

Abstract
Transgenic mice expressing human insulin-like growth factor 1 (IGF-1) in basal epithelial cells of prostate have been characterized. Transgene expression led to activation of the IGF-1 receptor and spontaneous tumorigenesis in prostate epithelium. Hyperplasia was evident in these mice by 2–3 months of age. Atypical hyperplasias and prostatic intraepithelial neoplasia were evident by 6–7 months of age. Well differentiated adenocarcinomas appeared in mice 6 months or older. Less differentiated tumors, diagnosed as small cell carcinomas, were also observed in two of the older mice. Both lobes of the mouse prostate gland (dorsolateral and ventral) presented preneoplastic and neoplastic changes. The incidence of tumors in mice ≥6 months of age (38 mice total) was 50%. The development of neoplasia in these transgenic mice appeared to follow a stepwise progression through early preneoplastic changes that ultimately culminated in frank neoplasia. These mice offer an animal model for prostate cancer that will allow study of the stepwise development of this disease and the mechanism(s) whereby IGF-1 mediates this process.
Prostate cancer is the most commonly diagnosed cancer in men in the United States (1). Progress in prostate disease research has been impaired by the lack of adequate animal models that reproduce the human disease. There are several established rat models of prostate cancer that are either hormonally and/or chemically induced, such as the Lobund Wistar or Nobel rat models (2–5). In these models, the time frame to adenocarcinoma is 12–24 months. Spontaneous adenocarcinomas develop in the Dunning model (R-3327 system), which is carried as both cell lines and transplantable tumors in syngeneic Copenhagen rats (4). All of these model systems have certain limitations that have hampered their utility. Recently, several laboratories have created transgenic models in which prostate adenocarcinomas develop with high frequency (6–10). All of these models are based on expression of SV40-T antigen in prostate epithelium. Thus, a potential limitation of these models is the use of a transgene not directly involved in human prostate cancer. In some of these models, tumors develop rapidly (in some cases by 10–12 weeks), are poorly differentiated or undifferentiated, and progress rapidly to metastatic disease (6–8, 10).
Recently, Chan et al. (11) reported a strong positive association between serum insulin-like growth factor 1 (IGF-1) levels and prostate cancer risk. The importance of IGF-1 receptor (IGF-1r) signaling in neoplastic transformation is clearly evident from a variety of studies (reviewed in refs. 12–16). Several transgenic models have been developed to explore the role of IGF-1r signaling in cellular growth and neoplasia (17–20). However, with the exception of transgenic mice in which IGF-2 expression was driven by the major urinary protein promoter (MUP), none developed spontaneous tumors in any tissue. The MUP/IGF-2 transgenic mice developed a variety of tumors, primarily hepatocellular carcinomas and lymphomas in very old animals, but no prostate phenotype was described (18).
As part of our ongoing studies of the role of IGF-1r signaling in epithelial tumorigenesis, we created transgenic mice in which human IGF-1 expression is targeted specifically to the basal layer of multiple epithelia, including prostate, using the bovine keratin 5 (BK5) promoter (21). The current results demonstrate that persistent expression of human IGF-1 in the basal epithelium of mouse prostate leads to spontaneous neoplasia in this tissue.
Materials and Methods
Production and Maintenance of Transgenic Mice.
The generation of transgenic mice expressing human IGF-1 driven by the BK5 promoter (referred to as BK5.IGF-1 transgenic mice) is described in detail elsewhere (21). In brief, human IGF-1 cDNA, encoding the prepro form of the IGF-1 polypeptide (22), was excised from the parent pGEM plasmid and was ligated into the BK5 vector described previously (23). Founder mice were mated to ICR animals for propagation of transgenic lines. Transgenic animals were genotyped by using human IGF-1-specific PCR on genomic DNA isolated from tails as described (20). All of the mice used in this study were from line II, which exhibited the strongest phenotype.
Analysis of Serum IGF-1 and IGF Binding Protein 3 (IGFBP-3) Levels.
Blood samples were collected via tail bleeding and were centrifuged at 1,000 × g for 5 min at 4°C. Serum samples were analyzed by using methods outlined in the Active Non-Extraction IGF-1 ELISA and IGFBP-3 ELISA kits (Diagnostic Systems Laboratories, Webster, TX).
Histochemistry, Immunohistochemistry, and Immunofluorescence.
