

Abstract
The RNA binding and export factor (REF) family of mRNA export adaptors are found in several nuclear protein complexes including the spliceosome, TREX, and exon junction complexes. They bind RNA, interact with the helicase UAP56/DDX39, and are thought to bridge the interaction between the export factor TAP/NXF1 and mRNA. REF2-I consists of three domains, with the RNA recognition motif (RRM) domain positioned in the middle. Here we dissect the interdomain interactions of REF2-I and present the solution structure of a functionally competent double domain (NM; residues 1–155). The N-terminal domain comprises a transient helix (N-helix) linked to the RRM by a flexible arm that includes an Arg-rich region. The N-helix, which is required for REF2-I function in vivo, overlaps the highly conserved REF-N motif and, together with the adjacent Arg-rich region, interacts transiently with the RRM. RNA interacts with REF2-I through arginine-rich regions in its N- and C-terminal domains, but we show that it also interacts weakly with the RRM. The mode of interaction is unusual for an RRM since it involves loops L1 and L5. NMR signal mapping and biochemical analysis with NM indicate that DDX39 and TAP interact with both the N and RRM domains of REF2-I and show that binding of these proteins and RNA will favor an open conformation for the two domains. The proximity of the RNA, TAP, and DDX39 binding sites on REF2-I suggests their binding may be mutually exclusive, which would lead to successive ligand binding events in the course of mRNA export.
Keywords: NMR, export, gene expression, mRNA, structure
INTRODUCTION
In eukaryotic cells mRNA has to be transported from the nucleus to the cytoplasm, where it is translated (Rodriguez et al. 2004). Genetic screens in yeast have identified Mex67p, Yra1p, and Sub2p as essential proteins for mRNA export (Segref et al. 1997; Strasser and Hurt 2001). Mex67p has the functional ortholog TAP/NXF in higher eukaryotes, which associate with nucleoporins, providing a link between mRNA and the nuclear pore (Katahira et al. 1999; Bachi et al. 2000). TAP forms a stable heterodimer with p15/NXT-1 and depletion of p15 by RNA interference (RNAi) leads to nuclear accumulation of poly(A)+RNA (Herold et al. 2001; Longman et al. 2003).
A synthetic lethal screen with mex67-6 identified Yra1p, which directly binds Mex67p and RNA with high affinity (Strasser and Hurt 2000). Yra1p belongs to a conserved family of proteins termed RNA binding and export factors (REFs) (Stutz et al. 2000), thought to facilitate the recruitment of Mex67p/TAP to mRNA and subsequently bridge the interaction between TAP and mRNA (Stutz and Izaurralde 2003). REF binds amino acids 1–202 of TAP, which includes the RNA binding domain (Huang et al. 2003), and studies in Xenopus oocytes showed that REF stimulates export of mRNAs that would otherwise be exported inefficiently (Zhou et al. 2000; Rodrigues et al. 2001). However, depletion of REFs in S2 cells or Caenorhabditis elegans by RNAi shows they are not essential for mRNA export in contrast to Yra1p in yeast, implying that there are alternative export adaptors in higher eukaryotes (Gatfield and Izaurralde 2002; Longman et al. 2003). Several other adaptors have been discovered, including U2AF35 (Zolotukhin et al. 2002), HuD (Saito et al. 2004), and the SR proteins SRp20, 9G8, and ASF (Huang et al. 2003).
The primary structure of the REF family consists of a 12-amino-acid REF-N motif followed by an Arg–Gly-rich variable region of between 0 and 95 amino acids, a central RRM, then a C-terminal variable region of between 0 and 91 amino acids and a REF-C motif of 17 amino acids (Fig. 1; Stutz et al. 2000). Deletion of the YRA1 C-variable region and REF-C motif induces a modest mRNA export defect in yeast, whereas deletion of the RRM, REF-N motif, or REF-N together with the N-variable region leads to substantial mRNA export defects (Zenklusen et al. 2001). The N- and C-variable regions of REF bind RNA and TAP, however, the RRM has no detectable RNA binding activity in electrophoretic mobility shift assays (EMSA) (Rodrigues et al. 2001; Zenklusen et al. 2001). The solution structure of ALY (REF1-I) RRM has been determined, revealing that a conserved Phe/Tyr within RNP-1, which normally binds RNA, was replaced with an Asp (Pérez-Alvarado et al. 2003). However, antibodies to the REF RRM block RNA binding in vitro and mRNA export in vivo, suggesting a role for this domain in RNA interactions and export (Rodrigues et al. 2001).
FIGURE 1.

Primary and secondary structures for REF2-I and alignment with ALY. The secondary structural elements for murine REF2-I determined using NMR are shown above the sequence. The regions of each protein analyzed using NMR techniques are shown in bold. A schematic of the principal regions of REF2-I referred to in the text is shown below the sequence alignment. Flexible hinge regions are shown as blue loops above the sequence alignment on the basis of 15N[1H] NOEs (Fig. 6B).
Sub2p and its mammalian ortholog, UAP56, share homology with DECD box helicases and are required for spliceosome assembly (Kistler and Guthrie 2001; Libri et al. 2001). Sub2p/UAP56 interact physically with Yra1p/REF1-I, and Mex67p binding to Yra1p displaces Sub2p in vitro (Luo et al. 2001; Strasser and Hurt 2001). RNAi and overexpression confirmed that UAP56 is required for mRNA export, however, its precise role remains unclear (Gatfield et al. 2001; MacMorris et al. 2003). Humans have a UAP56 paralog, which shares 90% sequence identity, known as DDX39/URH49, and both proteins can rescue Sub2p loss in yeast (Pryor et al. 2004).
The TREX complex that couples transcription and export consists of the transcriptional elongation complex THO, Tex1p, UAP56/Sub2p, and REF/Yra1p (Strasser et al. 2002). The association of TREX with transcribed genes provides a means to recruit UAP56/Sub2p and REF/Yra1p to spliced and intronless genes to promote their export. Yeast Npl3p provides a further link between transcription and export, since it is cotranscriptionally recruited to mRNAs and binds Mex67p (Gilbert and Guthrie 2004). REF proteins are also found in the exon junction complex (Le Hir et al. 2000), deposited on mRNAs during splicing, although recent data indicate that splicing plays a relatively minor role in mRNA export. Instead, it leads to enhanced gene expression by stimulating 3′ end formation and translation (Lu and Cullen 2003; Nott et al. 2003).
Although the general domain architecture of REF2-I, based on homology sequence analysis, is known to consist of three domains, N, RRM, and C (Stutz et al. 2000), and the solution structure of the RRM domain of ALY has been reported previously (Pérez-Alvarado et al. 2003), little is known about the structural properties of N and C domains, how and whether they interact with each other, and how these functionally important domains behave when REF2-I interacts with various ligands. Here we show that in the free state the REF2-I N domain, which includes a transient helix, interacts with the RRM. Together, the N and RRM domains of REF2-I provide a binding platform for DDX39, RNA, and TAP. The close proximity of the DDX39 and TAP binding sites on REF2-I revealed by this analysis provides an explanation for the displacement of Sub2p from Yra1p by Mex67p observed previously (Strasser and Hurt 2001).
