

Abstract
Adherence of Helicobacter pylori to inflamed gastric mucosa is dependent on the sialic acid–binding adhesin (SabA) and cognate sialylated/fucosylated glycans on the host cell surface. By in situ hybridization, H. pylori bacteria were observed in close association with erythrocytes in capillaries and post-capillary venules of the lamina propria of gastric mucosa in both infected humans and Rhesus monkeys. In vivo adherence of H. pylori to erythrocytes may require molecular mechanisms similar to the sialic acid–dependent in vitro agglutination of erythrocytes (i.e., sialic acid–dependent hemagglutination). In this context, the SabA adhesin was identified as the sialic acid–dependent hemagglutinin based on sialidase-sensitive hemagglutination, binding assays with sialylated glycoconjugates, and analysis of a series of isogenic sabA deletion mutants. The topographic presentation of binding sites for SabA on the erythrocyte membrane was mapped to gangliosides with extended core chains. However, receptor mapping revealed that the NeuAcα2–3Gal-disaccharide constitutes the minimal sialylated binding epitope required for SabA binding. Furthermore, clinical isolates demonstrated polymorphism in sialyl binding and complementation analysis of sabA mutants demonstrated that polymorphism in sialyl binding is an inherent property of the SabA protein itself. Gastric inflammation is associated with periodic changes in the composition of mucosal sialylation patterns. We suggest that dynamic adaptation in sialyl-binding properties during persistent infection specializes H. pylori both for individual variation in mucosal glycosylation and tropism for local areas of inflamed and/or dysplastic tissue.
Synopsis
Helicobacter pylori infections are very common worldwide and cause chronic inflammation in the stomach (gastritis), which may progress to peptic ulcer disease and stomach cancer. In the gastric epithelium, H. pylori infections induce expression of inflammation-associated “sialylated” carbohydrates. The ability to bind to the glycosylated epithelial cells is considered to be essential for H. pylori to cause persistent infection and disease. Here the authors show that during established infection, H. pylori also binds to red blood cells in gastric mucosal blood vessels in both infected humans and Rhesus monkeys. The authors found that “sialic acid–binding adhesin” (SabA), is the bacterial surface protein that mediates binding of H. pylori to red blood cells. Furthermore, they show that clinical H. pylori isolates demonstrate “polymorphism” in their abilities to bind various sialylated carbohydrates, and that the variation in binding properties depends on the sialic acid–binding adhesin protein itself. This variability may adapt the binding properties of H. pylori both to individual hosts and the changing epithelial glycosylation patterns during chronic inflammation. Continuous adaptation to inflamed tissue during persistent infections is probably a general feature of microbial pathogens, although their binding properties have not yet been explored in detail.
Introduction
The gastric pathogen Helicobacter pylori exhibits specific tropism for gastric mucosa in human populations worldwide [1]. Adherence to gastric epithelium may benefit the bacterium by placing it in close contact with epithelial surfaces and nutrients leaching from host cells that are damaged by local inflammation processes. The size of the H. pylori genome is only one-third of that of Escherichia coli, with ensuing limitations in metabolic pathways [2] and adoption of an adhesive and intracellular parasitic lifestyle. In addition, binding to highly glycosylated mucins in the mucus layer closest to the epithelium may stabilize H. pylori colonization and thus avoid clearance of infection caused by high epithelial turnover and shedding of the mucus layer [3]. H. pylori has been shown to adhere to erythrocytes and neutrophils in vitro [4,5], and virulence-associated cag+ H. pylori strains have been shown to invade both the gastric mucosa and individual cells [6–10]. Thus, the ability to adhere may also affect the outcome of H. pylori infection by facilitating focused delivery of effector molecules into the host cell [11,12]. Consequently, during infection, tissue invasion and migration of H. pylori bacterial cells through the endothelial lining of capillaries and post-capillary venules followed by adherence to blood cells may result in transfer and systemic dissemination of H. pylori.
H. pylori adapts to the gastric environment by binding to oligosaccharides (glycans) of various complexities, so-called receptors or binding epitopes for establishment of infection in different parts of the mucosa. These glycans are presented on cell surfaces by glycoproteins and glycosphingolipids, and in the gastric mucus by MUC5AC and MUC6 mucin molecules [13]. The H. pylori glycan receptors include fucosylated ABO blood group antigens [14], glycans with charged modifications such as sialic acid [15] and sulfate [16], and, in addition, unsubstituted core chain glycans [17]. The many different receptor structures described for mucosal adherence suggest that, similar to multiadhesive pathogens such as Pseudomonas aeruginosa, H. pylori expresses a range of different attachment proteins, so-called adhesins [18]. The best-characterized interaction between H. pylori adhesins and host cell receptors is that between the blood group antigen–binding adhesin (BabA) and the ABO blood group and Lewis b (Leb) antigens [14]. ABO/Leb blood group antigens are best known from erythrocytes, but they are also highly expressed in the epithelium and mucus lining in the oro-gastrointestinal tract [19], where they are known as histo–blood group antigens [20].
As shown in a previous study, a H. pylori babA deletion mutant that cannot bind ABO/Leb blood group antigens nevertheless binds to gastric epithelium [15]. Further analysis showed that the babA mutant preferentially bound to inflamed gastric mucosa, and that binding was mediated by sialylated glycans such as sialyl-Lewis x (sLex) and sialyl-Lewis a (sLea). The sLex and sLea glycans are better known as the glycan binding sites for the selectin family of cell adhesion molecules. Expression of selectin molecules is activated by inflammatory responses, and they have important roles in recruitment of white blood cells from circulation to the tissue in need (reviewed in [21]). Infection of the gastric mucosa by H. pylori results in inflammatory responses, with concomitant expression of sialylated glycans. H. pylori has been suggested to exploit mechanisms of “selectin mimicry” to “home in” on inflamed gastric tissue by binding to epithelial sLex and sLea. By analysis of both gastric biopsy material from individuals with gastritis or peptic ulcer disease and experimentally infected Rhesus monkeys, strong correlations were found between expression of sialylated Lewis antigens, gastritis, and H. pylori infection. Similar to BabA, the sialic acid–binding adhesin, SabA, was purified and identified by the retagging technique based on its affinity for sLex [15].
Sialylated glycoconjugates are common binding sites for both Gram-negative and Gram-positive bacteria [22], influenza and adenoviruses [23,24], and parasites [25]. Hemagglutination analysis is an established method for characterization of microbial adherence to host cell surfaces. Analysis of sialic acid–dependent hemagglutination (sia-HA) has been facilitated by the easy removal of sialylated epitopes by sialidase enzyme, and has been refined in some cases by complementary enzymatic resialylation of cell surfaces [22,26]. Soon after its discovery, H. pylori was shown to agglutinate erythrocytes [4]. The activity was suggested to be dependent on sialic acid since the HA activity was lost by prior sialidase treatment of erythrocytes [27]. About one-third of fresh clinical H. pylori isolates demonstrate sialidase-sensitive HA [28]. This figure is similar to the prevalence of sLex binding among clinical isolates [15]. The sialic acid–binding epitope was characterized as NeuAcα2–3Gal since sialyl-lactose could competitively inhibit sia-HA [27]. The N-acetyl-neuraminyl-lactose–binding hemagglutinin was originally affinity purified by use of a sialylated serum protein and denoted H. pylori adhesin A (HpaA) [29]. However, results from later studies have questioned the role of HpaA in sia-HA; first, a hpaA deletion mutant demonstrated no reduction in sia-HA activity; [30] and second, immunogold localization analysis suggested that HpaA is most likely a flagellar sheath protein [31]. Similarly, the H. pylori neutrophil-activating protein (HP-NAP) has been described to exhibit sialic acid–binding properties [32]. However, a J99 HP-NAP depleted mutant was no different from the parent strain in sialic acid–binding or in sia-HA properties [33].
Here we report that H. pylori can be found on erythrocytes in capillaries and post-capillary venules in gastric mucosa of infected humans and Rhesus monkeys. These results extend our earlier findings that H. pylori is a facultative intracellular bacterium that can leach from the lumen of the stomach into epithelial cells and the lamina propria [9], indicating that the bacterium may disseminate into the circulation by way of gastric mucosal capillaries. Our results also demonstrate that the SabA adhesin is the sought-after sialyl-dependent hemagglutinin of H. pylori. The preferred binding sites for SabA on the erythrocyte cell surface were mapped to extended ganglioside glycans. We also found a high level of polymorphism in sialyl-binding properties among clinical isolates, which suggest functional adaptation of SabA both to individual and disease-related differences in mucosal sialylation patterns.