The expression of IGF-1 and IGF-1r was determined by indirect immunofluorescence analysis. For detection of IGF-1 expression, 5-μm frozen sections were incubated with 10% non-immunized goat serum for 30 min to block the nonspecific Fc receptor in the tissue and then were washed with PBS containing BSA (BSA/PBS). Sections were incubated in a 1:100 dilution of the primary sheep polyclonal antibody against human IGF-1 (Chemicon) in BSA/PBS or with preimmune sheep serum (negative control) overnight at 4°C. Sections were washed with BSA/PBS and were incubated with the secondary FITC affinity pure F(ab′)s fragment goat anti-chicken IgG (1:200) (Jackson ImmunoResearch) for 40 min at room temperature. Expression of the IGF-1r was determined by the method described in ref. 41, with the exception that a chicken polyclonal antibody (Upstate Biotechnology, Lake Placid, NY) and Cy-3 conjugated affinity pure F(ab′)2 fragment goat anti-sheep IgG (Jackson ImmunoResearch) were used as the primary and secondary antibodies. To detect neuroendocrine cells, slides were treated with protease and were stained with mouse monoclonal anti-seratonin antibody (Dako) according to the manufacturer's instructions.
For the analysis of labeling index, paraffin sections were stained with anti-BrdUrd antibody as described (24). The individual apoptotic cells in frozen tissue sections were identified by the TUNEL assay (ApopTag In Situ Apoptosis Detection kit, Intergen, Purchase, NY). Three mice, 8–10 weeks of age, were used for each group.
Results
Previous studies have shown that keratin 5 is expressed in basal cells of rat and mouse prostate (ref. 25; C.C., unpublished studies); therefore, the BK5 promoter was expected to drive gene expression to this epithelial subpopulation. However, this had not been previously demonstrated experimentally (26–28). Indirect immunofluorescence staining for human IGF-1 in basal epithelial cells of the ventral prostate from a 7-week-old BK5.IGF-1 mouse is shown in Fig. 1A. In addition, we determined that the IGF-1r was expressed in basal epithelial cells of mouse prostate (Fig. 1B). As can be seen in Fig. 1C, IGF-1r levels (IGF-1r β chain at molecular mass of 94 kDa) appeared to be similar in both transgenic and nontransgenic mice. In contrast, the IGF-1r was hyperphosphorylated in the prostate of BK5.IGF-1 mice, indicating it was constitutively activated. Another protein band with an apparent molecular mass similar to that of IRS-1 (≈165 kDa) was dramatically hyperphosphorylated in the prostate of transgenic mice compared with nontransgenic mice. Fig. 1D shows that phosphotidylinositol 3 (PI3) kinase activity was elevated nearly four-fold in prostate tissue of transgenic mice compared with nontransgenic mice.
Figure 1.

Indirect immunofluorescence localization of human IGF-1 (A) and mouse IGF-1r (B) in ventral prostate of an 8-week-old BK5.IGF-1 mouse. Arrowheads in A point to the brightly labeled basal epithelial cells, and asterisks are placed on top of luminal cells of the prostatic acini. (A and B, ×300.) (C) Western blot analysis of IGF-1r in prostate of BK5.IGF-1 mice. IGF-1r levels were determined in tissue lysates prepared as described (21) from pooled ventral and dorsolateral prostate of 8- to 10-week-old nontransgenic (NTg) and transgenic (Tg) mice. Protein bands were visualized by enhanced chemiluminescence (ECL, Amersham). (D) Analysis of PI3 kinase activity in prostate tissue (ventral and dorsolateral combined) lysates of 8-week-old nontransgenic (NTg) and transgenic (Tg) mice. The PI3 kinase assay was performed as described (21). In brief, PI3 kinase activity was determined in immunoprecipitates (anti-PI3 kinase antibody, Upstate Biotechnology) by the incorporation of [γ-32P]ATP. Radioactivity was assayed by liquid scintillation counting. Note that nearly identical results were obtained in repeat experiments for both IGF-lr phosphorylation and PI3 kinase activity.
BK5.IGF-1 mice also had measurable serum levels of human IGF-1 and elevated levels of endogenous IGFBP-3. Serum levels of human IGF-1 were 477 ± 44 ng/ml in transgenic mice and not detectable (as expected) in nontransgenic mice. Serum levels of IGFBP-3 were 11.3 vs. 5.3 ng/ml in transgenic compared with nontransgenic mice, respectively.