RESULTS
Domain structure for REF2-I
The sequences and secondary structural elements for REF2-I and REF1-I/ALY are shown in Figure 1. The structure of a fragment of ALY (bold sequence in Fig. 1) was determined recently using NMR spectroscopy (Pérez-Alvarado et al. 2003). However, the N and C domains of ALY were largely removed to improve protein solubility and spectra, and this structural analysis revealed little about the mechanism of the export adaptor function. In the presence of the N- or C-terminal domains, REF2-I has poor solubility in standard buffers. However, we found REF2-I can be solubilized at concentrations suitable for NMR analysis with 50 mM L-Arg and L-Glu and still interacts with proteins and RNA (Golovanov et al. 2004). Using such media, we compared 2D 1H-15N heteronuclear single quantum coherence (HSQC) spectra of REF2-I (amino acids 1–218) with spectra of NM (amino acids 1–155) and C domains (amino acids 156–218) (Fig. 2). Spectra of the C domain and REF2-I overlay well, and neither deletion of the C domain (Fig. 2B) nor addition of the nonlabeled C domain to 15N,2H-labeled NM (data not shown) altered the amide signal positions of residues in the N (amino acids 1–74) and RRM (amino acids 75–155) domains. This indicates that NM and C domains do not interact noticeably. Much of the C domain is poorly folded, as revealed by small chemical shift dispersion and the presence of negative heteronuclear 15N[1H] nuclear Overhauser effects (NOEs) (data not shown). In light of this, we focused the structural analysis on the double domain NM, which binds RNA, UAP56/DDX39, and TAP-p15. By using a functional fragment of REF2-I we have been able to investigate its mode of action, in particular relating to the functionally important N domain, which was not possible with the earlier ALY structure.
FIGURE 2.

The REF2-I, NM, and C domain HSQC spectra overlay. (A) The HSQC spectrum for full length REF2-I is shown in black. (B) An overlay of the HSQC spectra for NM (amino acids 1–155; green) with the spectra for full-length REF2-I (black). (C) An overlay of the HSQC spectra for full-length REF2-I (black), NM (green), and the C domain (red).
To assess whether the solubilization protocol modifies the structural properties of NM, we compared the HSQC spectra of 15N,2H-NM at low protein concentration (<0.07 mM) collected either in the presence of 50 mM L-Arg, L-Glu, and β-mercaptoethanol or in the presence of 5 mM L-Arg and L-Glu without β-mercaptoethanol. The spectra collected in these two buffers were very similar (not shown), with signal shifts of δ < 0.03 ppm for residues belonging to the RRM domain, and shifts of δ < 0.05 ppm for residues from the flexible N domain. The general similarity of these spectra confirms that the additives used to increase the solubility of the protein do not modify its structural properties. To eliminate even the small influence of buffer solution on chemical shifts of amide signals, all other comparisons between spectra were done using identical buffers and experimental conditions.
Structural overview of REF2-I (NM)
The NMR data statistics and structural quality analysis for a final set of 14 structures of NM are presented in Table 1 and secondary structure elements were identified (Figs. 1, 3A). The results of structural calculation revealed that the N-terminal residues 9–18 form an α-helix (N-helix), as predicted previously (Stutz et al. 2000). The helical structure is supported by the chemical shift index (Wishart and Sykes 1994), strong sequential HN-HN NOE contacts, and several weak NOEs between residue side chains (Fig. 3A,B). One side of the N-helix is formed by hydrophobic residues, and the opposite side is polar. The N-helix is likely to be transient, as the values of heteronuclear 15N[1H] NOEs for this region were significantly reduced compared with those for the RRM (Fig. 6B, see below), and the intensities of Hα/Hβ(i) to HN(i+3/i+4) NOE cross-peaks were smaller than expected for stable α-helices. The remainder of the N domain appears largely unstructured, although some regions have restricted mobility (see below). The RRM has a typical βαββαβ topology conserved in other RRM domains. The backbone positions of the final set of 14 structures of NM are well defined for the RRM regions with secondary structure, with root-mean-square deviations (RMSDs) of 0.47 Å and 0.98 Å for the backbone and all heavy atoms, respectively. The best-fit superposition of the final 14 RRM structures and a ribbon representation of the RRM of NM are shown in Figures 3C,D.
TABLE 1.
Input for the structure calculations and quality analysis of final 14 best NMR conformers of REF2-I (1-155)

FIGURE 3.

The secondary and tertiary structure for NM. (A) Secondary structure of NM derived from NMR data. The intensities of NOE cross-peaks between HN protons of adjacent amino acid residues dNN(i,i+1) are classified as strong, medium, weak, or absent (represented by the height of the bars). Asterisks denote cases when HN protons of subsequent amino acid residues have the same chemical shift; hence no information about their NOE cross-peak intensity can be obtained. Consensus chemical shift indexes based on analysis of Cα, Cβ, CO, and Hα chemical shifts for each residue are shown as bars, above the central line for β-structure and below the line for α-structure. Positions of secondary structure elements identified in the process of analysis and structure calculation are shown at the bottom. The N-terminal T7 tag sequence is unstructured and is not shown. The figure was prepared using the Vince program from the Rowland NMR Toolkit. (B) The transient N-helix is shown as a ribbon and side chains are shown in red lines. Backbone HN and CO bonds are shown as red thin lines. The NOE upper-limit distance constraints derived from 3D 13C-edited NOESY-HSQC and 15N-edited NOESY-HSQC are shown in blue. (C) Superpositon of the 14 best structures of the RRM domain of NM. (D) Ribbon representation of the RRM domain of NM.
FIGURE 6.

NMR chemical shift mapping of RNA binding to NM. (A) Overlay of 2D HSQC spectra of 15N,2H-NM in the absence (blue) or in the presence (red) of RNA. Spectra on the left were collected in high-salt buffer (in the presence of 50 mM L-Arg, 50 mM L-Glu, and 100 mM NaCl). Spectra on the right were collected in low-salt buffer (in the presence of 2.5 mM L-Arg, 2.5 mM L-Glu, and 50 mM NaCl). Other components of the buffer were the same. Signal shifts induced by RNA binding to the flexible domain become more pronounced. (B) The top panel shows the chemical shift differences (δ > 0.015 ppm) between free and RNA-bound 15N,2H-NM (red dots) in the presence of high Arg/Glu (50 mM). Black arrows at the bottom mark residues whose RNA-induced amide signal shifts are codirectional with those caused by removal of amino acids 1–53 from NM. The bottom panel presents heteronuclear 15N[1H] NOEs measured for 15N,2H-NM in the absence (green squares) or presence (red triangles) of RNA.
Despite the presence of the N-terminal domain, the RRM structures for REF2-I and ALY are very similar; the RMSD of backbone atom positions between the mean structures is 1.03 Å for residues within the secondary structure elements (excluding loops) and 2.26 Å for the whole RRM (residues 75–152 including loops). The sequences for the RRM domains of REF2-I and ALY are similar (Fig. 1), although there are minor differences: (1) Asp107 of NM is replaced with His in ALY, which may affect the stability of loop L3; and (2) at the base of loop L5, Lys136 and Asp146 are replaced with asparagines in ALY, which may affect the stability of L5. The other three amino acid substitutions affect solvent-exposed residues and are unlikely to account for the differences between these domains. The loop region L5 of REF2-I is less well defined than that of ALY, and this is probably because it transiently interacts with the N domain (see below), which in turn reduces the number of local NOE restraints due to signal broadening.
Interaction between N and RRM domains
The absence of long-range proton–proton NOEs and reduced 15N[1H] NOEs indicates that the N domain is flexible (Fig. 6B, see below) yet has restricted mobility in the following regions: amino acids 6–23, 30–34, and 46–62, mapping to the N-helix, and Arg- and aromatic-rich regions, respectively (Fig. 1). This suggests that regions of the N domain may have the propensity to form local structure and/or interact with other parts of REF2-I. However, no proton–proton NOEs were found between the N and RRM domains, indicating that any intramolecular interactions were probably transient. Many of the resonances within the RRM domain of NM, in particular from residues at or near loop regions L1, L3, and L5, show significant line broadening (data not shown), which we considered may be due to conformational exchange induced by the interaction of the N domain with these regions of the RRM. Consistent with this hypothesis, we found that removal of the first 53 amino acids of REF2-I, generating a construct similar to that used for ALY structure determination, significantly improves the HSQC spectrum and eliminated the differential signal broadening for residues from the RRM domain (Fig. 7B, see below).