Results
Adherence of H. pylori to Erythrocytes in Capillaries and Post-Capillary Venules in Gastric Mucosa of Infected Humans and Rhesus Monkeys
Rhesus monkeys and humans have very similar gastric anatomy, histology, and mucosal glycosylation patterns, and they can be naturally infected by H. pylori. In addition, H. pylori infection is associated with mucosal inflammation, gastritis [6], and sialylated mucosal glycosylation pattern [15]. Biopsies harvested from the gastric mucosa of humans and experimentally infected Rhesus monkeys were analyzed for spatial localization of H. pylori cells. Genta-stained and toluidine blue–stained sections of gastric mucosa (Figure 1A and 1B, respectively) revealed the presence of a few H. pylori bacterial cells tightly associated with erythrocytes within capillaries and post-capillary venules in addition to the previously reported distribution of the bacterial cells in the lumen and foveolar epithelium [9]. To test if these bacterial cells were H. pylori that had invaded the gastric tissue of Rhesus monkeys and entered the microcirculation, in situ hybridization was performed. Thus, bacterial cells attached to the erythrocyte surfaces were identified by use of probes specific for H. pylori 16S RNA (Figure 1C). Next, the possibility that H. pylori can also invade blood vessels and adhere to erythrocytes in humans was analyzed. In situ hybridization in biopsies of infected human gastric mucosa revealed a similar localization of H. pylori bacterial cells (Figure 1D). These results suggest that H. pylori can reach the gastric mucosa capillaries, attach to erythrocytes, and perhaps disseminate throughout the body of both humans and Rhesus monkeys. Importantly, the number of bacterial cells in the endothelial lining was drastically lower compared to the foveolar epithelium (illustrated in Figure 1A). This difference in H. pylori bacterial cell density is consistent with the absence of clinical cases with overt sepsis caused by H. pylori infection.
Figure 1. H. pylori Adheres to Erythrocytes in Capillaries and Post-Capillary Venules of Infected Humans and Rhesus Monkeys.

(A) Genta-stained section of human gastric biopsy. Black spiral- and comma-shaped bacteria are observed in the lumen of the stomach, adherent to gastric epithelial cells, within the mucus globule of the cells. Bacterial cells (arrow) are also present in close contact to an erythrocyte within a capillary located in the supporting connective tissue of lamina propria of the mucosa.
(B) Section of human gastric biopsy stained with toluidine blue. A capillary vessel lined by endothelial cells is visible in the lamina propria of the mucosa. It contains several erythrocytes to which H. pylori are attached. Insert: higher magnification of two H. pylori (arrows) in close approximation to erythrocytes.
(C) Section of a Rhesus monkey gastric biopsy. In situ hybridization was performed using probes specific for H. pylori 16S rRNA, demonstrating the presence of several H. pylori apparently attached to erythrocyte surfaces of a post-capillary venule located in the lamina propria of submucosa. Inserts: higher magnification of H. pylori bacterial cells (arrows) in close approximation to erythrocytes.
(D) Section of a human gastric biopsy. In situ hybridization was performed using probes specific for H. pylori 16S rRNA. This high magnification of a capillary immediately adjacent to a gastric gland (on the top-right corner of the picture) demonstrates the presence of several H. pylori bacterial cells, stained blue, apparently attached to the surfaces of erythrocytes.
Bars = 5 μm.
Correlation between sia-HA and sLex Binding
A series of 99 Swedish clinical H. pylori isolates were tested for both sia-HA and for binding to sLex glycoconjugate. Sia-HA was assessed by desialylation of human erythrocytes with sialidase from Clostridium perfringens, a glycosidase that hydrolyses α2–3–, α2–6–, and α2–8–linked sialic acid in oligosaccharide chains of natural glycoconjugates, such as glycoproteins and glycosphingolipids. Enzymatic desialylation of the erythrocyte surfaces and subsequent removal of sialylated bacterial binding sites would result in reduced erythrocyte aggregation, and thus reduced HA titers. For the series of clinical isolates, sialidase-dependent shifts in HA titers ranged from −2 to ≥ 2, where positive values relate to the expected effect of sialidase on HA (i.e., the reduction in sialyl-dependent binding by one to three titer shifts). Negative values correspond to strains that, in contrast, increase HA titers due to removal of sialic acid (i.e., sialyl-independent HA). HA titers (1 to ≥ 2) were found for 27 isolates (27%), whereas 31 isolates (31%) showed increased HA titers (−1 to −2). The remaining 41 isolates (41%) displayed no change in sia-HA titers (Figure 2A).
Figure 2. Characterization of Binding Properties of the SabA Adhesin, and Its siaHA Properties.

(A) A panel of 99 Swedish clinical H. pylori isolates was tested for sia-HA properties and for binding to 125I-sdiLex conjugate. The numbers on the x axis indicate the shifts in HA titers after sialidase treatment: positive values indicate lowered sia-HA titers (i.e., sia-HA, whereas negative values indicate increased HA titers (i.e., sialic acid–independent HA). No change in HA titer is indicated by 0. The y axis gives the percentage of bound sLex-conjugate.
(B) SabA was affinity-adsorbed to erythrocytes from a cell-surface protein extract of strain J99. Immunostaining using SabA antibodies confirmed the presence of SabA adsorbed onto the erythrocyte surfaces as a result of binding to sialylated glycans (lane 1), whereas SabA was completely absent when erythrocytes had been depleted of sialic acid by sialidase treatment prior to the test (lane 2). Molecular weight markers (in kDa) are indicated.
(C) Binding of H. pylori strains J99 and J99sabA to human erythrocyte glycosphingolipids. (i) Chemical detection by anisaldehyde. (ii–iii) Autoradiograms obtained by binding of 35S-labeled H. pylori strain J99 and the J99sabA mutant, respectively, to separated glycosphingolipids. The lanes contain non-acid glycosphingolipids of human erythrocytes, 40 μg (lane 1); gangliosides of human erythrocytes, 40 μg (lane 2); GM3 ganglioside (NeuAcα2–3Galβ4Glcβ1Cer), 4 μg (lane 3); NeuAcα2–3-neolactotetraocylceramide
(NeuAcα2–3Galβ4GlcNAcβ3Galβ4Glcβ1Cer), 4 μg (lane 4); NeuAcα2–6-neolactotetraocylceramide
(NeuAcα2–6Galβ4GlcNAcβ3Galβ4Glcβ1Cer), 4 μg (lane 5); G-10 ganglioside (NeuAcα2–3Galβ4GlcNAcβ6 (NeuAcα2–3Galβ4GlcNAcβ3)Galβ4GlcNAcβ3Galβ4Glcβ1Cer), 1 μg (lane 6); G9-B ganglioside (Galα3(Fucα2)Galβ4GlcNAcβ6
(NeuAcα2–3Galβ4GlcNAcβ3)Galβ4GlcNAcβ3Galβ4Glcβ1Cer), 1 μg (lane 7); and reference gangliotriaosylceramide (GalNAcβ4Galβ4Glcβ1Cer) of mouse feces, 4 μg (lane 8).
For the series of 99 clinical isolates, HA titers were compared with prevalence of sLex binding. This was assessed by the use of 125I-labeled sialyl-dimeric Lex (sdiLex) antigen conjugate. The sdiLex antigen consists of two repetitive and fucosylated Lex antigens terminally substituted with α2–3–linked sialic acid (Table 1). A total of 37 of 99 isolates (37%) could bind to sdiLex, and 22 of 37 (60%) of these isolates proficient in sdiLex binding also demonstrated reduction in sialyl-dependent binding (sia-HA properties). In comparison, among the 62 isolates that did not bind sdiLex, only five isolates (8%) demonstrated sia-HA properties. In the group of 31 isolates that instead increased their HA titers due to sialidase treatment, only one single isolate showed distinct sdiLex binding, two isolates bound weakly, and the great majority (28 isolates) completely lacked sdiLex-binding properties (Figure 2A).
Table 1.
Soluble Glycoconjugates Used in This Study

A strong correlation was found between bacterial binding to sdiLex and HA titers resulting from sialidase treatment (correlation of rank = 0.547; p < 0.001) (Figure 2A). These results demonstrate that clinical isolates that are reduced in HA activity due to desialylation of erythrocytes by sialidase treatment constitute the group of H. pylori strains in which the great majority can bind sialylated Lewis antigens.