To evaluate the effect of deregulated IGF-1 expression on prostate epithelial homeostasis, apoptosis and cell proliferation rates were examined in the ventral prostate. The effect of castration on these parameters was also examined. Eight- to ten-week-old male transgenic and nontransgenic mice were surgically castrated, and the prostates were removed 4 and 10 days after castration. Prostates were taken from intact animals as well. In the noncastrated mice, the apoptotic rate in the ventral prostate was very low for both the BK5.IGF-1 mice and age-matched nontransgenic littermates (0.01 ± 0.02 and 0.03 ± 0.05, respectively). On androgen withdrawal (i.e., castration), the apoptotic rate increased >10-fold by day 4 after castration for both the transgenic mice and nontransgenic mice (0.42 ± 0.17 and 0.49 ± 0.3, respectively). On day 10, a slight, but insignificant, decrease was observed in both genotypes (0.11 ± 0.11, transgenic; 0.29 ± 0.35, nontransgenic). There was no significant difference in the prostate labeling index between intact transgenic and nontransgenic mice (0.62 ± 0.10 and 0.51 ± 0.06, respectively). After castration, the labeling index decreased similarly in both transgenic and nontransgenic mice by day 4 (0.07 ± 0.07 and 0.06 ± 0.04, respectively).
A number of pathologic changes were observed in the prostate and other male accessory glands of the BK5.IGF-1 mice. The urogenital tracts of 40 transgenic males ranging from 2 months to 18 months of age were analyzed histologically. Significant hyperplasia was observed in the male accessory glands of the two youngest transgenic mice (2–3 months; data not shown). In addition, all of the older male transgenic mice (38 total; ≥6 months of age) had pronounced hyperplasia of different components of the male accessory glands. In the younger transgenic mice (2–3 months), the structure of the acini appeared normal, and overexpression of the transgene did not appear to have differentially affected either the morphology of the acini or the ratio of basal to luminal cells. The stroma also appeared relatively normal. In contrast, prostate tissue of older transgenic mice (≥6 months of age) presented atypical hyperplastic changes in all lobes of the prostate gland. Comparison of the ventral prostate from a nontransgenic male with that of a transgenic male, both aged 7 months, shown in Fig. 2 illustrates that the degree of hyperplasia was sufficient to increase the overall size of the gland (A and B, respectively). The lesions observed in prostate of older transgenic mice consisted of crowded acini with irregular shapes and moderate cellular atypia. Areas of frank dysplasia were frequently observed in these lesions and particularly in the dorsolateral and ventral prostate, where some of the acini resembled human prostatic intraepithelial neoplasia (PIN) (29, 30). An atypical acinus from the ventral prostate of a 7-month-old transgenic mouse illustrating epithelial stratification and both cellular and nuclear atypia is shown in Fig. 2C. Using criteria for diagnosis of human prostate cancer, this lesion exhibited features resembling human PIN. A lower magnification of a ventral prostate lesion that illustrates the disorganized growth typical of the prostate glands from transgenic mice is shown in Fig. 2D. In addition, this section shows some invasion of the periglandular adipose tissue.
Figure 2.

Preneoplastic and neoplastic changes in the ventral and dorsolateral prostate in BK5.IGF-1 mice. Five-micrometer sections of formalin-fixed tissue embedded in paraffin were stained with hematoxylin and eosin. (A) Ventral prostate of a 7-month-old nontransgenic littermate. (B) A transgenic mouse of the same age. Note that the hyperplasia in the transgenic mouse is sufficient to increase the overall size of the gland in B. (A and B, ×37.5.) (C) Atypical acinus at higher magnification illustrating epithelial stratification (boxed area) as well as both cellular and nuclear atypia. (×300.) (D) Ventral prostate adenocarcinoma from a transgenic mouse illustrating the disorganized growth typical of these lesions. Arrows indicate atypical acini invading periglandular adipose tissue. (×75.) (E) Acini from dorsolateral prostate of a nontransgenic mouse at 14 months of age showing normal appearance. (×150.) (F) Atypical acinus from dorsolateral prostate of a 6-month-old transgenic mouse. Note the presence of medium grade PIN lesions (see long arrow). Neural ganglia (ng) compression caused by gland enlargement and stromal vascularization are also evident in this section. Short arrow points to a highly vascularized area of this lesion. (×150.) (G and H) Focal areas of an adenocarcinoma from a 14-month-old transgenic mouse showing atypical glandular structures and vascularized stroma. Note in H the invasion into the pelvic muscle mass (pm). (G and H, ×150.)
Representative sections of dorsolateral prostate from transgenic mice are shown in Fig. 2 as is a representative section of dorsolateral prostate of a 14-month-old nontransgenic mouse (E). Fig. 2F shows a representative section from a 7-month-old transgenic mouse while Fig. 2 G and H show representative sections from a 14-month-old transgenic mouse. A medium-grade PIN-like lesion similar to those seen in the ventral prostate is shown in Fig. 2F. This section also shows compression of the neural ganglia by the enlarged (hyperplastic) gland and vascularization of the surrounding stroma. Focal areas of adenocarcinoma with atypical glandular structures and a vascularized stroma are shown in Fig. 2 G and H, and PIN-type lesions are evident adjacent to the focal area of adenocarcinoma. Invasion into the pelvic muscle mass is also evident in Fig. 2H.