FIGURE 7.

The RRM of REF2-I binds RNA. (A) Enlarged fragments of the 1H-15N HSQC spectra of 15N,2H-NM in the absence (black) or presence (red) of RNA, overlayed with fragments of the 15N-Δ53 spectra (green). The top panel depicts shifts due to RNA binding and N-terminal truncation that are codirectional and, hence, may both be caused by displacement of the N-terminal arm. The bottom panel shows signals with noncodirectional shifts, implicating residues directly involved in RNA binding. Arrows show the direction of the shifts. (B) 2D 1H-15N HSQC spectra of 15N-Δ53 in the absence (black) or in the presence (red) of RNA. Top panel displays enlarged parts of the spectra where signals shift. (C) UV cross-linking of a 32P-continuously labeled RNA probe to GST fusions of REF2-I. Proteins were analyzed by SDS-PAGE stained with Coomassie (left panel) and phosphorimaging (right panel). The asterisk shows the position of RNase A.
To investigate potential intra- and interdomain interactions in REF2-I we used GST pull-down assays with GST-N together with radiolabeled N, RRM, and C domains (Fig. 4A,B). These experiments showed that the N domain interacts with itself and with the RRM but fails to interact with the C domain. Further insights into the nature of the intra- and interdomain interactions were gained by comparing the chemical shift changes in the HSQC spectra associated with deletion of the first 15, 37, 53, and 70 amino acids from NM (Fig. 4C), using 15N-labeled NM, Δ15, Δ37, Δ53, and Δ70 protein constructs (Fig. 4A). Deletion of amino acids 1–15 removes a major part of the N-helix, causing severe changes in chemical shifts that span nine amino acids of the adjacent Arg-rich region. Since the N-helix has a significant number of negative charges on one side, we propose that these residues may bind the adjacent positively charged region to form a hairpin structure, which would account for the intradomain interaction seen using GST pull-downs with the N domain (Fig. 4B). This was further investigated using GST pull-downs with the N domain and truncation construct lacking the N-helix (Fig. 4D). Whereas GST-REF-N bound the N domain well, which could be accounted for by reciprocal binding of the positive and negatively charged regions on both N domains, GST-REF-N Δ15 bound the N domain weakly. Presumably, this was because the Arg-rich region from GST-REF-N Δ15 could still interact weakly with the negative charged side of the N-helix in the free radiolabeled N domain. When the N-helix was deleted from both the GST-fused N domain and the free N domain, the intramolecular N-domain interaction was no longer detectable, indicating that the N-helix is normally required for this interaction.
FIGURE 4.

Interdomain interactions in REF2-I. (A) Schematic representation of REF2-I truncation constructs used. (B) Pull-down assays between GST-N and 35S-labeled N, RRM, or C domains of REF2-I (lanes 1–3). Lane 4 contains purified GST-REF2-I. Lanes 5, 7, and 9 are GST controls. Lanes 6, 8, and 10 used GST-N. Eluted proteins (lanes 5–10) were analyzed by SDS-PAGE stained with Coomassie (left panel) and phosphorimaging (right panel). (C) Chemical shift differences (δ > 0.012 ppm) induced by successive deletions in NM, measured in high-salt NMR100 buffer. The protein constructs whose spectra are compared in each box is shown on the left. Arrows indicate residues for which the lower estimates for δ were larger than 0.1 ppm. The positions of secondary structural elements are shown above. (D) Pull-down assays between GST-N or GST –NΔ15 with 35S-labeled N or NΔ15 domains of REF2-I. The left panel shows the input samples. The central and right panels show the results of the pull-down assay.
Following deletion of amino acids 1–15 or 1–37 from NM, small signal shifts were observed in the RRM, which mapped to loop L5 and helix α1 where a large negatively charged surface is formed by the side chains of Asp 88, 90 and Glu 93, 97 (Figs. 4C, 9B, see below). This suggests that both the N-helix and adjacent Arg-rich region interact weakly with this surface patch of the RRM. Interestingly, further removal of amino acids 37–53 causes virtually no additional signal shifts in the RRM, suggesting this section does not interact with the RRM. This observation is supported by an increased mobility within amino acids 37–53, as indicated by negative 15N[1H] NOEs (Fig. 6B, see below). The removal of the first 53 amino acids, generating a construct seven amino acids shorter than that used for the ALY structure determination (Fig. 1), reduced the differential line-broadening apparent in the spectra of NM with the intensities of signals from the RRM becoming more uniform. However, removal of amino acids 1–70 of NM caused widespread signal shifts and broadening in the RRM, suggesting significant perturbation of the RRM fold and the existence of interconverting backbone conformations on the intermediate timescale (Fig. 4C, 9B, see below). This suggests the aromatic-rich region, which according to the heteronuclear NOEs has restricted mobility (Fig. 6B, see below), may also transiently interact with the RRM, contributing to its conformational stability. Finally, the glycine-rich region (amino acids 64–70), which displays negative NOE values (Fig. 6B, see below) and hence is very mobile, possibly acts as a hinge for N-domain movements. A model summarizing the structural features of NM is presented in Figure 9A (see below).
FIGURE 9.

Structural overview for NM and models for interaction with export factors. (A) A structural model for NM. The transient N-helix associates with the adjacent Arg-rich region (shown in dark green) and together they associate with the RRM. (B) Ligand-binding surfaces of the RRM domain of REF2-I identified by NMR signal mapping. (i) Structure of the RRM is shown in two orientations related by 180° rotation around the x-axis. Molecular surfaces are presented in the same orientations colored by (ii) electrostatic charge, negative (red) and positive (blue); (iii) residues whose amide chemical shifts are affected by deletion of residues 1–15 (Δ15 green) and further deletion of residues 16–37 (Δ37 magenta) or by deletion of residues 1–70 (Δ70 cyan; iv); residues affected (red) by RNA binding to NM (v) and Δ53 (vi). Residues affected (red) by complex formation with DDX39 (vii) or TAP-p15 (viii). Structural models for the interaction of DDX39 (C) and TAP-p15 (D) with NM. The Arg-rich region is shown in green and the aromatic rich region in blue.
The signal shifts for residues from the RRM domain caused by the successive removal of N-terminal fragments are relatively small (δ < 0.1 ppm). As the 50 mM L-Arg, L-Glu present in the buffer could weaken interactions between the domains, we confirmed that the small magnitudes in shifts were not caused by the presence of these additives; a parallel set of spectra from the same truncated proteins obtained at lower protein concentrations and 5 mM L-Arg, L-Glu gave similar small magnitudes of shift changes (data not shown). In addition, the spectrum of a 1:1 mixture of 15N-labeled samples of both NM and Δ53 revealed that the signals from the RRM domains from both samples were in identical positions to those found when separate samples were used. This direct experiment excluded the possibility that the relatively small signal shifts were systematic artifacts. In summary, both the chemical shift data and the GST pull-down assay pointed toward an interaction between the N and the RRM domains, this being transient as revealed by the absence of long-range proton–proton NOEs and the magnitude of the shift changes.