A sabA Deletion Mutant Identified SabA as the Sialyl-Dependent Hemagglutinin
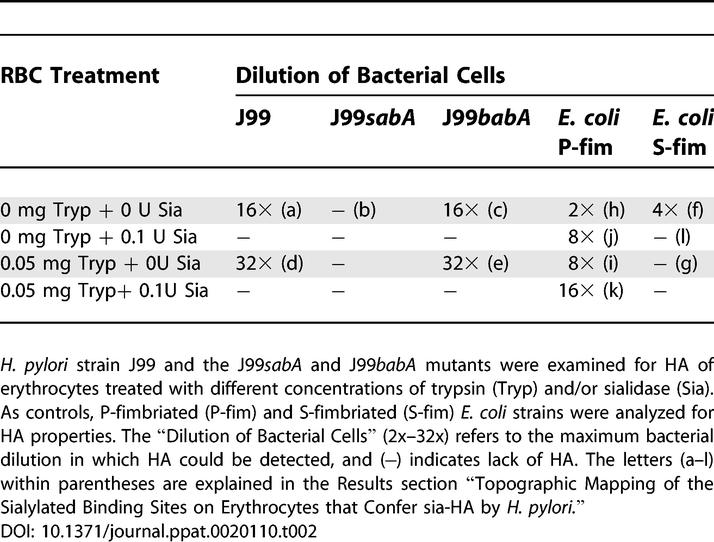
Strain J99 has previously been shown to express the SabA adhesin and to bind sialylated Lewis glycans such as sdiLex, sLex, and sLea (Table 1). By comparison, the J99 sabA deletion mutant (J99sabA) has lost the ability to bind sialyl-Lewis antigens [15]. Here, the J99 and J99sabA strains were compared for sia-HA properties. The results showed that while strain J99 was positive for sia-HA, the J99sabA mutant is fully devoid of all sia-HA properties (Table 2, 16× [a] versus 0 [b]). This result is most consistent with SabA being the sialyl-dependent hemagglutinin of H. pylori. In comparison, the J99 babA deletion mutant (J99babA) was not affected in sia-HA, but instead behaved most similarly to the J99 parent strain (Table 2, 16x [c] versus [a]). Taken together, the results also demonstrate that binding by BabA to fucosylated blood group antigens does not confer erythrocyte aggregation and HA.
Table 2.
Bacterial HA of Enzyme-Treated Erythrocytes
SabA Is Adsorbed to Sialylated Erythrocyte Surfaces
Binding of solubilized SabA protein was analyzed by affinity adsorption to sialylated erythrocyte surfaces. Here, a cell-surface protein extract from strain J99 was mixed with naive or sialidase-treated erythrocytes. Immunoblot analysis with antibodies against SabA showed that SabA was affinity absorbed onto intact and sialylated erythrocytes, whereas no SabA bound to sialidase-treated and sialic acid–depleted erythrocytes (Figure 2B).
Topographic Mapping of the Sialylated Binding Sites on Erythrocytes that Confer sia-HA by H. pylori
Sialylated erythrocyte antigens have been described in glycoproteins such as glycophorin A [22], in glycosphingolipids (gangliosides), and in polyglycosylceramides [34]. Sialylated bacterial binding sites were topographically localized on erythrocytes by functional discrimination between sialylated glycoproteins, which reach above the membrane level, and tight membrane–associated gangliosides. Erythrocytes were first treated with protease (trypsin) to destroy most glycoproteins at the erythrocyte surface, followed by treatment with sialidase to test for sialic acid–dependent binding sites. Proteolytic removal of cell-surface glycoproteins resulted in a distinct increase in sia-HA for both the J99 wt strain and the J99 babA mutant (Table 2, [d] and [e]). In contrast, S-fimbriated E. coli, which is known to hemagglutinate by binding to the sialylated erythrocyte glycoprotein glycophorin A (Table 2, [1]) [22], was most sensitive to protease treatment of erythrocytes, which even at low concentrations completely abrogated sia-HA (Table 2, [f] versus [g]). Similar to H. pylori, protease treatment of erythrocytes conferred stronger HA of P-fimbriated E. coli, which binds Galα4Gal antigens that are only present in glycolipids. This is most likely due to increased accessibility of the adhesive P-fimbriae for the Galα4Gal receptor epitopes present in membrane-close glycolipids (Table 2, [h] versus [i]). In addition, sialidase treatment and removal of the charged sialic acid residues could confer better exposure of the Galα4Gal receptor epitopes in glycan core chains and increased sia-HA (Table 2, [j] and [k] versus [h]). Taken together, the results show that the main part of sia-HA is conferred by sialyl-dependent binding of SabA to the tight membrane–associated and sialylated glycosphingolipids (i.e., gangliosides).
SabA Binds to Gangliosides from Human Erythrocytes
The major ganglioside structures found in human erythrocytes are the GM3 ganglioside and NeuAcα2–3-neolactotetraosylceramide (Table 3; NeuAcα3SPG). In addition, a number of minor complex gangliosides have been described [35]. Binding of strain J99 to non-acid and sialylated glycosphingolipid fractions of human erythrocytes immobilized on thin-layer chromatography plates (i.e., solid phase presentation) was analyzed (Figure 2Cii, lanes 1 and 2, respectively). The NeuAcα2–3-neolactotetraosylceramide region of the acid fraction (lane 2) was positive for binding, while no binding by strain J99 to the non-acid glycosphingolipids was observed. A higher sensitivity was obtained when purified human erythrocyte gangliosides was used (lanes 3–7). As for the acid glycosphingolipid fraction (lane 2), strain J99 bound to purified NeuAcα2–3-neolactotetraosylceramide (lane 4). In addition, the bacteria bound the purified complex gangliosides G-10 and G9-B (lanes 6 and 7). These structures are minor components of the erythrocyte gangliosides, which may explain their lack of binding in the crude fraction (lane 2). Strain J99 did not bind the shorter GM3 ganglioside (lane 3), nor did it bind the α2–6–linked NeuAcα2–6-neolactotetraosylceramide (lane 5 and Table 3; NeuAcα6SPG). Interestingly, NeuAcα2–8 modifications to the NeuAcα2–6–substituted GD3 and DPG glycans seemingly interfered with SabA-mediated binding. Similarly, the GD1a glycan, which is similar to the NeuAcα2–3-neolactotetraosylceramide (NeuAcα3SPG), but with an additional NeuAcα2–3 residue linked to the core, had lost its binding epitope for SabA (Table 3), which further points to the importance of the steric presentation of the NeuAcα2–3Gal epitope for the SabA-mediated binding mode. The absolute requirement for SabA was demonstrated by the sabA deletion mutant strain J99sabA, which did not bind to any of the gangliosides tested (Figure 2Ciii), although there was consistent SabA-independent binding to the reference non-acid gangliotriaosylceramide (lane 8; [36]).
Table 3.
Results from Binding of H. pylori to Human Erythrocyte Gangliosides

Characterization of H. pylori Binding Specificities to Sialylated Glycans
The sialylated receptor epitope for SabA was further characterized by use of structurally defined sialylated glycoconjugates of various complexities in terms of fucosylation and core chain and spacer unit lengths (Figure 3). The extended sdiLex antigen was recently shown to be the best receptor for SabA, which further suggests that fucosylation, sialylation, and length of the core chain are parameters that together form a high-affinity binding epitope for SabA. NeuAcα2–3-lactose and NeuAcα2–6-lactose were first compared for bacterial binding. Both structures are based on purified sialyl-lactose structures, but the glucose ring has been opened by reductive amination and used for conjugation to albumin, which leaves only the NeuAc-Gal disaccharide intact and available for bacterial binding. Strains J99, CCUG17875 (17875), 17875babA1A2, CCUG17874 (17874), SMI65, and SMI27 all bound the NeuAcα2–3-lactose conjugate, while none of the strains tested bound the NeuAcα2–6-lactose conjugate. The results suggest that presentation of the sialic acid residue by the α2–3 linkage is essential for SabA-mediated binding both to soluble glycoconjugates and for binding to gangliosides in solid phase. Since binding by SabA to sialyl-lactose was rather weak for all strains, the influence of chain length and steric flexibility in promotion of binding was investigated by use of sialyl-lactosamine (sLn; NeuAcα3Galβ4GlcNAc) attached to albumin, by either the short three-atom [sLn(3)] or the extended 14-atom [sLn(14)] spacer molecules. All strains that bind to sialylated glycans bound ≥4-fold better to sLn(14) than to sLn(3). Strains J99, SMI65, and SMI27 bound strongly to both sLea and sLex, whereas strains 17875, 17874, and the 17875babA1A2 mutant demonstrated much weaker binding to sLea compared to sLex/sdiLex. In keeping with our previous results, strains 26695, J99sabA, and 17875/Leb (a spontaneous mutant which does not bind sialylated antigens) appear to lack binding properties for sLex/sdiLex, sLea [15], and for the sialyl-lactose/lactosamine conjugates tested here.
Figure 3. Binding of H. pylori Strains and Clinical Isolates to Fucosylated and Sialylated Glycans of Various Complexities.

H. pylori reference strains, mutants, and clinical isolates were tested for binding to the fucosylated Leb antigen and to a series of sialylated antigens, all presented by 125I-labeled albumin conjugates. The y axis gives the percentage of bound conjugate.