The basal layer was conserved in most of the hyperplastic acini from dorsolateral and ventral prostate but in the more dysplastic areas the basal layer was disrupted or totally absent. Loss of the basal layer in some of these areas was confirmed by the absence of endogenous keratin 5 staining. A dysplastic acinus from the ventral prostate of a 7-month-old transgenic mouse presenting loss of keratin 5 staining is shown in Fig. 3A. In four of the older mice (>8 months of age), areas of necrosis of the dorsolateral prostate were observed accompanied by infiltration of the prostate fluid into the adjacent connective tissue. Glandular cells presenting atypical features were also observed in some cases floating in the prostatic fluid. Although most of the lesions were very well differentiated, they frequently invaded adjacent tissues, particularly periglandular fat and peritoneum, and in some cases they invaded the wall of seminal vesicles. In addition, two poorly differentiated tumors were observed in older mice. A section (stained with hemotoxylin and eosin) from one of these tumors, diagnosed as small cell carcinoma, is shown in Fig. 3B. The neuroendocrine nature of this tumor was confirmed by positive staining for serotonin (Fig. 3C). In this section, also note the highly vascularized nature of this lesion. All of the other lesions showing either infiltration or substantial loss of basal cells were considered well differentiated adenocarcinomas.
Figure 3.
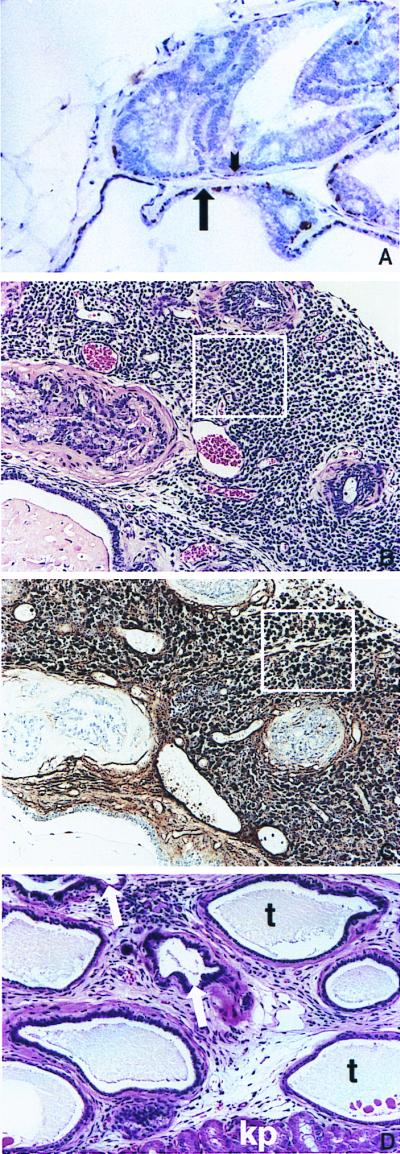
(A) Staining for endogenous K5 in the basal cell layer in the ventral prostate of a BK5.IGF-1 mouse. Note the lack of staining for K5 in the dysplastic acinus in the upper half of the picture (short arrow points to area devoid of K5 staining) whereas the more normal acinus in the lower portion of this panel (long arrow) still retains K5 staining. Keratin 5 was detected by using a rabbit polyclonal anti-mouse K5 antibody (Covance, Berkeley, CA) (described in ref. 47). (B) Small cell carcinoma from a 14-month-old BK5.IGF-1 mouse. Area enclosed in box is representative of this lesion. (C) Immunohistochemical staining for serotonin in a section from the small cell carcinoma shown in B. Area enclosed in box shows representative cells darkly stained for serotonin. To detect neuroendocrine cells, slides were treated with protease and were stained with mouse monoclonal anti-seratonin antibody (Dako) according to the manufacturer's instructions. (D) Atypical acini growing in the kidney capsule of an athymic mouse. Arrows point to atypical acini. Also note the unusual tubular structures (t) and the kidney parenchyma (kp) present in this section. (A–D, ×150.)