The role of the N-helix in interactions between the N and RRM domains was investigated further by site-directed mutagenesis of hydrophobic and polar sides of the helix. Three types of mutations (Fig. 5A) were introduced into the N-helix within a construct expressing the N domain (amino acids 1–73). The rationale for making these mutations was: (1) to disrupt the helical structure of the region 9–18 by introduction of two prolines (I12P, L15P, N-α proline mutant); (2) to modify the charged/polar side of the helix (D10A, D11A, K14A, N18A , N-α charged mutant); and (3) to modify the hydrophobic side of the helix (L9A, I12A, I13A, L15A, N-α hydrophobic mutant). The pull-down assays using radiolabeled constructs (Fig. 5B) revealed that the structural integrity of the N-helix and the presence of hydrophobic side chains on one side of the N-helix is required for interaction between domains N and RRM, as both the proline mutations and the mutation of the hydrophobic face of the helix weaken this interaction beyond detection. The removal of charged/polar face of the N-helix does not affect the binding of N to RRM, which identifies the hydrophobic side of the N-helix formed by the side chains of residues L9, I12, I13, and L15 as the binding site for the transient interaction with the RRM. Hence, while the NMR chemical shift changes revealed the presence of interactions between N and RRM domains, the mutagenesis data show that the N helix is involved in these interactions. A model for NM is proposed in which the N-terminal domain interacts transiently with the RRM domain via the hydrophobic side of the N-helix, and the positively charged Arg-rich region interacts both with the negatively charged patch on the surface of RRM and the negatively charged side of the N-helix (Fig. 9A,B, see below).
FIGURE 5.

The N-helix is functionally important. (A) Schematic of REF-N 1-73 highlighting mutations on two opposite sides of the transient N-helix. (B) Pull-down assays. Lanes 1–5 show the 35S-REF fragments used. GST (control, lanes 6, 8) or GST-TAP-p15 (lanes 7, 9–12) expressed in E. coli were first immobilized on glutathione-coated beads. Various 35S-radiolabeled REF2-I domains synthesized in rabbit reticulocytes were added to the binding reactions and eluted proteins were analyzed on SDS-PAGE by phosphorimaging (left, middle panels) and Coomassie blue (right panel). (C) Schematic representation of the tethered mRNA export reporter assay: splice donor (SD), splice acceptor (SA). (D) α-Myc Western blot of total 293T extracts expressing the MS2 fusions used for the tethered export assay. (E) Luciferase activity generated by the MS2 fusions in the tethered export assay. Error bars represent standard deviations from four independent sets of assays, each carried out in triplicate.
To investigate the functional importance of the N-helix in vivo we used a tethered mRNA export assay (Fig. 5C,D,E; Williams et al. 2005; Hargous et al. 2006), in which export factors were directly tethered via MS2 operators to an inefficiently spliced and exported pre-mRNA containing a luciferase gene within an intron. The tethering of TAP to the reporter mRNA leads to constitutive export and very high levels of luciferase activity, whereas REF2-I gives a more modest activation of luciferase (Fig. 5E). Using this assay we found that when the hydrophobic face of the N-helix is mutated to alanine, which prevents the interaction with the RRM (Fig. 5B), it also drastically reduces the ability of the MS2-REF2-I fusion to promote mRNA export, yet does not alter the expression levels of the MS2 fusion (Fig. 5D). Whether this reduction is a direct result of the loss of the N-RRM interdomain interaction or due to disruption of other protein–protein interactions with REF2-I is not possible to distinguish in this experiment; nevertheless, the results indicate an important role for the N-helix region in the function of REF2-I.
Mapping the RNA binding sites on REF2-I
Earlier EMSA experiments with REF proteins have shown that the N- and C-terminal domains are involved in RNA binding (Rodrigues et al. 2001), and studies of Yra1p indicate that the arginine-rich variable regions (amino acids 14–77 and 167–210 in Yra1p) within the N and C domains are required for RNA binding (Zenklusen et al. 2001). However, in all these studies using EMSA, there was no detectable RNA interaction with the RRM.
The RNA binding activity of NM was investigated here using NMR chemical shift and dynamic mapping. In 50 mM L-Arg, L-Glu a number of residues from the N domain of NM displayed signal shifts on addition of a 15-mer RNA oligonucleotide (Fig. 6A, left panel, B) with changes for amino acids 7–24 and 29–47 being most pronounced, consistent with EMSA data (Rodrigues et al. 2001; Zenklusen et al. 2001). The values of these chemical shifts were relatively small, which may have been caused by the high concentrations of L-Arg and L-Glu. Therefore, we carried out similar experiments using low salt/L-Arg-LGlu buffer (2.5 mM L-Arg, 2.5 mM L-Glu, 50 mM NaCl in 20 mM phosphate buffer) and lower (∼50 μM) protein concentration. The signal shifts caused by RNA binding became more pronounced (Fig. 6A, right panels) and increased up to twofold in value; however, the same set of signals was affected as for high salt/L-Arg-L-Glu buffer. These control experiments show that although L-Arg, L-Glu influences the magnitude of signal shifts on RNA binding, it does not affect which residues show shifts.
To investigate further which residues participate in RNA binding we measured the changes in heteronuclear 15N[1H] NOEs that provide a quantitative parameter for mobility of polypeptide chain. Residues 15–58 show significantly increased heteronuclear 15N[1H] NOEs upon addition of RNA, indicating reduced mobility and direct involvement in RNA binding (Fig. 6B), even in high-salt buffer, yet the heteronuclear 15N[1H] NOEs for amino acids 7–14 were virtually unchanged, implying these residues are not directly involved in RNA binding, despite the changes in their amide signal positions. These data agree with the finding that deletion of amino acids 1–15 does not affect RNA binding for Yra1p (Zenklusen et al. 2001). We speculate that the signal shifts observed for amino acids 7–14, following RNA binding, may be caused by the displacement of the N-helix from the Arg-rich region and/or the surface of the RRM domain. If the N-terminal domain is displaced from the RRM following RNA binding, then this may lead to indirect changes in the spectra for residues of the RRM, similar to those caused by N-terminal deletion. Consistent with this, the signals for RRM residues affected by N-terminal truncation also change their chemical shifts in complex with RNA, and these signals move in the same direction (codirectionally); their position in the sequence is marked with downward black arrows in Figure 6B. Therefore, we propose that once the arginine-rich region binds RNA, the N domain is displaced from and no longer associates with the RRM.
Interestingly, some signal shifts induced by addition of RNA implicated additional residues within the RRM, which were unaffected by the N-terminal deletion, namely, 82LDFGV86 from loop L1 and R143 from loop L5 (red dots without downward pointing arrows underneath, Figs. 6B, 7A). We called these shifts “noncodirectional” and suggest that they are induced by the direct interaction of these regions with the RNA. As an additional test, to separate the direct and indirect effects of RNA binding on the signal shifts within the RRM, we added an excess of RNA to 15N-labeled REF-Δ53, which lacks much of the N domain, under the same experimental conditions as for REF-NM (in high-salt buffer). This time only the signals from the same L1 and L5 loop regions were affected; however, the values of these signal shifts were very small even at fivefold excess of RNA. This is consistent with very weak binding to the isolated RRM domain. In contrast, when the N domain is present, which binds RNA well in EMSA, the chemical shifts on the RRM residues affected by RNA binding are larger (Fig. 7, cf. A and B), suggesting that the RNA bound via the N domain has an increased probability of interacting with the RRM domain residues, causing larger chemical shifts.
Since EMSAs failed to detect interactions between the REF/Yra1p RRM and RNA (Rodrigues et al. 2001; Zenklusen et al. 2001), we used UV cross-linking, which is more sensitive, to confirm the REF2-I RRM binds RNA. As shown in Figure 7C, full-length REF2-I together with N- and C-terminal domains showed a strong UV cross-link whereas the RRM showed a weaker UV cross-link with RNA, confirming that this domain interacts with RNA. We suggest that the similarity of the effect of RNA addition and N-terminal deletion on parts of the RRM domain can be interpreted as RNA binding favoring an open conformation for NM in which the N terminus is displaced from the surface of the RRM domain, providing an extended RNA binding interface, consisting of the Arg-rich region from the N domain and L1 and L5 from the RRM.