Analysis of Binding Affinities for Sialylated Antigens
The binding affinities of strains J99, 17874, SMI65, SMI27, and the mutant 17875babA1A2 for the series of sialylated conjugates described above were analyzed according to Scatchard [37] (Figure 4). Strains J99, SMI65, and SMI27 demonstrated similar profiles in binding affinities for the series of sdiLex, sLex, sLea, and sLn(14) in the range of 9.6 × 108 M−1 to 6.4 × 109 M−1. By analogy with the results above, all three strains demonstrated lower binding affinity for sialyl-lactose (5.1 × 108 M−1 to 6.1 × 108 M−1). However, sialyl binding is not uniform among strains, since strains 17874 and 17875babA1A2 demonstrated a different binding pattern, with 10-fold lower affinity for sLea (9.7 × 107 M−1 and 1.0 × 108 M−1, respectively) and reduced affinity for sialyl-lactose (2.0 × 108 M−1 and 1.9 × 108 M−1, respectively). In the present experimental series, the 17875babA1A2 mutant exhibited binding affinities, which are approximately 10-fold higher than previously reported [15]. The stronger binding affinity reported relates to the improved bovine serum albumin (BSA) preparation used here as blocking agent, because we found BSA to contain sialyl-competitive constituents (unpublished data). Interestingly, binding to nonsialylated glycans such as Leb was not affected by the improved BSA preparation, which might be due to lack of the human/primate-specific antigen, Leb, in bovine serum constituents. To remove endogenous sialylated glycans, periodate oxidation of the BSA preparation was performed. This treatment resulted in >10-fold higher binding affinities of H. pylori for sLex, and consequently, the deglycanated blocking agent was used for all binding analyses in this study.
Figure 4. Binding Affinities Analyzed According to Scatchard of H. pylori Reference Strains, a babA Deletion Mutant, and Clinical Isolates for Sialylated Glycans.

Polymorphism in Binding to Sialylated Glycans among Clinical Isolates
The 99 Swedish clinical isolates were all analyzed for binding to sdiLex, sLea, and sLn(14) glycans (Figure 5). Of those, 39 (39%) could bind to sialylated glycans, and the great majority, 34 (87%) of 39 of these isolates, could bind all three glycans. Three isolates exclusively bound sdiLex and two isolates only bound sLea, but isolates with such unusual binding properties were generally poor binders (<1.5% of bound conjugate). Multiple different sialic acid–dependent adherence modes were found among the 39 sialyl-binding isolates, and these were classified as follows: (1) isolates that bind to all three sialylated glycans, with preferential binding to sdiLex (12 of 39 isolates, or approximately 30%); (2) isolates that bind to sLea better than sdiLex and sLn(14) (i.e., strains with an “Λ-shaped” binding mode) (seven of 39 isolates, or approximately 20%); (3) isolates that bind sdiLex and sLn(14) the best, whereas binding to sLea is lower (i.e., isolates with a “V-shaped” binding mode [similar to the binding mode of strain 17874; Figure 3]) (three of 39 isolates, or approximately 10%); (4) isolates that bind sLex but show only modest binding to sLea and sLn(14) (seven of 39 isolates, or approximately 20%); and (5) isolates generally modest in binding (in the interval 1%–5%) for all sialyl conjugates (ten of 39 isolates, or approximately 20%). As shown in Figure 5, preferential binding to sdiLex was the most common binding mode among the clinical isolates tested (24 of 39 isolates, or approximately 60% of strains tested).
Figure 5. Different Sialyl-Dependent Binding Modes of H. pylori Identified by Use of sdiLex, sLea, and sLn Conjugates.

A total of 39 Swedish clinical isolates were investigated for detailed sialyl-dependent binding properties. Representatives of the different binding modes for 125I-labeled sialylated glycans are illustrated in the diagram: (i) (in thick line) binds efficiently to all three sialylated glycans with preferential binding to sdiLex; (ii) binds to all three sialylated glycans, with better binding to sLea (“Λ-shaped” hatched line); (iii) binds preferentially to sdiLex and sLn(14) (“V-shaped” dotted line); (iv) binds preferentially to sdiLex but exhibits only modest binding for sLea and sLn(14) sialyl conjugates; and (v) binds modestly for all sialyl conjugates (<5% bound conjugate). The y axis gives the percentage of bound conjugate.
Polymorphism in Binding to Sialylated Glycans Is an Inherent Feature of SabA
To test if SabA alone accounts for the polymorphic binding modes to sialylated glycans, sabA was deleted in five clinical isolates, which together are representative of the main modes of sialyl-dependent binding (as analyzed in Figure 5). All five isogenic sabA deletion mutants were tested both for binding to 125I-labeled sdiLex, sLea, and sLn glycan conjugates and for expression of SabA. The results showed that the series of sabA deletion mutants could no longer bind to sialylated antigens, which suggests that SabA is the main factor responsible for polymorphism in binding to sialylated antigens (Figure 6). To thoroughly investigate if the local environment, such as the outer membrane and lipopolysaccharide composition of the individual strain, could influence sialyl-dependent binding properties of SabA, a complementation test was performed. The test was based on the Swedish clinical isolate SMI9 and the reference strain J99, which exhibits low and high proficiency in binding to sLea, respectively, whereas both strains bind sdiLex and sLn the most similarly (Figure 6). The deletion mutant SMI9sabA::kan, which cannot bind to sialylated antigens, was used as a background strain for complementation with the sabA open reading frame of strain J99. Transformant clones with gained sialyl antigen–binding properties were isolated by an enrichment procedure based on HA and identified by colony screening using SabA antibodies. SabA-positive transformants were analyzed for binding to sdiLex, sLea, and sLn(14) glycans, and the SMI9sabAJ99 complementation mutant was identified. The donor strain J99 exhibits the high sLea/sdiLex binding affinity ratio of 1.1, whereas the recipient strain SMI9 exhibits the low sLea/sdiLex ratio of 0.3. Interestingly, the SMI9sabAJ99 complementation mutant demonstrated a sLea/sdiLex binding affinity ratio of 0.93 (i.e., SabAJ99 expressed in the background strain SMI9 is most similar in affinity to that of the donor strain J99) (Figure 7). The sdiLex-binding capacity, which reflects the number of functional SabA adhesins on the bacterial surface, remained the same (1.2 nM) in strain SMI9 after introduction of the sabAJ99 gene. In comparison, strain J99 exhibits a binding capacity of 0.5 nM for sdiLex. Sequence analyses demonstrated that full-length J99 sabA had recombined into the sabA locus of SMI9 sabA::kan and formed a functional sabA gene. To verify that the change to a high sLea/sdiLex binding ratio depends exclusively on the sabAJ99 complementation, a SMI9sabAJ99::cam deletion mutant was made. Immunoblot and binding analyses of the SMI9sabAJ99::cam mutant demonstrated both absence of SabA expression and sialic acid binding properties (unpublished data). Taken together, these results conclude that the sialyl polymorphism binding is an inherent characteristic of the SabA adhesin protein itself.
Figure 6. Analyses of SabA-Dependent Binding among Clinical Isolates.

(A) Binding characteristics of H. pylori clinical isolates and SabA-deficient isogenic mutants were studied by use of soluble 125I-labeled sdiLex, sLea, and sLn(14) conjugates.
(B) Immunoblot analysis of H. pylori clinical isolates and mutant strains using SabA antibodies. There were no SabA bands detected among the sabA deletion mutants; actually, there were no additional bands on the blots.
Figure 7. Analyses of Binding Affinities for Sialylated Antigens by SabA from Strain J99 Complementarily Expressed in Background Clinical Strain SMI9.

Binding affinities of H. pylori strains J99, SMI9, and the complementation mutant SMI9sabAJ99 were analyzed according to Scatchard. Binding affinities and the percentage ratio of sLex/sLea binding affinities are indicated.
Discussion
Epithelial adherence should benefit H. pylori by providing better access to nutrient-rich tissues, and may contribute to delivery of bacterial toxins and various effector molecules through the type IV secretion mechanisms. Tight adherence may also be deleterious for H. pylori whenever robust host responses confront the bacterium with bactericidal agents. This suggests that persistence of H. pylori in the gastric mucosa depends on the maintenance of a balanced mode of mucosal adherence and the continuous adaptation of its binding properties to functionally match shifts in host glycosylation patterns during chronic inflammation. Mechanisms of rapid adaptation to local inflammation responses may be conferred by rapid on/off phase variation in SabA expression [15] and recombination events involving babA and babB [38,39].