To further confirm the neoplastic nature of the lesions observed in prostate of BK5.IGF-1 mice, three individual enlarged prostates (one ventral; two dorsolateral) from transgenic mice were grafted into the renal capsule of male athymic mice. All three renal capsule grafts gave rise to development of prostate tissues. In all of the cases, the grafted tissue appeared to have increased in size, and in one case a large peritoneal mass formed that became abscessed and produced a renal-spleen adherence. Another renal capsule transplant generated a tumor mass formed by dysplastic acini, and tubular prostate structures were observed infiltrating the parenchyma of the kidney (Fig. 3D). The third graft also produced an adherence with the spleen formed by inflammatory tissues and some poorly developed prostate acini. Collectively, the results obtained indicated that the changes in the prostate of BK5.IGF-1 transgenic mice were of a neoplastic nature and could be considered homologous to human preneoplastic lesions (atypical hyperplasia) and well differentiated adenocarcinomas.
Of the 38 male transgenic mice examined in the current study ≥6 months of age, a total of 7 had ventral prostate adenocarcinomas and 11 had dorsolateral prostate adenocarcinomas. These numbers represent independent animals with tumors. In most cases, where adenocarcinomas were present in one lobe, the corresponding prostate lobe also was atypical but could not be conclusively diagnosed as adenocarcinoma. In a few cases, we did not have the corresponding prostate lobe for analysis (because of processing errors or poor tissue quality). Thus, the overall incidence of tumors (adenocarcinoma plus small cell carcinoma) in mice ≥6 months of age was 50% (19/38) (Table 1).
Table 1.
Occurrence of prostate tumors in BK5.IGF-1 transgenic mice
| Prostate lesions | Mice 6–9 months old, n = 6
|
Mice >9 months old, n = 32
|
||
|---|---|---|---|---|
| VP | DLP | VP | DLP | |
| Adenocarcinoma | 1 | 1 | 6 | 10 |
| Small cell carcinoma | −* | − | 1† | 1 |
VP, ventral prostate; DLP, dorsolateral prostate.
*None detected.
†This animal had a ventral prostate adenocarcinoma as well.
Discussion
The current data demonstrate that constitutive signaling through the IGF-1r alone is sufficient to induce tumorigenesis in the mouse prostate. The development of prostatic adenocarcinomas in BK5.IGF-1 transgenic mice appeared to follow a stepwise progression through characteristic preneoplastic changes involving atypical hyperplasia, PIN-like lesions, and adenocarcinomas exhibiting local invasive properties, a process that resembles the development of prostate cancer in humans. There are few reports in the literature showing that elevated levels of IGF-1 or other IGF-1r ligands (i.e., IGF-2) can induce tumorigenesis. Rogler et al. (18) reported that IGF-2 transgenic mice, where IGF-2 expression was driven by the MUP promoter, developed hepatocellular carcinomas and lymphomas in addition to several other tumors at a higher frequency than controls after 18 months of age. However, no tumors were reported in prostate of these transgenic mice. The MUP promoter expresses at high levels in liver and preputial glands; however, these mice did have elevated serum levels of IGF-2. Because IGF-2 binds to and activates the IGF-1r (14), it is likely that the mechanism for tumorigenesis in the MUP/IGF-2 transgenic mice involves signaling through the IGF-1r. The fact that the MUP/IGF-2 transgenic mice did not develop prostate tumors indicates that the tissue-specific expression of IGF-2 was essential for inducing neoplasia in these mice. The BK5.IGF-1 transgenic mice also had elevated serum levels of human IGF-1 as well as endogenous IGFBP-3. Nevertheless, tumors developed only in the prostate and skin (21) where the transgene was expressed. Thus, it is concluded that the tissue-specific expression of IGF-1 in BK5.IGF-1 transgenic mice was essential for the observed phenotype in the prostate gland.
The IGF-1r is expressed on the surface of many cell types, including epithelial cells, and IGF-1 has been shown to possess mitogenic effects on numerous epithelial cells, including prostate epithelial cells (refs. 31–37; reviewed in 14). In mouse prostate, in vivo, the IGF-1r was found to be expressed primarily on the surface of basal epithelial cells (Fig. 1). The data shown in Fig. 1 also demonstrate that the IGF-1r is constitutively activated and that PI3 kinase activity is elevated in the prostate of BK5.IGF-1 transgenic mice, showing a direct correlation between phenotype, transgene expression, and receptor activation in vivo. The mechanism(s) whereby constitutive activation of the IGF-1r leads to hyperplasia and eventually neoplasia in the prostate of BK5.IGF-1 mice remains unknown at present. Several studies were performed to initially address this question. Because IGF-1 can have both mitogenic and anti-apoptotic actions (14, 16), we examined these parameters in intact and castrated BK5.IGF-1 transgenic mice. Surprisingly, there were no clear differences in cell proliferation rates or apoptotic rates in the intact transgenic mice compared with nontransgenic littermates under the conditions of our experiments. Thus, it is not clear at present what the cellular mechanisms are that lead to hyperplasia of the prostate gland in these mice. Because the BrdUrd labeling and apoptosis indices were very low, it is conceptually possible that subtle differences in these parameters, below the level of detection of the methods used, had a cumulative effect over a period of several weeks or months in generating the type of hyperplasia observed in the transgenic animals. In the epidermis of adult BK5.IGF-1 transgenic mice, the labeling index was elevated ≈2- to 3-fold compared with nontransgenic mice whereas no significant differences were observed in basal apoptotic index (21). After exposure to UV light, however, epidermal proliferation was exaggerated and apoptosis was suppressed in the epidermis of BK5.IGF-1 mice. One possible explanation for the current results is that endogenous or exogenous stimuli that induce proliferation/apoptosis in the prostate gland epithelium will lead to similar differential responses in the BK5.IGF-1 transgenic mice and ultimately contribute to prostate epithelial hyperplasia.