Identification of the DDX39 and TAP/p15 binding sites on REF2-I NM
To investigate how mRNA export factors might interact with REF2-I we used a combination of GST pull-down assays and NMR signal mapping experiments. For NMR studies we found that DDX39, which has 90% sequence identity with UAP56, expressed much better in Escherichia coli and was more stable in NMR experiments. We investigated the changes in the HSQC spectra on addition of nonlabeled DDX39 to 15N,2H-labeled NM. A significant number of NM signals in the NM:DDX39 disappear from the spectra due to line broadening, suggesting that both the N and RRM domains are involved in complex formation (Fig. 8A,C). The interaction observed was specific since addition of excess lysozyme as a control caused no significant changes in the 2D HSQC spectra of NM (not shown). Regions of the RRM most affected by DDX39 binding indicated by amide signals disappearing include loop L1, helix α1, and the first β-strand (Figs. 8C,9B). We assessed the contribution of the N and RRM domains to the DDX39 interaction using GST pull-down assays (Fig. 8D). The Δ53 construct was used in these assays, since further truncation of the protein leads to extensive perturbation of the HSQC spectrum for the RRM (Fig. 4C). Whereas NM showed a strong interaction with DDX39, the Δ53 construct encompassing the RRM showed a weak interaction. Thus, both the N and RRM domains are required for strong interaction with DDX39, which is consistent with the NMR signal mapping, which showed peaks disappearing in both domains. The weaker interactions between the isolated RRM and DDX39 observed in pull-down assays may be due to loss of cooperativity when the N domain DDX39 binding site is removed. In the N domain, signals disappear within amino acids 5–16, which includes a major part of the N-helix, and additionally within amino acids 29–41 from the Arg-rich region. Together the biochemical and NMR data indicate that NM presents an extended binding interface for DDX39 involving both the N and RRM domains.
FIGURE 8.

Interaction of NM with DDX39 and TAP. Overlay of the 2D HSQC spectra of 15N,2H-NM in the absence (black) and in the presence (red) of (A) DDX39 and (B) TAP-p15. (C) Amide chemical shift differences between free 15N,2H-NM and the complexes formed with DDX39 or TAP-p15. Arrows on top mark residues where signals disappeared from the spectra of complexes. Asterisks indicate residues with broadened amide signals in NM:TAP-p15 complex. (D) GST pull-downs using various truncations and peptides derived from REF2-I with 35S-labeled DDX39 and TAP. The left panel is stained with Coumassie blue and the right panel shows a phosphorimage of the same gel.
Analysis of HSQC spectra of 15N,2H-NM upon addition of TAP-p15 revealed that signals from amino acids 8, 9, 17 (N-helix), and 23–48 (Arg-rich) of the N domain are broadened and disappear from the spectra (Fig. 8B,C). In contrast to the complex of NM with DDX39, no signals belonging to the RRM disappear completely nor are shifted significantly in the NM:TAP-p15 spectra. However, signals from residues denoted with asterisks, which cluster on α1 and α2 helices, are significantly broadened (Figs. 8C, 9B), indicating that they might be involved in transient interactions. This is consistent with the GST pull-down data, which show that TAP interacts weakly with the RRM and more strongly with NM (Fig. 8D). Earlier studies failed to detect the interaction between TAP/MEX67p and the REF/Yra1p RRM (Rodrigues et al. 2001; Zenklusen et al. 2001). This may have resulted from truncation of the RRM constructs used beyond the equivalent of residue 53, which leads to widespread perturbation of the HSQC spectrum for the RRM region of REF2-I and probably disrupts its structure (Fig. 4C).
DISCUSSION
REF2-I structure and interdomain interactions
Structural studies on the REF2-I-related protein ALY (Pérez-Alvarado et al. 2003) utilized the RRM domain with a short N-terminal extension, which lacked the functionally important N- and C-terminal domains and thus provided limited information about the function of this family of proteins. In the presence of the N- and C-terminal domains, REF2-I displays very poor solubility in standard buffers. We, therefore, devised a solubilization protocol involving the addition of 50 mM L-Arg/L-Glu to overcome the solubility problems (Golovanov et al. 2004), which has allowed us to analyze the structural properties of a functional fragment of REF2-I. We carried out extensive controls to demonstrate these additives do not disrupt the overall structure of REF2-I. These results are borne out by our observation that the structures of the highly related SR proteins 9G8 (determined in the presence of 50 mM L-Arg/L-Glu) and SRp20 (no L-Arg/L-Glu) are very similar (Hargous et al. 2006). Together these results show that these additives do not interfere with the process of structure determination and do not affect the protein structure itself. Furthermore, we have shown that while these additives influence the magnitude of chemical shifts associated with ligand binding, the residues implicated in ligand interactions do not alter. Therefore, this solubilization strategy provides a means to analyze protein–ligand interactions with targets that were previously inaccessible due to solubility problems.
The analysis of the structural properties of REF2-I revealed that the conserved REF-N motif overlaps a region that can form a transient helix and that this helix, together with the adjacent arginine-rich region, bind to the RRM of REF2-I. The interdomain interaction in free REF2-I may prime the protein for interactions with RNA and protein ligands that involve both domains. It may also explain the propensity of REF proteins to aggregate and form multimers (Virbasius et al. 1999; Rodrigues et al. 2001). The REF2-I C-terminal domain does not interact with the N or RRM domains but does contain a conserved REF-C motif, which, for ALY, is involved in the interaction with UAP56 (Luo et al. 2001). The REF-C motif shows sequence homology with the REF-N motif (Zenklusen et al. 2001) and thus may form a transient helix as previously predicted (Stutz et al. 2000).
RNA binding to REF2-I
The N- and C-terminal arginine-rich domains of REF proteins bind RNA well, and this has been demonstrated using both EMSA and UV cross-linking in this and previous studies (Zenklusen et al. 2001). However, the values of changes in NMR chemical shifts (<0.2 ppm even in low-salt buffer) of amide signals from the N-domain residues interacting with RNA are still much smaller than those often reported for stable protein–RNA complexes (e.g., Auweter et al. 2006). However, unlike conventional folded RNA binding domains, the Arg-rich region of REF2-I is unstructured. Since REF2-I is likely to bind RNA without sequence specificity, the local flexibility of its RNA binding region may be important to allow interaction with a wide range of RNA molecules. As a consequence, one cannot expect the presence of unique conformations in the complexed form, as the binding site on RNA is not unique. Hence, for a sufficiently long RNA fragment, i.e., the 15mer used in these studies, the complex will exist as a heterogeneous mixture of individual complexes bound at slightly different sites on the RNA. We suggest that if the exchange between these different states is slow or intermediate on the NMR timescale, the protein signals would likely broaden up and multiply and/or disappear if the concentration of individual conformational states falls beyond the detection level. If the exchange is fast the signals from the bound form will be visible. However, their chemical shifts will be averaged over a great range of different conformations, in a similar way as chemical shifts of amides forming part of random coil are averaged to typical “random coil shifts” due to fast conformational exchange. Therefore, we predict that non-sequence-specific RNA binding to the unstructured Arg-rich region would cause smaller chemical shift perturbations than those occurring when forming a unique specific complex.