Once established in the gastric epithelium, H. pylori tends to invade the gastric mucosa by slippage through the tight-cell junctions, which is a process that requires reorganization of the cytoskeleton and induced cell morphology [7]. Within the gastric tissue there are several alternative routes available for H. pylori, such as SabA-mediated adherence to neutrophils that have invaded the gastric mucosa [5]. Alternatively, H. pylori can enter into an intracellular lifestyle, and can invade gastric epithelia cells, whether normal, metaplastic, dysplastic, or cancerous [9], and possibly also gastric stem cells [10]. The inflammatory processes also confers upregulation of sialylated and sulfated carbohydrates in the local vessel lining (i.e., addressins), which act to “home in” on neutrophils from the peripheral circulation (i.e., recruits and activates the inflammatory cells from circulation and directs them to local areas of inflamed and infected tissue [40]). The sialylated parts of the addressins are most similar to the sialylated glycans that are bound by SabA. Thus, invasion of the high-endothelial venule–like vessels might be mediated by SabA-dependent adherence, first to the endothelial lining, and later also to the sialylated erythrocytes. In such a putative scenario, the results presented here are intriguing since systemic dissemination of microbial pathogens might be related to systemic disease. Indeed, chronic dental gum infections, such as periodontal infection in dental pockets, have been shown to frequently leach bacterial cells into circulation, although most bacterial cells are quickly killed by the acquired immunity and complement systems. However, persistent periodontal disease is associated with arteriosclerosis of the carotid arteries and coronary heart disease [41,42]. Interestingly, H. pylori infection is also associated with coronary heart disease, suggesting the influence of the disseminated bacterial cells in the long-term development of this pathology [43]. Although the erythrocyte-binding sia-HA phenotype is prevalent among clinical isolates, it has not yet been recognized as a virulence property of H. pylori, since sia-HA has not explicitly been shown to correlate with gastric disease [28,44]. However, the prevalence of sialic acid binding may have been underestimated among clinical isolates since: (1) H. pylori exhibits on/off phase variation in sialyl binding, and sia-HA could be rapidly lost during passage in culture [15]; (2) almost half of the strains that bound sLex did not cause sia-HA (Figure 2), which suggests that many strains require complex sialylated glycans for binding (such structures are limited on erythrocytes, but are present both in gastric epithelium and on neutrophils [45]); and (3) culture conditions have been shown to influence binding specificity and affinity for sialylated glycoconjugates [34]. In this report, we show that clinical isolates exhibit three distinct HA patterns in response to sialidase treatment (Figure 2): (1) reduced HA titers; (2) increased HA titers; or (3) unaltered HA titers. Reduced HA titers would be the expected effect of sialidase-dependent depletion of sialic acid, whereas an increase in HA titers suggests that depletion of sialic acid may better expose (cryptic) binding sites (i.e., less-accessible glycan epitopes). The third pattern, with no change in HA titers, probably relates to combinations of reduced and increased titers (i.e., strains with yet additional adhesins. The HA properties were found to be highly correlated with sLex binding activity, which implicates SabA as the causative agent in sia-HA. Thus, by the use of deletion mutants, we were able to unambiguously identify SabA as the H. pylori sialic acid–dependent hemagglutinin.
To further our understanding of SabA binding, H. pylori strains and mutants were tested for binding to sialylated glycans of various lengths and complexities. The results suggest that the NeuAcα2–3Gal disaccharide constitutes the minimal binding epitope for SabA binding, which is in agreement with previous reports on the binding specificity for the H. pylori sialyl-dependent hemagglutinin [27]. However, the core chain length was shown to affect SabA-mediated binding to erythrocyte gangliosides, where the NeuAcα2–3-neolactooctaocylceramide bound much better than the shorter NeuAcα2–3-neolactotetraocylceramide. Similarly, semisynthetic glycoconjugates with sialylated glycans presented on 14-atom spacers bound better than short three-atom spacers, which suggests that the sialylated binding epitope is best presented by extended and flexible core chains. Furthermore, all strains demonstrated increased binding for sLex, sdiLex, and sLea compared to sialylated structures lacking fucose constituents. These results are in keeping with our previous results on high-affinity binding of SabA to extended gangliosides with repetitive Lex motifs [15]. In the present study, we also showed by protease treatment analysis of host cell surfaces that H. pylori HA is mainly mediated by binding to glycosphingolipids. Nevertheless, the sialylated protein (albumin) conjugates used in this study are also efficient receptors for SabA-mediated H. pylori binding. This might in part relate to the uniform coating and presentation of sialylated glycans on the globular albumin molecule, which mimic the host cell–surface presentation of receptor glycans. In addition, the soluble albumin conjugates are probably more efficient for bacterial binding due to pretty flexible presentation of the sialylated antigens compared to bacterial binding to surfaces presented glycolipids (i.e., solid-phase interactions). Thus, strong and multivalent (“Velcro”-type) binding to sialylated epitopes on glycosphingolipids (gangliosides) with extended core chains would promote membrane-tight binding of SabA during experimental HA and in the inflamed gastric epithelium.
In the present study, the great majority of sialyl-binding H. pylori isolates bound the full series of sdiLex, sLea, and sLn glycans. This is a distinct difference compared to previous results, where merely half of sLex-binding strains also bound sLea [15]. The higher prevalence of sLea binders reported here is probably best explained by the presence of competitive sialylated antigens in the albumin-based blocking agent used previously. This is also supported by the 10-fold increased binding affinity for sLex, 1 × 109 M−1, revealed by strain 17875babA1A2 (Figure 4). Interestingly, clinical isolates demonstrated several distinct binding modes for sialylated glycans, although most isolates bound best to sLex. The differences in binding affinities to various sialylated glycans relate both to complexities in fucosylation and to the type of lacto-series core chains. Subtle differences in binding specificities have been described for urinary tract infectious Galα4Gal-binding P-fimbriated E. coli. For urinary tract infectious isolates the length of the globoseries glycolipids, required for bacterial binding, sorted the cognate adhesins into different functional subtypes that recognize human or canine kidney tissue [46,47]. In addition, single amino acid changes in type-I fimbriated E. coli urinary tract infectious isolates can change the normal binding mode for antennary mannosylated structures into a high-affinity binding mode that also accepts mono-mannosylated structures [48].
Hence, detailed differences in binding properties often relate to the adhesin polypeptide itself, but the binding mode can sometimes be distinctly influenced by physical constraints imposed by associated proteins in the local environment (e.g., the detailed binding specificity of Salmonella type I fimbriae is dependent on the fimbrial shaft on which the FimH adhesin is presented [49]). In contrast to pilus-associated adhesins, SabA exhibits a C-terminal putative ß-barrel domain, and hence is most likely instead a membrane-integrated protein. Our studies point to the fact that the detailed binding properties of SabA described here, similar to BabA [14], are inherent features of the adhesin proteins themselves.
The polymorphic binding to sialylated conjugates probably relates to small differences in SabA, which is similar to BabA in that positive selection for nonsynonymous codon substitutions have generated variant BabA adhesins that demonstrate specialist and generalist binding modes for ABO blood group antigens [14]. The high mutation rates in H. pylori [50,51] will promote formation of derivative strains with modified binding properties. Polymorphic binding properties could be of utmost importance during mixed infections, when recombination and exchange of genetic information might promote formation of transformants with chimeric binding properties that together behaves like a quasi-species. The occurrence of H. pylori strains that express SabA adhesins with different subtypes of binding modes as described here could therefore reflect such ongoing molecular evolution.
H. pylori infection and gastritis have been found to enhance gastric mucosal expression of the inflammation associated sLex/sLea antigens [15]. These sialylated glycans are dynamically expressed in competition with fucosylated blood group antigens [52]. Furthermore, malignant transformation has been reported to confer a pronounced expression of Lea, sLea, and sdiLex in the gastric mucosa [19,53]. Such changes to the differentiation programs of the gastric epithelial cell lineages will promote expression of a wide range of sialylated antigens. Taken together, these reports suggest that H. pylori infection and the associated chronic inflammation responses continuously change the availability of sialylated glycans of different complexities. Such changes in the gastric sialylation patterns would select for SabA clones that evolve with new or modified binding properties for sialylated epitopes. Thus, the bacteria–host crosstalk selects for polymorphic clones with optimal fitness for both the sialyl archipelago of the local environment (gastric mucosa) and the individual's phenotype for balanced, lifelong infection.
Materials and Methods
Bacterial strains.
The H. pylori strains used in this study were CCUG17875, CCUG17874 [54], 26695 [55], J99 [2], the sabA deletion mutant J99sabA(JHP662)::cam, the babA deletion mutant J99babA::cam, and the babA “double” mutant 17875babA1::kan babA2::cam (the series of mutants are abbreviated J99sabA, J99babA, and 17875babA1A2, respectively). The 17875/Leb strain is a spontaneous mutant that binds Leb but does not bind to sialylated antigens [15]. The panel of 99 clinical H. pylori isolates (including the series of SMI strains) came from Uppsala University Hospital, Sweden, and have been described previously [54]. Bacteria were grown on Brucella agar supplemented with 10% bovine blood and 1% IsoVitalex (Svenska LABFAB, http://www.labfab.se), for 43–48 h at 37 °C in 10% CO2 and 5% O2 before harvest in phosphate-buffered saline (PBS) containing 0.05% Tween 20 and 1% BSA (Sigma-Aldrich, http://www.sigmaaldrich.com). E. coli strains HB101/pPAP5 [47] and HB101/pAZZ50 [56] were cultured overnight on Luria broth plates supplemented with chloramphenicol and tetracycline.