It is well established that castration impairs the development of the prostate gland in immature animals (38) and induces atrophy of the prostate in the adult (39–42). Atrophy of the prostate by androgen deprivation in adult mice is believed to be the result of massive apoptosis of the glandular epithelium that occurs after castration (43). The trophic action of androgen appears to be mediated, in part, by the stroma through the secretion of paracrine factors, including IGF-1 as well as other growth factors (44, 45). Because IGF-1 is considered a cell survival factor (i.e., anti-apoptotic), we examined whether overexpression of IGF-1 could prevent apoptosis after castration. However, both transgenic and nontransgenic mice behaved very similarly, indicating that IGF-1 did not prevent the castration-induced atrophy in the prostate. Further, when we measured the number of apoptotic cells after castration, there were again no differences between the transgenic and nontransgenic mice. These results demonstrate that overexpression of IGF-1 alone cannot compensate for the lack of androgen after castration and that tumor development in this animal model likely depends on the continued presence of androgens. Colombel et al. (40) reported that functional p53 protein was not essential for prostate epithelial cells to undergo castration-induced apoptosis in mice. This observation is interesting in light of our results with UV-induced apoptosis in skin of BK5.IGF-1 mice (21). UV light-induced apoptosis in epidermis is mediated by p53 because mice lacking p53 exhibit significantly reduced apoptotic rates (46). A possible explanation for the lack of effect of IGF-1 on castration-induced apoptosis is that this process does not require functional p53. This hypothesis predicts that p53-mediated apoptosis in prostate epithelial cells of BK5.IGF-1 mice would be suppressed. Future experiments are designed to address this possibility.
In conclusion, the BK5.IGF-1 transgenic mice appear to represent a new animal model for prostate cancer. It appears that prostate cancer development can be followed from early stages of hyperplasia through more dysplastic lesions and adenocarcinomas in these transgenic mice. The current results also provide a direct link between IGF-1r signaling and tumor development in prostate, confirming an important role for this growth factor in prostate cancer development (11). The further study of these mice should not only provide insight into the role of IGF-1r signaling in epithelial tumorigenesis but should also provide a greater understanding of its role in prostate cancer.
Acknowledgments
This work was supported by U.S. Public Health Service Grant CA37111 (J.D.), University of Texas M. D. Anderson Cancer Center Core Grant CA16672, and National Institute on Environmental Health Sciences Center Grant ES07784. E.W. is supported by Training Grant ES07247.
