therapy today for HIV is likely to involve a combination of antiretroviral and immunomodulatory drugs. CCR5 and CXCR4 ligands engineered to be devoid of inflammatory effects may be the immunomodulators of choice; CCR5–02 mAb may be a candidate.
Acknowledgments
We thank Drs. S. Baik and R.W. Doms for JFRL gp120:CCR5 binding assays and critical reading of the manuscript, L. Gómez and J. Martín-Caballero for animal treatment, and C. Bastos and C. Mark for secretarial and editorial assistance, respectively. CCR5-transfected MT2 cells and R5 HIV-1 primary isolates were donated by Dr. J. Alcamí (Hospital 12 Octubre, Madrid). SF2 dual-tropic HIV-1 strain was a gift of Dr. C. López-Galíndez (Instituto de Salud Carlos III, Madrid). This work was supported by grants from the Spanish Comision Interministerial de Ciencia y Tecnologia, European Union Structural Fund, and Comunidad Autónoma de Madrid, and Pharmacia & Upjohn. The Department of Immunology and Oncology was founded and is supported by the Spanish Research Council (CSIC) and Pharmacia & Upjohn.
Abbreviations
- PBMC
peripheral blood mononuclear cell
- RANTES
regulated upon activation, normal T cell expressed and secreted
- SCID
severe combined immunodeficient
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.050457797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.050457797
References
- 1.Baggiolini M. Nature (London) 1998;392:565–568. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- 2.Rollins B J. Blood. 1997;90:909–928. [PubMed] [Google Scholar]
- 3.Kunkel S L, Strieter R M, Lindley I J D, Westwick J. Immunol Today. 1995;16:559–561. doi: 10.1016/0167-5699(95)80076-X. [DOI] [PubMed] [Google Scholar]
- 4.Murphy P M. Annu Rev Immunol. 1994;12:593–633. doi: 10.1146/annurev.iy.12.040194.003113. [DOI] [PubMed] [Google Scholar]
- 5.Littman D R. Cell. 1998;93:677–680. doi: 10.1016/s0092-8674(00)81429-4. [DOI] [PubMed] [Google Scholar]
- 6.Hoffman T L, Doms R W. AIDS. 1998;12, Suppl. A:S17–S26. [PubMed] [Google Scholar]
- 7.Doranz B J, Rucker J, Yi Y, Smyth R J, Samson M, Peiper S C, Parmentier M, Collman R G, Doms R W. Cell. 1996;85:1149–1158. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- 8.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, DiMarzio P, Marmon S, Sutton R E, Hill C M, et al. Nature (London) 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 9.Alkhatib G, Combadiere C, Broder C C, Feng Y, Kennedy P E, Murphy P M, Berger E A. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 10.Berger E A, Doms R W, Fenyš E-M, Korber B T M, Littman D R, Moore J P, Sattentau Q J, Schuitemaker H, Sodroski J, Weiss R A. Nature (London) 1998;391:240. doi: 10.1038/34571. [DOI] [PubMed] [Google Scholar]
- 11.Liu R, Paxton W A, Choe S, Ceradini D, Martin S R, Horuk R, MacDonald M E, Stuhlmann H, Koup R A, Landau N R. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 12.Samson M, Libert F, Doranz B J, Rucker J, Liesnard C, Farber C M, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, et al. Nature (London) 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 13.Kostrikis L G, Huang Y, Moore J P, Wolinsky S M, Zhang L, Guo Y, Deutsch L, Phair J, Neumann A U, Ho D D. Nat Med. 1998;4:350–353. doi: 10.1038/nm0398-350. [DOI] [PubMed] [Google Scholar]
- 14.Smith M W, Dean M, Carrington M, Winkler C, Huttley G A, Lomb D A, Goedert J J, O'Brien T R, Jacobson L P, Kaslow R, et al. Science. 1997;277:959–965. doi: 10.1126/science.277.5328.959. [DOI] [PubMed] [Google Scholar]
- 15.Smith M W, Carrington M, Winkler C, Lomb D, Dean M, Huttley G, O'Brien S J. Nat Med. 1997;3:1052–1053. doi: 10.1038/nm1097-1052c. [DOI] [PubMed] [Google Scholar]
- 16.Mellado M, Rodríguez-Frade J M, Vila-Coro A J, Martín de Ana A, Martínez-A C. Nature (London) 1999;400:723–724. doi: 10.1038/23382. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez-Frade J M, Vila-Coro A, Martín de Ana A, Nieto M, Sánchez-Madrid F, Proudfoot A E I, Wells T N C, Martínez-A C, Mellado M. J Cell Biol. 1999;144:755–765. doi: 10.1083/jcb.144.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee B, Sharron M, Blanpain C, Doranz B J, Vakili J, Setoh P, Berg E, Liu G, Guy H R, Durell S R, et al. J Biol Chem. 1999;274:9617–9626. doi: 10.1074/jbc.274.14.9617. [DOI] [PubMed] [Google Scholar]
- 19.Baik S W S, Doms R W, Doranz B J. Virology. 1999;259:267–273. doi: 10.1006/viro.1999.9779. [DOI] [PubMed] [Google Scholar]