Biopsies.
Gastric biopsies harvested at endoscopy in human and monkeys were fixed in Z-fix (10% paraformaldehyde + 1% ionized Zn; Anatech LTD, http://www.anatechltd.com), dehydrated, and embedded in paraffin. The protocol involving human subjects was approved by the Institutional Review Boards of the Veterans Affairs New York Harbor Health Care System and the Uniformed Services University of the Health Sciences, and written informed consent was obtained from all patients before study entry. All animal experiments were approved by the Armed Forces Radiobiology Research Institute Institutional Animal Care and Use Committee and monitored and reapproved at yearly intervals. All experiments were conducted according to the principles set forth in the Guide for the Care and Use of Laboratory Animals, Institute of Laboratory Animal Resources, National Research Council, (Washington, D. C.: National Academy Press, 1996).
In situ hybridization.
Sections (5 μm) were processed for in situ hybridization [9]. Sections were first deparaffinized in xylene, rehydrated in graded ethanol, and treated with proteinase K. They were then covered with hybridization mixture as described [9]. A specific cDNA probe was designed using a sequence database for H. pylori 16S rRNA [9], and the 5′ end of the oligonucleotide was labeled with biotin. The probe was denatured by heating for 10 min at 65 °C; a drop of probe was then placed on the tissue, and the reaction was incubated overnight at 37 °C for 18 h. The unbound probe was removed by successive washes in decreasing concentrations of Na citrate at room temperature and at 65 °C for the last wash. The biotin-labeled probe was detected by streptavidin-conjugated alkaline phosphatase (2 h of incubation at room temperature), and a chromogenic substrate (nitro-blue-tetrazolium, NBT/BCIP kit; Vector Labs, http://www.vectorlabs.com). After washing, sections were counterstained with Nuclear Fast Red and mounted with permount. Positive controls were as described [9] and included H. pylori pure cultures streaked onto precleaned microscope slides. Negative controls included E. coli and Shigella flexneri cultures, and gastric biopsy specimens from a 62-y-old H. pylori–negative patient with dyspepsia were also used as negative controls [9]. Finally, control for nonspecific binding was performed by using sense instead of antisense probe, hybridization buffer instead of antisense probe, unlabeled antisense probe, digoxigenin- or biotin-labeled probe for the scorpion Buthus martensi Karsch neurotoxin sequence (5′-GGC CAC GCG TCG ACT AGT AC-3′), RNase A pretreatment (Roche, http://www.roche.com), DNase I pretreatment (Roche), and RNase plus DNase I pretreatment [9].
Epoxy (Spurr low viscosity) semithin sections stained with toluidine blue.
Biopsies were processed for transmission electron microscopy using the conventional standard method [9]. Semithin sections of 0.5 μm were stained with 1% toluidine blue.
Image acquisition.
Sections were viewed using a light microscope Nikon Eclipse E300 (http://www.nikon.com) and pictures were captured and digitized using a QCapture camera (QImaging, http://www.qimaging.com) and Micropublisher 5.0 RTV (QImaging).
HA conditions and reagents.
Fresh human blood from a healthy donor of blood group A phenotype was used for the HA assays. The erythrocytes were washed twice with PBS. A 20% erythrocyte suspension was treated at 28 °C with 0.05 or 0.1 mg/ml trypsin (Sigma, http://www.sigmaaldrich.com) for 2 h at neutral pH [57] followed by 1 mM phenylmethylsulfonylfluoride (PMSF; Sigma) for 15 min, and then gently washed five times with PBS. A 4% suspension of trypsin-treated or untreated erythrocytes was incubated with sialidase type VI from C. perfringens (Sigma) at 0.1 U/ml [26]. After washing, a 0.75% erythrocyte suspension was mixed with a 2-fold dilution series of H. pylori for estimation of sia-HA titers (i.e., the reduction in HA titers as a result of sialidase treatment of erythrocytes). Round-bottomed enzyme-linked immunosorbent assay (ELISA) plates were used for the analysis, and the aggregation of erythrocytes was determined visually after 1 h at room temperature (23 °C).
Neoglycoproteins.
All glycan conjugates used were semisynthetic glycoproteins constructed of purified or chemically synthesized oligosaccharides conjugated to either human serum albumin (HSA) or to BSA (see Table 1). Leb, sLex, sdiLex, sLea, 3′-sialyl-lactose, 6′-sialyl-lactose (IsoSep AB, http://www.isosep.com), and sLn (with three-atom or 14-atom spacer, sLn(3) and sLn(14), respectively) glycoconjugates (Dextra Laboratories, http://www.dextra-labs.co.uk) were labeled with 125I by the chloramine-T method [54]. The glycan densities of the conjugates were 24 mol Leb oligosaccharides/mol HSA, 13 mol sLex oligosaccharides/mol HSA, 11 mol sdiLex oligosaccharides/mol HSA, 12 mol sLea oligosaccharides/mol HSA, 6 mol 3′-sialyl-lactose oligosaccharides/mol HSA, 15 mol 6′-sialyl-lactose oligosaccharides/mol HSA, 11 mol sLn(3) oligosaccharides/mol BSA, and 11 mol sLn(14) oligosaccharides/mol BSA.
Periodate oxidation of BSA blocking agent to destroy competitive carbohydrate receptors for H. pylori binding.
BSA (A3294; Sigma), 15% wt/vol in Milli-Q water (MQ; Millipore, http://www.millipore.com), was treated with 10 mM periodate for 1 h at pH 4.5. The reaction mixture was then reduced with 20 mM Na2S2O5 (pH 6.7). Reagents were removed by dialysis against MQ followed by PBS containing 0.05% Tween 20 (PBS-Tween). Treated BSA was stored in aliquots at −20 °C. BSA treated by periodate oxidation did not quench sialyl binding, whereas the untreated BSA preparation substantially reduced binding of the 17875babA1A2 mutant.
Analysis of H. pylori binding to soluble neoglycoproteins by radioimmunoassay.
For radioimmunoassay analyses, 1 ml of bacteria (OD 600 nm = 0.1) was mixed with 5 ng of 125I-labeled conjugate. After 2 h at room temperature, the bacteria were pelleted and bound conjugate was measured by gamma scintillation counting (Wallac, http://las.perkinelmer.com) [54]. Binding experiments were carried out in duplicate. To test the periodate-treated BSA for presence of competitive carbohydrate receptors, bacteria of strain SMI65 (OD 600 nm = 0.1) were mixed with 2 ng of 125I-labeled sdiLex conjugate and a dilution series of 0, 0.2, 0.4, 0.6, 0.8, and 1% (wt/vol) periodate-treated or untreated BSA in PBS. After 2 h of incubation at room temperature, bacteria were pelleted and bound conjugate was quantified as described above. The results revealed that periodate-treated BSA did not inhibit sLex binding, whereas the untreated BSA showed strong inhibition. The full series of binding experiments reported in this study have been performed by use of this deglycanated BSA-blocking agent.
Affinity adsorption of SabA to erythrocyte surfaces.
Bacteria were harvested from overnight cultures, washed once with PBS, and then treated with 500 μl deionized water for 15 min at room temperature with gentle shaking. After centrifugation for 30 min at 10,000g at 4 °C, 10× PBS was added to the supernatant to bring it to 1× PBS. A total of 200 μl of 4% erythrocyte suspension (with or without prior sialidase treatment) was mixed 1:1 with bacterial protein extract for 1 h at room temperature. The erythrocytes were washed three times with PBS, mixed with SDS-PAGE sample buffer, and heated for 5 min at 95 °C. Electrophoresis was carried out using a 7.5% Tris-HCl Ready gel (Bio-Rad Laboratories, http://www.bio-rad.com). For immunoblot analysis, proteins were transferred to immunoblot polyvinylidene difluoride (PVDF) membranes (Bio-Rad). Membranes were blocked for 1 h in PBS-Tween containing 5% nonfat dried milk. Antibodies were raised in rabbit against full-length recombinant SabA as previously described for BabA [58]. Blots were incubated with horseradish peroxidase–conjugated goat anti-rabbit antibodies (Dako, http://www.dako.com) diluted in PBS-Tween containing 1% nonfat dried milk, and finally developed with SuperSignal West Pico Chemiluminiscent Substrate (Pierce, http://www.piercenet.com).
Glycosphingolipids.
Total non-acid and acid glycosphingolipid fractions from human erythrocytes were obtained by standard procedures [59]. Individual glycosphingolipids were isolated by acetylation of total glycosphingolipid fractions and repeated chromatography on silicic acid columns. Acid glycosphingolipid fractions were separated by DEAE-Sepharose chromatography, followed by repeated silicic acid chromatography, and final separation was achieved by high-performance liquid chromatography. The identity of the purified glycosphingolipids was confirmed by mass spectrometry [60], proton nuclear magnetic resonance spectroscopy [61], and degradation studies [62,63]. A detailed description of the isolation and characterization of the H. pylori–binding gangliosides has been given elsewhere [45].