On the basis of the ALY structure determination and sequence analysis (Pérez-Alvarado et al. 2003) it was expected that the RRM would not bind RNA since it lacked critical residues in the RNP-1 and RNP-2 motifs within the RRM normally involved in RNA binding, and these predictions were consistent with the existing EMSA data, which failed to detect an interaction between the RRM and RNA. It was, therefore, somewhat surprising to observe NMR signal shifts in loops L1 and L5 of the REF2-I RRM on RNA binding. Since the chemical shift changes within this folded domain were also small, this would be consistent with weak binding, which would account for the failure to observe this interaction using EMSA. The mode of RNA binding by the REF2-I RRM is not unprecedented, since the direct contribution of loops L1 and L5 to RNA binding was recently observed for the RRM domain of Fox-1 (Auweter et al. 2006), with Phe126 in loop 1 playing a crucial part. The equivalent Loop 1 residue of REF2-I, which shows amide signal shifts on RNA binding is Phe84, which is conserved in REF proteins across species. The coordinate involvement of the RRM and N domains in RNA binding may in part explain why antibodies raised to the RRM block RNA binding (Rodrigues et al. 2001).
mRNA export factor binding to REF2-I
Previous work has shown that TAP/Mex67p can interact with both the N and C domains of REF/Yra1p proteins, which both contain arginine-rich regions (Rodrigues et al. 2001; Zenklusen et al. 2001), and the DDX39 ortholog, UAP56, interacts with the REF-C motif of ALY (Luo et al. 2001), which is completely conserved in REF2-I. We now show that for both TAP and DDX39 their interaction with REF2-I NM involves both the N domain and the RRM. In the case of TAP the N domain interaction may well be driven by conserved arginines present in the Arg-rich region (Stutz et al. 2000), which show signal shifts on TAP binding, since for two other export adaptors, 9G8 and SRp20, TAP recognition involves arginines within a flexible peptide adjacent to their RRM motifs (Hargous et al. 2006). The interaction of DDX39 with the REF N domain may well involve the REF-N motif, which shows signal shifts on addition of DDX39 but also shares sequence homology with the REF-C motif, which is involved in the recognition of ALY. The observation that the REF2-I RRM provides a protein interaction site is not unprecedented. The Herpes simplex virus ICP27 protein, which is involved in the post-transcriptional control of viral RNAs including export, interacts with REF2-I via the RRM motif and can, in fact, form a ternary complex with REF2-I and TAP (Koffa et al. 2001).
Whereas the N domain of free REF2-I interacts with the RRM, the C domain does not. This may lead to functional differences between these two domains with respect to ligand binding. Indeed, for Yra1p, the interaction with Mex67p is much stronger with the N+RRM domain than the RRM+C domain, which is very weak (Zenklusen et al. 2001). Furthermore, deletion of the REF-N motif, which is likely to prevent the N-RRM domain interaction, reduces binding to Mex67p, suggesting the N-RRM interaction primes the protein for interaction with Mex67p. However, we cannot exclude the possibility that Mex67p interacts weakly with the REF-N motif. In fact, the N-RRM domain interaction may prevent robust RRM+C domain interactions with export factors, or at least lead to preferential use of the N-domain binding sites in conjunction with the RRM. Furthermore, the use of the RRM for binding proteins might ensure that only a single molecule of TAP or DDX39/UAP56 associates with REF2-I in vivo despite the identification of N- and C-terminal binding sites using protein truncations. Consistent with this, reports showing an interaction between purified TAP/Mex67p and REF/Yra1p proteins have never demonstrated a stoichiometry greater than 1:1 (Stutz et al. 2000; Strasser and Hurt 2001). Depletion of the REF-N motif in Yra1p leads to a more significant mRNA export defect than depletion of the REF-C motif, which once again may in part be attributed to the ability of this motif to bind the RRM. However, the REF-N motif is also implicated in the interaction between Yra1p and Mlp proteins in yeast since mutations in this motif generate a temperature-sensitive protein when fused to GFP (GFP-yra1-8), which shows nuclear retention of poly(A)+RNA at the permissive temperature. Mlp2p functions as a high copy suppressor of GFP-yra1-8 and copurifies with Yra1p (Vinciguerra et al. 2005), thus, loss of these interactions may contribute to the mRNA export defect.
The pincerlike grip of mRNA export factors by REF2-I (Fig. 9C,D) involving the N-terminal arm and RRM together with the juxtaposition of potential binding sites in the C-terminal arm suggests that mRNA export factors such as UAP56 and TAP are unlikely to be able to simultaneously bind to REF2-I, which is consistent with the earlier observation that Mex67p displaces Sub2p from Yra1p (Strasser and Hurt 2001). Similarly, the overlapping binding sites for RNA and TAP suggests that their binding to REF2-I may also be mutually exclusive.
MATERIALS AND METHODS
Expression and purification of proteins
Plasmids construction and mutagenesis used standard methods, and details of the plasmids used in this study are shown in Supplementary Table 1, which can be found at http://www.shef.ac.uk/mbb/staff/wilson. Proteins were expressed in E. coli BL21(DE3)-RP cells following induction and overnight growth at 20°C. 6× His-tagged protein complexes were purified on TALON beads. TAP-p15 was further purified on a Hi-Trap heparin column. NMR protein samples were labeled by expression in minimal medium containing 15NH4Cl and/or 13C6-glucose and/or 2H2O and purified on TALON resin. Eluted proteins were supplemented with 50 mM β-mercaptoethanol, 10 mM EDTA, and 50 mM L-Arg and L-Glu (Golovanov et al. 2004) before dialysis against “high-salt” NMR100 buffer (20 mM Na phosphate buffer at pH 6.3, 100 mM NaCl, 50 mM L-Arg, 50 mM L-Glu, 5 mM EDTA, 50 mM β-mercaptoethanol). Ten millimolar DTT was added to the dialyzed extracts, and proteins were concentrated to 1 mM. Samples for NMR analyses were prepared in the NMR100 buffer using the procedures outlined by Golovanov et al. (2004). In a set of control mapping experiments a “low-salt” NMR50 buffer containing decreased amounts of L-Arg, L-Glu and NaCl was used (20 mM Na phosphate buffer at pH 6.3, 50 mM NaCl, 5 mM L-Arg, 5 mM L-Glu, 5 mM EDTA, 1 mM DTT).
NMR structure determination
Experiments used for sequence-specific assignments (Golovanov et al. 2006) and structure calculation were run on Bruker DRX600 and Varian Inova 800 MHz spectrometers at 30°C using 13C,15N-labeled NM. Spectral data were processed with NMRPipe (Delaglio et al. 1995) and visualized with NMRView (Johnson and Blevins 1994). NOE cross-peaks were obtained from 3D 15N-resolved NOESY (Varian Inova 800 MHz, in 1H2O), 13C-resolved NOESY (Bruker DRX600, in 1H2O), and 2D NOESY spectrum in 2H2O acquired with a mixing time of 150 msec. Structure calculations employed ARIA (Nilges et al. 1997) using as input NOE distance restraints, 1 DNH residual dipolar coupling restraints measured in a 5% solution of polyoxyethylene-5-lauryl ether (C12E5)/hexanol (Ruckert and Otting 2000), and dihedral angle constraints for the backbone angles φ and ψ derived from the analysis of chemical shift indexes (Wishart and Sykes 1994) and TALOS (Cornilescu et al. 1999). The involvement of amide groups in hydrogen bonds was deduced from temperature coefficients, as the exchange rate of amide hydrogens to deuterons was very fast. The final 14 structures with the lowest target function minimized in water (Nilges et al. 1997) were analyzed with PROCHECK (Morris et al. 1992), and drawings were prepared using MolMol (Koradi et al. 1996) and GRASP (Nicholls et al. 1991). The structure of NM has been deposited in the Protein Data Bank, accession number 2F3J.
Chemical shift mapping
All chemical shift mapping experiments were done on a Bruker DRX600 spectrometer using CryoProbe or TXI probeheads. All pairs of HSQC spectra used for interactions mapping were acquired under identical experimental and buffer conditions to exclude signal shifts caused by artifacts. For RNA interaction analysis, a 1.5 M excess of a 15mer CAGUCGCAUAGUGCA was predissolved in buffer and mixed with 15N, 2H-NM, present either at 0.5 mM in high-salt NMR100 buffer or at 0.05 mM in low-salt NMR50 buffer. The signal shifts observed are classified either as codirectional or noncodirectional with the shifts of correspondent signals observed upon removal of the N-terminal 53 amino acids. For Figure 7B, a fivefold molar excess of RNA was added to 15N-Δ53 in a high-salt NMR100 buffer.