- 20.Wu L, LaRosa G, Kassam N, Gordon C J, Heath H, Ruffing N, Chen H, Humblias J, Samson M, Parmentier M, et al. J Exp Med. 1997;186:1373–1381. doi: 10.1084/jem.186.8.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez-Frade J M, Vila-Coro A J, Martín de Ana A, Albar J P, Martínez-A C, Mellado M. Proc Natl Acad Sci USA. 1999;96:3628–3633. doi: 10.1073/pnas.96.7.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aragay A, Frade J M R, Mellado M, Serrano A, Martínez-A C, Mayor F., Jr Proc Natl Acad Sci USA. 1998;95:2985–2990. doi: 10.1073/pnas.95.6.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frade J M R, Llorente M, Mellado M, Alcamí J, Gutierrez-Ramos J C, Zaballos A, del Real G, Martínez-A C. J Clin Invest. 1997;100:497–502. doi: 10.1172/JCI119558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.del Real G, Llorente M, Boscá L, Hortelano S, Serrano A, Lucas P, Torán J L, Redondo C, Martínez-A C. AIDS. 1998;12:865–872. doi: 10.1097/00002030-199808000-00008. [DOI] [PubMed] [Google Scholar]
- 25.Nieto M, Rodríguez-Frade J M, Sancho D, Mellado M, Martínez-A C, Sánchez-Madrid F. J Exp Med. 1997;186:153–158. doi: 10.1084/jem.186.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vila-Coro A J, Mellado M, Martín de Ana A, Martínez-A C, Rodríguez-Frade J M. J Immunol. 1999;163:3037–3044. [PubMed] [Google Scholar]
- 27.Vila-Coro A J, Rodríguez-Frade J M, Martín de Ana A, Moreno-Ortíz M C, Martínez-A C, Mellado M. FASEB J. 1999;13:1699–1710. [PubMed] [Google Scholar]
- 28.Mosier D E. Adv Immunol. 1996;63:79–125. doi: 10.1016/s0065-2776(08)60855-x. [DOI] [PubMed] [Google Scholar]
- 29.Amara A, Gall S L, Schwartz O, Salamero J, Montes M, Loetscher P, Baggiolini M, Virelizier J L, Arenzana-Seisdedos F. J Exp Med. 1997;186:139–146. doi: 10.1084/jem.186.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mack M, Luckow B, Nelson P J, Cihak J, Simmons G, Clapham P R, Signoret N, Marsh M, Stangassinger M, Borlat F, et al. J Exp Med. 1998;187:1215–1224. doi: 10.1084/jem.187.8.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arenzana-Seisdedos F, Virelizier J L, Rousset D, Clark-Lewis I, Loetscher P, Moser B, Baggiolini M. Nature (London) 1996;383:400. doi: 10.1038/383400a0. [DOI] [PubMed] [Google Scholar]
- 32.Simmons G, Clapham P R, Picard L, Offord R E, Rosenkilde M M, Schwartz T W, Busser R, Wells T N C, Proudfoot A E I. Science. 1997;276:276–279. doi: 10.1126/science.276.5310.276. [DOI] [PubMed] [Google Scholar]
- 33.Rucker J, Samson M, Doranz B J, Libert F, Berson J F, Yi Y, Smyth R J, Collman R G, Broder C C, Vassart G, et al. Cell. 1996;87:437–446. doi: 10.1016/s0092-8674(00)81364-1. [DOI] [PubMed] [Google Scholar]
- 34.Atchison R E, Gosling J, Monteclaro F S, Franci C, Digilio L, Charo I F, Goldsmith M A. Science. 1996;274:1924–1926. doi: 10.1126/science.274.5294.1924. [DOI] [PubMed] [Google Scholar]
- 35.Samson M, LaRosa G, Libert F, Paindavoine P, Detheux M, Vassart G, Parmentier M. J Biol Chem. 1997;272:24934–24941. doi: 10.1074/jbc.272.40.24934. [DOI] [PubMed] [Google Scholar]
- 36.Blanpain C, Lee B, Vakili J, Doranz B J, Govaerts C, Migeotte I, Sharron M, Dupriez V, Vassart G, Doms R W, Parmentier M. J Biol Chem. 1999;274:18902–18908. doi: 10.1074/jbc.274.27.18902. [DOI] [PubMed] [Google Scholar]
- 37.Dragic T, Trkola A, Lin S W, Nagashima K A, Kajumo F, Zhao L, Olson W C, Wu L, Mackay C R, Allaway G P, et al. J Virol. 1998;72:279–285. doi: 10.1128/jvi.72.1.279-285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rabut G E, Konner J A, Kajumo F, Moore J P, Dragic T. J Virol. 1998;72:3464–3468. doi: 10.1128/jvi.72.4.3464-3468.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farzan M, Choe H, Vaca L, Martin K, Sun Y, Desjardins E, Ruffing N, Wu L, Wyatt R, Gerard N, et al. J Virol. 1998;72:1160–1164. doi: 10.1128/jvi.72.2.1160-1164.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siciliano S J, Kuhmann S E, Weng Y, Madani N, Springer M S, Lineberger J E, Danzeisen R, Miller M D, Kavanaugh M P, DeMartino J A, Kabat D. J Biol Chem. 1999;274:1905–1913. doi: 10.1074/jbc.274.4.1905. [DOI] [PubMed] [Google Scholar]
- 41.Baggiolini M, Moser B. J Exp Med. 1997;186:1189–1191. doi: 10.1084/jem.186.8.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ward S G, Bacon K, Westwick J. Immunity. 1998;9:1–11. doi: 10.1016/s1074-7613(00)80583-x. [DOI] [PubMed] [Google Scholar]