Chromatogram binding assay.
The conditions used for culture, 35S-labeling of H. pylori, and the chromatogram binding assays were as described previously [36]. Mixtures of glycosphingolipids (40 μg/lane) or pure compounds (1–4 μg/lane) were separated on aluminum-backed silica gel 60 high-performance thin-layer chromatography plates (Merck, http://merck.com) using chloroform/methanol/0.25% KCl in water (50:40:10 by volume) as a solvent system. Thereafter, a suspension of 35S-labeled bacteria (diluted in PBS to 1 × 108 CFU/ml and 1–5 × 106 cpm/ml) was sprinkled over the chromatograms and incubated for 2 h at room temperature. After washing with PBS and drying, the thin-layer plates were autoradiographed using XAR-5 X-ray film (Eastman Kodak, http://www.kodak.com).
Analysis of binding affinities for soluble glycoconjugates.
The affinity for H. pylori SabA-mediated binding to sialylated glycoconjugates was measured by Scatchard analysis [37]. Each H. pylori strain exhibits distinct binding affinities for sialylated receptor structures. Thus, for the affinity analyses, the bacterial cells were diluted to ascertain a free-to-bound ratio of conjugate binding close to 1.0 (equivalent to an equilibrium of ~50% bound glycoconjugate). Unlabeled glycoconjugate was then added in a dilution series of seven different concentrations, where the highest concentration was predicted to block approximately 80% of glycoconjugate binding. The bacterial cell–glycoconjugate mixture was incubated at room temperature for 2 h in PBS-Tween with 1% of the periodate-treated BSA as blocking agent. Bound conjugate was analyzed as described above by gamma scintillation counting for 5 min.
Construction of sabA deletion mutants.
To construct SMI14sabA::kan, SMI38sabA::cam, SMI100sabA::kan, and SMI31sabA::cam sabA deletion mutants, the Swedish clinical isolates SMI14, SMI38, SMI100, and SMI31 were first single-colony purified and transformed with sabA::kan or sabA::cam deletion vectors (previously described in [15]). For construction of SMI9sabA::kan, sabA was first PCR-amplified using the primers M3F and M4R and cloned in the pBluescript SK+/− EcoRV site. The plasmid clone was then linearized with M7F and M5R and ligated with the kanR gene, as described in [54]. H. pylori transformants were analyzed for binding to 125I-labeled sLex conjugate and for expression of SabA using anti-SabA antibodies, as described above. The primer sequences were as follows: M3F (5′-CGCTAGTGTCCAGGGTAAC-3′); M4R (5′-TTGATCGTAAGGCAGTGTGATA-3′); M7F (5′-TCCCTAAAGATCAGTATCGT-3′); and M5R (5′-CCGCGTATTGCGTTGGGTAG-3′)
Construction of SMI9 sabAJ99 recombinants.
The sabA gene of strain J99 was amplified by PCR using primers J99-4F and J99-7R. The resulting DNA fragment was transformed into strain SMI9sabA::kan. Transformants expressing recombinant SabA were isolated by the HA method as described below. Sequencing of sabA was performed using primers sabA-278F and sabA-1136R. The primer sequences were the following: J99-4F (5′-GAATACGCAATCTTGTGGAGT-3′); J99-7R (5′-CCAAATCACCCAATTACTTTG-3′); sabA-278F (5′-TACAACAGCACCACCCAA-3′); and sabA-1136R (5′-CATCTTTAGCCACGCTTAA-3′).
HA method for enriching sialic acid–binding sabA recombinants.
For isolation of sialyl-binding transformants fresh human blood from a healthy donor was used. The erythrocytes were washed three times with PBS, and a 4% erythrocyte suspension was added onto 1 ml of bacterial transformation mixture (OD600 = 1). The bacterial-erythrocyte suspension was incubated with gentle rocking for 30 min at 37 °C in 10% CO2 and 5% O2. For separation of transformants with gained erythrocyte-associated (sialyl-binding) binding properties from the nonbinding (parent) SMI9sabA deletion mutant, aggregated erythrocytes were allowed to sediment for 25 min. After removal of the supernatant and three rinses with PBS, 200 μl Brucella broth was added and the suspension was spread on a Brucella agar plate. Clones that express full-length SabA were identified by colony screening as described below. The HA and colony-screening procedure was repeated three times for efficient enrichment of clones of background strain SMI9, which had gained in complementary expression of SabA from donor strain J99.
Colony screening for sialic acid–binding sabA recombinants.
For identification of SabA-expressing clones the bacteria were spread in serial dilutions onto Brucella agar plates and cultured until single colonies appeared. The bacterial colonies were printed onto nitrocellulose membranes (Bio-Rad), which subsequently were soaked in boiling hot 1× sample buffer (4 ml 10% SDS, 1.6 ml Tris [pH 6.8], and 14.4 ml MQ H2O, with 500 μl β-mercaptoethanol) for 5 min. The membranes were blocked overnight in TBS with 0.05% Tween 20 (TBS-Tween) containing 5% nonfat dried milk at 4°C. SabA-expressing clones were detected by using anti-SabA antibodies (described above) and horseradish peroxidase–conjugated goat anti-rabbit antibodies and finally visualized by using 4-chloro-1-naphtol (4C1N) tablets (Sigma).
Construction of SMI9sabAJ99 sabA deletion mutants.
In order to construct SMI9sabAJ99::cam, SMI9sabAJ99 was transformed with a sabA::cam deletion vector as described [14]. Chloramphenicol-resistant transformants were analyzed for binding and expression of SabA as described above.
Acknowledgments
Gastric biopsies from human subjects provided by Dr. V. Simko (Brooklyn VA Medical Center, Veterans Administration New York Harbor Healthcare System, Brooklyn, New York, USA).
Abbreviations
- BabA
blood group antigen–binding adhesin
- BSA
bovine serum albumin
- HpaA
H. pylori adhesin A
- Leb
Lewis b
- SabA
sialic acid–binding adhesin
- sdiLex
sialyl-dimeric Lewis x
- sia-HA
sialic acid–dependent hemagglutination
- sLex
sialyl-Lex
- sLn
sialyl-lactosamine
Footnotes
¤a Current address: Institute of Molecular Biosciences, University of Oslo, Oslo, Norway
¤b Current address: Environmental Health Institute, National Environment Agency, Ministry of Environment, Singapore
Competing interests. In 2002 TB filed a patent application for the use of SabA as a vaccine candidate, International PCT pending number PCT/SE02/00301/ Helicobacter pylori sialic acid binding adhesin, SabA, and sabA gene.
Author contributions. MA, FOO, JN, BS, CL, RS, SA, SO, CSM, HL, AD, ST, and AA conceived, designed and performed the experiments. MA, FOO, JN, BS, RS, AD, ST, and TB analyzed the data. SO, RH, AD, ST, and TB contributed reagents/materials/analysis tools. MA, AD, and TB wrote the paper with the assistance of RH, TW, LE, ST, and AA.
Funding. This work was supported by the Umeå University Biotechnology Fund, the County Council of Västerbotten, the J. C. Kempe and Seth M. Kempe Foundation (TB), the Swedish Medical Research Council (TB, ST, LE, AA, TW), the Swedish Cancer Society (TB, ST, LE, AA), Swedish Foundation for Strategic Research SSF programs “Glycoconjugates in Biological Systems” (T.B., S.T.) and “Infection and Vaccinology” (MA, TB, LE), the Swedish Medical Society (ST), the Swedish Society for Medical Research (MA), the National Institutes of Health (CA82312/AD), Region Scania/ALF (TW), Åke Wiberg Foundation (AA), Nanna Svartz Foundation (AA), and EMBO Long-Term Fellowship (MA).