For protein–protein complexes, a 1.5 M excess of DDX39 or TAP-p15 was mixed with 0.5 mM of 15N,2H-NM before dialysis against NMR100 buffer. Ten millimolar DTT was added to the samples before concentrating. Protein ratios were checked by SDS-PAGE (data not shown).
The weighted amide chemical shift differences δ between HSQC spectra 1 and 2 caused by mutations or complex formation were measured as 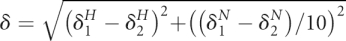 , where δH and δN are proton and nitrogen chemical shifts, respectively. In Figure 4C the peaks that remained in the spectra but were shifted for δ > 0.1 ppm, are mainly those from amino acid residues close to the N-terminal deletion site. The assignment of such signals from the truncation constructs required for precise measurement of δ could not be made reliably. Therefore, we used the lower estimate for their shifts by measuring the distance to the closest candidate peak (Williamson et al. 1997).
, where δH and δN are proton and nitrogen chemical shifts, respectively. In Figure 4C the peaks that remained in the spectra but were shifted for δ > 0.1 ppm, are mainly those from amino acid residues close to the N-terminal deletion site. The assignment of such signals from the truncation constructs required for precise measurement of δ could not be made reliably. Therefore, we used the lower estimate for their shifts by measuring the distance to the closest candidate peak (Williamson et al. 1997).
GST pull-down assays
GST fusions were bound to GSH beads. Nine microliters of 35S-Met N, M, C constructs made in reticulocyte lysates were added to the washed beads in 900 μL of RB100 buffer (25 mM HEPES-KOH at pH 7.5, 100 mM KOAc, 10 mM MgCl2, 1 mM DTT, 10% glycerol, 0.05% Triton) for 30 min at 4°C. Bound proteins were eluted from washed beads and analyzed on SDS-PAGE stained with Coomassie and by phosphorimaging. The input lane corresponds to 5% of the protein used in the binding reactions and 25% of the bound material was loaded on gels.
RNA analysis
For UV cross-linking assays, a 32mer 32P continuously labeled RNA was synthesized in vitro from 1 μg XbaI-restricted pBluescript-KS using T7 polymerase. Five-microgram GST fusions of REF, N, RRM, or C in 18 μL RNA-CL buffer (15 mM HEPES at pH 7.9, 8 mM NaCl, 100 μM KCl, 0.2 mM EDTA, 5 mM MgCl2, 0.05% Tween 20, 10% glycerol) were mixed with 2 μL of RNA for 10 min on ice and 10 min at room temperature. Reactions were treated with 5 μg RNase A for 30 min at 37°C and incubated for 15 min with 10 μL GSH beads. Purified complexes were analyzed by SDS-PAGE stained with Coomassie and phosphorimaging.
ACKNOWLEDGMENTS
We thank G. Kelly for help at the UK National 800 MHz NMR Facility and V. Porteous for technical support. This work was supported by grants from the BBSRC.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.212106.
REFERENCES
- Auweter, S.D., Fasan, R., Reymond, L., Underwood, J.G., Black, D.L., Pitsch, S., Allain, F.H. Molecular basis of RNA recognition by the human alternative splicing factor Fox-1. EMBO J. 2006;25:163–173. doi: 10.1038/sj.emboj.7600918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachi, A., Braun, I.C., Rodrigues, J.P., Pante, N., Ribbeck, K., von Kobbe, C., Kutay, U., Wilm, M., Gorlich, D., Carmo-Fonseca, M., et al. The C-terminal domain of TAP interacts with the nuclear pore complex and promotes export of specific CTE-bearing RNA substrates. RNA. 2000;6:136–158. doi: 10.1017/s1355838200991994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornilescu, G., Delaglio, F., Bax, A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., Bax, A. Nmrpipe—A multidimensional spectral processing system based on unix pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Gatfield, D., Izaurralde, E. REF1/Aly and the additional exon junction complex proteins are dispensable for nuclear mRNA export. J. Cell Biol. 2002;159:579–588. doi: 10.1083/jcb.200207128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatfield, D., Le Hir, H., Schmitt, C., Braun, I.C., Kocher, T., Wilm, M., Izaurralde, E. The DExH/D box protein HEL/UAP56 is essential for mRNA nuclear export in Drosophila . Curr. Biol. 2001;11:1716–1721. doi: 10.1016/s0960-9822(01)00532-2. [DOI] [PubMed] [Google Scholar]
- Gilbert, W., Guthrie, C. The Glc7p nuclear phosphatase promotes mRNA export by facilitating association of Mex67p with mRNA. Mol. Cell. 2004;13:201–212. doi: 10.1016/s1097-2765(04)00030-9. [DOI] [PubMed] [Google Scholar]
- Golovanov, A.P., Hautbergue, G.M., Wilson, S.A., Lian, L.-Y. A simple method for improving protein solubility and long-term stability. J. Am. Chem. Soc. 2004;126:8933–8939. doi: 10.1021/ja049297h. [DOI] [PubMed] [Google Scholar]
- Golovanov, A.P., Hautbergue, G.M., Wilson, S.A., Lian, L.Y. Assignment of (1)H, (13)C, and (15)N resonances for the REF2-I mRNA export factor. J. Biomol. NMR. 2006 doi: 10.1007/s10858-006-9012-2. (in press) [DOI] [PubMed] [Google Scholar]
- Hargous, D., Hautbergue, G.M., Tintaru, A.M., Skrisovska, L., Golovanov, A.P., Stevenin, J., Lian, L.-Y., Wilson, S.A., Allain, F.H.-T. Molecular basis of RNA recognition and TAP binding by the SR proteins SRp20 and 9G8. EMBO J. 2006 doi: 10.1038/sj.emboj.7601385. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold, A., Klymenko, T., Izaurralde, E. NXF1/p15 heterodimers are essential for mRNA nuclear export in Drosophila . RNA. 2001;7:1768–1780. [PMC free article] [PubMed] [Google Scholar]
- Huang, Y., Gattoni, R., Stevenin, J., Steitz, J.A. SR splicing factors serve as adapter proteins for TAP-dependent mRNA export. Mol. Cell. 2003;11:837–843. doi: 10.1016/s1097-2765(03)00089-3. [DOI] [PubMed] [Google Scholar]
- Johnson, B.A., Blevins, R.A. Nmr View—A computer-program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- Katahira, J., Strasser, K., Podtelejnikov, A., Mann, M., Jung, J.U., Hurt, E. The Mex67p-mediated nuclear mRNA export pathway is conserved from yeast to human. EMBO J. 1999;18:2593–2609. doi: 10.1093/emboj/18.9.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kistler, A.L., Guthrie, C. Deletion of MUD2, the yeast homolog of U2AF65, can bypass the requirement for sub2, an essential spliceosomal ATPase. Genes & Dev. 2001;15:42–49. doi: 10.1101/gad.851301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffa, M.D., Clements, J.B., Izaurralde, E., Wadd, S., Wilson, S.A., Mattaj, I.W., Kuersten, S. Herpes simplex virus ICP27 protein provides viral mRNAs with access to the cellular mRNA export pathway. EMBO J. 2001;20:5769–5778. doi: 10.1093/emboj/20.20.5769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., Wuthrich, K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:51. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- Le Hir, H., Izaurralde, E., Maquat, L.E., Moore, M.J. The spliceosome deposits multiple proteins 20–24 nucleotides upstream of mRNA exon–exon junctions. Embo J. 2000;19:6860–6869. doi: 10.1093/emboj/19.24.6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libri, D., Graziani, N., Saguez, C., Boulay, J. Multiple roles for the yeast SUB2/yUAP56 gene in splicing. Genes & Dev. 2001;15:36–41. doi: 10.1101/gad.852101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longman, D., Johnstone, I.L., Caceres, J.F. The Ref/Aly proteins are dispensable for mRNA export and development in Caenorhabditis elegans . RNA. 2003;9:881–891. doi: 10.1261/rna.5420503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, S., Cullen, B.R. Analysis of the stimulatory effect of splicing on mRNA production and utilization in mammalian cells. RNA. 2003;9:618–630. doi: 10.1261/rna.5260303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, M.L., Zhou, Z., Magni, K., Christoforides, C., Rappsilber, J., Mann, M., Reed, R. Pre-mRNA splicing and mRNA export linked by direct interactions between UAP56 and Aly. Nature. 2001;413:644–647. doi: 10.1038/35098106. [DOI] [PubMed] [Google Scholar]