Abbreviations
- IGF-1
insulin-like growth factor 1
- IGF-1r
insulin-like growth factor-1 receptor
- MUP
major urinary protein
- BK5
bovine keratin 5
- IGFBP-3
IGF binding protein 3
- PIN
prostatic intraepithelial neoplasia
- PI3
phosphatidylinositol 3
References
- 1.Parker S L, Tong T, Bolden S, Wingo P A. Ca Cancer J Clin. 1997;47:5–27. doi: 10.3322/canjclin.47.1.5. [DOI] [PubMed] [Google Scholar]
- 2.Bosland M C. Urol Oncol. 1996;2:117–119. doi: 10.1016/s1078-1439(97)82840-2. [DOI] [PubMed] [Google Scholar]
- 3.Ho S, Lane K. Urol Oncol. 1996;2:110–115. doi: 10.1016/s1078-1439(97)82841-4. [DOI] [PubMed] [Google Scholar]
- 4.Isaacs J T. Urol Oncol. 1996;2:115–116. doi: 10.1016/s1078-1439(97)82842-6. [DOI] [PubMed] [Google Scholar]
- 5.Thompson T C. Urol Oncol. 1996;2:117–119. doi: 10.1016/s1078-1439(97)82843-8. [DOI] [PubMed] [Google Scholar]
- 6.Maroulakou I G, Anver M, Garrett L, Green J E. Proc Natl Acad Sci USA. 1994;91:11236–11240. doi: 10.1073/pnas.91.23.11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shibata M-A, Ward J M, Devor D E, Liu M-L, Green J E. Cancer Res. 1996;56:4894–4903. [PubMed] [Google Scholar]
- 8.Greenberg N M, DeMayo F, Finegold M J, Medina D, Tilley W D, Aspinall J O, Cunha G R, Donjacour A A, Matusik R J, Rosen J M. Proc Natl Acad Sci USA. 1995;92:3439–3443. doi: 10.1073/pnas.92.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perez-Stable C, Altman N H, Mehta P P, Deftos L J, Roos B A. Cancer Res. 1997;57:900–906. [PubMed] [Google Scholar]
- 10.Garabedian E M, Humphrey P A, Gordon J I. Proc Natl Acad Sci USA. 1998;95:15382–15387. doi: 10.1073/pnas.95.26.15382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan J M, Stampfer M J, Giovannucci E, Gann P H, Ma J, Wilkinson P, Hennekens C H, Pollak M. Science. 1998;279:563–566. doi: 10.1126/science.279.5350.563. [DOI] [PubMed] [Google Scholar]
- 12.Baserga R. Cancer Res. 1995;55:249–252. [PubMed] [Google Scholar]
- 13.Baserga R, Hongo A, Rubini M, Prisco M, Valentinis B. Biochem Biophys Acta. 1997;1332:F105–F126. doi: 10.1016/s0304-419x(97)00007-3. [DOI] [PubMed] [Google Scholar]
- 14.Jones J I, Clemmons D R. Endocr Rev. 1995;16:3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- 15.Aaronson S A. Science. 1991;254:1146–1153. doi: 10.1126/science.1659742. [DOI] [PubMed] [Google Scholar]
- 16.Le Roith D, Parrizas M, Blakesley V. Endocrine. 1997;7:103–105. doi: 10.1007/BF02778074. [DOI] [PubMed] [Google Scholar]
- 17.Mathews L S, Hammer R E, Behringer R R, D'Ercole J, Bell G I, Brinster R L, Palmiter R D. Endocrinology. 1988;123:2827–2833. doi: 10.1210/endo-123-6-2827. [DOI] [PubMed] [Google Scholar]
- 18.Rogler C E, Yang D, Rossetti L, Donohoe J, Alt E, Chang C J, Rosenfeld R, Neely K, Hintz R. J Biol Chem. 1994;269:13779–13784. [PubMed] [Google Scholar]
- 19.Ward A, Bates P, Fisher R, Richardson L, Graham C F. Proc Natl Acad Sci USA. 1994;91:10365–10369. doi: 10.1073/pnas.91.22.10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bol D, Kiguchi K, Gimenez-Conti I, Rupp T, DiGiovanni J. Oncogene. 1997;14:1725–1734. doi: 10.1038/sj.onc.1201011. [DOI] [PubMed] [Google Scholar]
- 21.DiGiovanni, J., Bol, D. K., Wilker, E., Beltran, L., Carbajal, S., Moats, S., Ramirez, A., Jorcano, J. & Kiguchi, K. (2000) Cancer Research, in press. [PubMed]
- 22.Jansen M, van Schaik F M A, Ricker A T, Bullock B, Woods D E, Gabbay K H, Nussbaum A L, Sussenback J S, Van den Brande J L. Nature (London) 1983;306:609–611. doi: 10.1038/306609a0. [DOI] [PubMed] [Google Scholar]
- 23.Bol D, Kiguchi K, Beltrán L, Rupp T, Moats S, Gimenez-Conti I, Jorcano J, DiGiovanni J. Mol Carcinog. 1998;21:2–12. [PubMed] [Google Scholar]
- 24.Eldridge S R, Tilbury L F, Goldsworthy T L, Butterworth B E. Carcinogenesis. 1990;4:281–284. doi: 10.1093/carcin/11.12.2245. [DOI] [PubMed] [Google Scholar]
- 25.Verhagen A P M, Aalders T W, Ramackers F C S, DeBruyne F M J, Schalken J. Prostate. 1988;13:25–38. doi: 10.1002/pros.2990130104. [DOI] [PubMed] [Google Scholar]
- 26.Ramirez A, Bravo A, Jorcano J L, Vidal M. Differentiation. 1994;58:53–64. doi: 10.1046/j.1432-0436.1994.5810053.x. [DOI] [PubMed] [Google Scholar]
- 27.Casatorres J, Navarro J, Blessing M, Jorcano J. J Biol Chem. 1994;32:20489–20496. [PubMed] [Google Scholar]
- 28.Robles A I, Larcher F, Whalin R B, Murillas R, Richie E, Gimenez-Conti I B, Jorcano J L, Conti C J. Proc Natl Acad Sci USA. 1996;93:7634–7638. doi: 10.1073/pnas.93.15.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bostwick D G, Brawer M K. Cancer. 1987;59:788–794. doi: 10.1002/1097-0142(19870215)59:4<788::aid-cncr2820590421>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 30.Bostwick D G. Cancer. 1995;75:1823–1836. [Google Scholar]
- 31.Cohen P, Peehl D M, Rosenfeld R G. Horm Metab Res. 1994;26:81–84. doi: 10.1055/s-2007-1000777. [DOI] [PubMed] [Google Scholar]
- 32.Cohen P, Peehl D M, Lamson G, Rosenfeld R G. J Clin Endocrinol Metab. 1991;73:401–407. doi: 10.1210/jcem-73-2-401. [DOI] [PubMed] [Google Scholar]
- 33.Rajah R, Valentinis B, Cohen P. J Biol Chem. 1997;272:12181–12188. doi: 10.1074/jbc.272.18.12181. [DOI] [PubMed] [Google Scholar]
- 34.Barreca A, De Luca M, Del Monte P, Bondaza S, Damonte G, Cariola G, Di Marco E, Giordano G, Cancedda R, Minuto F. J Cell Physiol. 1992;151:262–268. doi: 10.1002/jcp.1041510207. [DOI] [PubMed] [Google Scholar]
- 35.Dlugosz A, Cheng C, Denning M F, Dempsey P, Coffey R, Yuspa S. Cell Growth Differ. 1994;5:1283–1292. [PubMed] [Google Scholar]
- 36.Neely E, Morhenn V, Hintz R, Wilson D, Rosenfeld R. J Invest Dermatol. 1991;96:104–110. doi: 10.1111/1523-1747.ep12515914. [DOI] [PubMed] [Google Scholar]
- 37.Tomsit J, Savino W, Safieh B, Chanson P, Gagnerault M-C, Bach J-F, Dardenne M. J Clin Endocrinol Metab. 1992;76:183–188. doi: 10.1210/jcem.75.1.1619008. [DOI] [PubMed] [Google Scholar]
- 38.Donjacour A A, Cunha G R. Dev Biol. 1988;128:1–14. doi: 10.1016/0012-1606(88)90260-6. [DOI] [PubMed] [Google Scholar]
- 39.English H F, Kyprianou N, Isaacs J T. Prostate. 1989;15:233–250. doi: 10.1002/pros.2990150304. [DOI] [PubMed] [Google Scholar]
- 40.Colombel M, Radvanyi F, Blanche M, Abbou C, Buttyan R, Donehower L A, Chopin D, Thiery J P. Oncogene. 1995;10:1269–1274. [PubMed] [Google Scholar]
- 41.Sinha A A, Bentley M D, Pomroy F E, Jr, Jamuar M P. Cell Tissue Res. 1981;215:547–561. doi: 10.1007/BF00233531. [DOI] [PubMed] [Google Scholar]
- 42.Buttyan R, Shabsigh A, Perlman H, Colombel M. Trends Endocrinol Metab. 1999;10:47–54. doi: 10.1016/s1043-2760(98)00104-0. [DOI] [PubMed] [Google Scholar]
- 43.Bannerjee P P, Banerjee S, Tilly K I, Tilly J L, Brown T R, Zirkin B R. Endocrinology. 1995;136:4368–4376. doi: 10.1210/endo.136.10.7664656. [DOI] [PubMed] [Google Scholar]
- 44.Culig Z, Hobisch A, Cronauer M V, Radmayr C, Hittmair A, Zhang J, Thurnher M, Bartsch G, Klocker H. Prostate. 1996;28:392–405. doi: 10.1002/(SICI)1097-0045(199606)28:6<392::AID-PROS9>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 45.Foster B A, Kaplan P J, Greenberg N M. Cancer Metastisis Rev. 1999;17:317–324. doi: 10.1023/a:1006162410436. [DOI] [PubMed] [Google Scholar]
- 46.Ziegler A, Jonason A S, Lefell D J, Simon J A, Sharma H W, Kimmelmann J, Remington L, Jacks T, Brash D E. Nature (London) 1994;372:773–776. doi: 10.1038/372773a0. [DOI] [PubMed] [Google Scholar]
- 47.Pierce A M, Schneider-Broussard R, Gimenez-Conti I B, Russell J L, Conti C J, Johnson D G. Mol Cell Biol. 1999;19:6408–6414. doi: 10.1128/mcb.19.9.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]