- MacMorris, M., Brocker, C., Blumenthal, T. UAP56 levels affect viability and mRNA export in Caenorhabditis elegans . RNA. 2003;9:847–857. doi: 10.1261/rna.5480803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, A.L., Macarthur, M.W., Hutchinson, E.G., Thornton, J.M. Stereochemical quality of protein–structure coordinates. Proteins. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K.A., Honig, B. Protein folding and association—Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- Nilges, M., Macias, M.J., Odonoghue, S.I., Oschkinat, H. Automated NOESY interpretation with ambiguous distance restraints: The refined NMR solution structure of the pleckstrin homology domain from β-spectrin. J. Mol. Biol. 1997;269:408–422. doi: 10.1006/jmbi.1997.1044. [DOI] [PubMed] [Google Scholar]
- Nott, A., Meislin, S.H., Moore, M.J. A quantitative analysis of intron effects on mammalian gene expression. RNA. 2003;9:607–617. doi: 10.1261/rna.5250403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Alvarado, G.C., Martinez-Yamout, M., Allen, M.M., Grosschedl, R., Dyson, H.J., Wright, P.E. Structure of the nuclear factor ALY: Insights into post-transcriptional regulatory and mRNA nuclear export processes. Biochemistry. 2003;42:7348–7357. doi: 10.1021/bi034062o. [DOI] [PubMed] [Google Scholar]
- Pryor, A., Tung, L., Yang, Z., Kapadia, F., Chang, T.H., Johnson, L.F. Growth-regulated expression and G0-specific turnover of the mRNA that encodes URH49, a mammalian DExH/D box protein that is highly related to the mRNA export protein UAP56. Nucleic Acids Res. 2004;32:1857–1865. doi: 10.1093/nar/gkh347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues, J.P., Rode, M., Gatfield, D., Blencowe, B.J., Carmo-Fonseca, M., Izaurralde, E. REF proteins mediate the export of spliced and unspliced mRNAs from the nucleus. Proc. Natl. Acad. Sci. 2001;98:1030–1035. doi: 10.1073/pnas.031586198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, M.S., Dargemont, C., Stutz, F. Nuclear export of RNA. Biol. Cell. 2004;96:639–655. doi: 10.1016/j.biolcel.2004.04.014. [DOI] [PubMed] [Google Scholar]
- Rückert, M., Otting, G. Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. J. Am. Chem. Soc. 2000;122:7793–7797. [Google Scholar]
- Saito, K., Fujiwara, T., Katahira, J., Inoue, K., Sakamoto, H. TAP/NXF1, the primary mRNA export receptor, specifically interacts with a neuronal RNA-binding protein HuD. Biochem. Biophys. Res. Commun. 2004;321:291–297. doi: 10.1016/j.bbrc.2004.06.140. [DOI] [PubMed] [Google Scholar]
- Segref, A., Sharma, K., Doye, V., Hellwig, A., Huber, J., Luhrmann, R., Hurt, E. Mex67p, a novel factor for nuclear mRNA export, binds to both poly(A)+ RNA and nuclear pores. EMBO J. 1997;16:3256–3271. doi: 10.1093/emboj/16.11.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser, K., Hurt, E. Yra1p, a conserved nuclear RNA-binding protein, interacts directly with Mex67p and is required for mRNA export. EMBO J. 2000;19:410–420. doi: 10.1093/emboj/19.3.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser, K., Hurt, E. Splicing factor Sub2p is required for nuclear mRNA export through its interaction with Yra1p. Nature. 2001;413:648–652. doi: 10.1038/35098113. [DOI] [PubMed] [Google Scholar]
- Strasser, K., Masuda, S., Mason, P., Pfannstiel, J., Oppizzi, M., Rodriguez-Navarro, S., Rondon, A.G., Aguilera, A., Struhl, K., Reed, R., et al. TREX is a conserved complex coupling transcription with messenger RNA export. Nature. 2002;417:304–308. doi: 10.1038/nature746. [DOI] [PubMed] [Google Scholar]
- Stutz, F., Izaurralde, E. The interplay of nuclear mRNP assembly, mRNA surveillance and export. Trends Cell Biol. 2003;13:319–327. doi: 10.1016/s0962-8924(03)00106-5. [DOI] [PubMed] [Google Scholar]
- Stutz, F., Bachi, A., Doerks, T., Braun, I.C., Seraphin, B., Wilm, M., Bork, P., Izaurralde, E. REF, an evolutionary conserved family of hnRNP-like proteins, interacts with TAP/Mex67p and participates in mRNA nuclear export. RNA. 2000;6:638–650. doi: 10.1017/s1355838200000078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinciguerra, P., Iglesias, N., Camblong, J., Zenklusen, D., Stutz, F. Perinuclear Mlp proteins downregulate gene expression in response to a defect in mRNA export. EMBO J. 2005;24:813–823. doi: 10.1038/sj.emboj.7600527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virbasius, C.M., Wagner, S., Green, M.R. A human nuclear-localized chaperone that regulates dimerization, DNA binding, and transcriptional activity of bZIP proteins. Mol. Cell. 1999;4:219–228. doi: 10.1016/s1097-2765(00)80369-x. [DOI] [PubMed] [Google Scholar]
- Williams, B.J., Boyne, J.R., Goodwin, D.J., Roaden, L., Hautbergue, G.M., Wilson, S.A., Whitehouse, A. The prototype γ-2 herpesvirus nucleocytoplasmic shuttling protein, ORF57, transports viral RNA via the cellular mRNA export pathway. Biochem. J. 2005;387:295–308. doi: 10.1042/BJ20041223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson, R.A., Carr, M.D., Frenkiel, T.A., Feeney, J., Freedman, R.B. Mapping the binding site for matrix metalloproteinase on the N-terminal domain of the tissue inhibitor of metalloproteinases-2 by NMR chemical shift perturbation. Biochemistry. 1997;36:13882–13889. doi: 10.1021/bi9712091. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S., Sykes, B.D. The C-13 chemical-shift index—A simple method for the identification of protein secondary structure using C-13 chemical-shift data. J. Biomol. NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- Zenklusen, D., Vinciguerra, P., Strahm, Y., Stutz, F. The yeast hnRNP-like proteins Yra1p and Yra2p participate in mRNA export through interaction with Mex67p. Mol. Cell. Biol. 2001;21:4219–4232. doi: 10.1128/MCB.21.13.4219-4232.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Z., Luo, M.J., Straesser, K., Katahira, J., Hurt, E., Reed, R. The protein Aly links pre-messenger-RNA splicing to nuclear export in metazoans. Nature. 2000;407:401–405. doi: 10.1038/35030160. [DOI] [PubMed] [Google Scholar]
- Zolotukhin, A.S., Tan, W., Bear, J., Smulevitch, S., Felber, B.K. U2AF participates in the binding of TAP (NXF1) to mRNA. J. Biol. Chem. 2002;277:3935–3942. doi: 10.1074/jbc.M107598200. [DOI] [PubMed] [Google Scholar]