References
- Cover TL, Berg DE, Blaser MJ, Mobley HLT. Helicobacter pylori pathogenesis. In: Groisman EA, editor. Principles of bacterial pathogenesis. San Diego (California): Academic Press; 2001. pp. 509–558. [Google Scholar]
- Alm RA, Ling LS, Moir DT, King BL, Brown ED, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- Schreiber S, Konradt M, Groll C, Scheid P, Hanauer G, et al. The spatial orientation of Helicobacter pylori in the gastric mucus. Proc Natl Acad Sci U S A. 2004;101:5024–5029. doi: 10.1073/pnas.0308386101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emody L, Carlsson A, Ljungh A, Wadström T. Mannose-resistant haemagglutination by Campylobacter pylori. Scand J Infect Dis. 1988;20:353–354. doi: 10.3109/00365548809032466. [DOI] [PubMed] [Google Scholar]
- Unemo M, Aspholm-Hurtig M, Ilver D, Bergström J, Borén T, et al. The sialic acid binding SabA adhesin of Helicobacter pylori is essential for nonopsonic activation of human neutrophils. J Biol Chem. 2005;280:15390–15397. doi: 10.1074/jbc.M412725200. [DOI] [PubMed] [Google Scholar]
- Dubois A, Fiala N, Heman-Ackah LM, Drazek ES, Tarnawski A, et al. Natural gastric infection with Helicobacter pylori in monkeys: A model for spiral bacteria infection in humans. Gastroenterology. 1994;106:1405–1417. doi: 10.1016/0016-5085(94)90392-1. [DOI] [PubMed] [Google Scholar]
- Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, et al. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300:1430–1434. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstrand L, Graham D, Scheynius A, Genta RM, El-Zaatari F. Is the sanctuary where Helicobacter pylori avoids antibacterial treatment intracellular? Am J Clin Pathol. 1997;108:504–509. doi: 10.1093/ajcp/108.5.504. [DOI] [PubMed] [Google Scholar]
- Semino-Mora C, Doi SQ, Marty A, Simko V, Carlstedt I, et al. Intracellular and interstitial expression of Helicobacter pylori virulence genes in gastric precancerous intestinal metaplasia and adenocarcinoma. J Infect Dis. 2003;187:1165–1177. doi: 10.1086/368133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JD, Karam SM, Gordon JI. Intracellular Helicobacter pylori in gastric epithelial progenitors. Proc Natl Acad Sci U S A. 2005;102:5186–5191. doi: 10.1073/pnas.0407657102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard M, Lehn N, Neumayer N, Borén T, Rad R, et al. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc Natl Acad Sci U S A. 1999;96:12778–12783. doi: 10.1073/pnas.96.22.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz C, Schoniger M, Rad R, Becker I, Keiditsch E, et al. Key importance of the Helicobacter pylori adherence factor blood group antigen binding adhesin during chronic gastric inflammation. Cancer Res. 2001;61:1903–1909. [PubMed] [Google Scholar]
- Lindén S, Nordman H, Hedenbro J, Hurtig M, Borén T, et al. Strain- and blood group-dependent binding of Helicobacter pylori to human gastric MUC5AC glycoforms. Gastroenterology. 2002;123:1923–1930. doi: 10.1053/gast.2002.37076. [DOI] [PubMed] [Google Scholar]
- Aspholm-Hurtig M, Dailide G, Lahmann M, Kalia A, Ilver D, et al. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science. 2004;305:519–522. doi: 10.1126/science.1098801. [DOI] [PubMed] [Google Scholar]
- Mahdavi J, Sondén B, Hurtig M, Olfat FO, Forsberg L, et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science. 2002;297:573–578. doi: 10.1126/science.1069076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slomiany BL, Piotrowski J, Samanta A, VanHorn K, Murty VL, et al. Campylobacter pylori colonization factor shows specificity for lactosylceramide sulfate and GM3 ganglioside. Biochem Int. 1989;19:929–936. [PubMed] [Google Scholar]
- Teneberg S, Leonardsson I, Karlsson H, Jovall PA, Angstrom J, et al. Lactotetraosylceramide, a novel glycosphingolipid receptor for Helicobacter pylori, present in human gastric epithelium. J Biol Chem. 2002;277:19709–19719. doi: 10.1074/jbc.M201113200. [DOI] [PubMed] [Google Scholar]
- Gerhard M, Sirmo S, Wadström T, Miller-Podraza H, Teneberg S, et al. Helicobacter pylori, an adherent pain in the stomach. In: Achtman M, Suerbaum S, editors. Helicobacter pylori: Molecular and cellular biology. Norfolk (United Kingdom): Horizon Scientific Press; 2001. pp. 297–309. [Google Scholar]
- Sakamoto S, Watanabe T, Tokumaru T, Takagi H, Nakazato H, et al. Expression of Lewisa, Lewisb, Lewisx, Lewisy, siayl-Lewisa, and sialyl-Lewisx blood group antigens in human gastric carcinoma and in normal gastric tissue. Cancer Res. 1989;49:745–752. [PubMed] [Google Scholar]
- Clausen H, Hakomori S. ABH and related histo-blood group antigens: Immunochemical differences in carrier isotypes and their distribution. Vox Sang. 1989;56:1–20. doi: 10.1111/j.1423-0410.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- Alper J. Searching for medicine's sweet spot. Science. 2001;291:2338–2343. doi: 10.1126/science.291.5512.2338. [DOI] [PubMed] [Google Scholar]
- Parkkinen J, Rogers GN, Korhonen T, Dahr W, Finne J. Identification of the O-linked sialyloligosaccharides of glycophorin A as the erythrocyte receptors for S-fimbriated Escherichia coli. Infect Immun. 1986;54:37–42. doi: 10.1128/iai.54.1.37-42.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter NK, Hanson JE, Glick GD, Brown JH, Crowther RL, et al. Binding of influenza virus hemagglutinin to analogs of its cell-surface receptor, sialic acid: Analysis by proton nuclear magnetic resonance spectroscopy and X-ray crystallography. Biochemistry. 1992;31:9609–9621. doi: 10.1021/bi00155a013. [DOI] [PubMed] [Google Scholar]
- Arnberg N, Kidd AH, Edlund K, Nilsson J, Pring-Akerblom P, et al. Adenovirus type 37 binds to cell surface sialic acid through a charge-dependent interaction. Virology. 2002;302:33–43. doi: 10.1006/viro.2002.1503. [DOI] [PubMed] [Google Scholar]
- Pandey KC, Singh S, Pattnaik P, Pillai CR, Pillai U, et al. Bacterially expressed and refolded receptor binding domain of Plasmodium falciparum EBA-175 elicits invasion inhibitory antibodies. Mol Biochem Parasitol. 2002;123:23–33. doi: 10.1016/s0166-6851(02)00122-6. [DOI] [PubMed] [Google Scholar]
- Hirmo S, Kelm S, Schauer R, Nilsson B, Wadström T. Adhesion of Helicobacter pylori strains to alpha-2,3–linked sialic acids. Glycoconj J. 1996;13:1005–1011. doi: 10.1007/BF01053196. [DOI] [PubMed] [Google Scholar]
- Evans DG, Evans DJ, Jr., Moulds JJ, Graham DY. N-acetylneuraminyllactose-binding fibrillar hemagglutinin of Campylobacter pylori: A putative colonization factor antigen. Infect Immun. 1988;56:2896–2906. doi: 10.1128/iai.56.11.2896-2906.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelwala-Guruge J, Ljungh A, Wadström T. Haemagglutination patterns of Helicobacter pylori. Frequency of sialic acid-specific and non-sialic acid-specific haemagglutinins. Apmis. 1992;100:908–913. [PubMed] [Google Scholar]
- Evans DG, Karjalainen TK, Evans DJ, Jr., Graham DY, Lee CH. Cloning, nucleotide sequence, and expression of a gene encoding an adhesin subunit protein of Helicobacter pylori. J Bacteriol. 1993;175:674–683. doi: 10.1128/jb.175.3.674-683.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole PW, Janzon L, Doig P, Huang J, Kostrzynska M, et al. The putative neuraminyllactose-binding hemagglutinin HpaA of Helicobacter pylori CCUG 17874 is a lipoprotein. J Bacteriol. 1995;177:6049–6057. doi: 10.1128/jb.177.21.6049-6057.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AC, Logan RP, Foynes S, Cockayne A, Wren BW, et al. A flagellar sheath protein of Helicobacter pylori is identical to HpaA, a putative N-acetylneuraminyllactose-binding hemagglutinin, but is not an adhesin for AGS cells. J Bacteriol. 1997;179:5643–5647. doi: 10.1128/jb.179.17.5643-5647.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teneberg S, Miller-Podraza H, Lampert HC, Evans DJ, Jr., Evans DG, et al. Carbohydrate binding specificity of the neutrophil-activating protein of Helicobacter pylori. J Biol Chem. 1997;272:19067–19071. doi: 10.1074/jbc.272.30.19067. [DOI] [PubMed] [Google Scholar]
- Petersson C, Forsberg M, Aspholm M, Olfat FO, Forslund T, et al. Helicobacter pylori SabA adhesin evokes a strong inflammatory response in human neutrophils which is down-regulated by the neutrophil-activating protein. Med Microbiol Immunol (Berl). Prepublished June 7, 2006. 2006. as DOI 10.1007/s00430–006-0018-x. [DOI] [PubMed]
- Miller-Podraza H, Bergström J, Milh MA, Karlsson KA. Recognition of glycoconjugates by Helicobacter pylori. Comparison of two sialic acid–dependent specificities based on haemagglutination and binding to human erythrocyte glycoconjugates. Glycoconj J. 1997;14:467–471. doi: 10.1023/a:1018599401772. [DOI] [PubMed] [Google Scholar]
- Stults CL, Sweeley CC, Macher BA. Glycosphingolipids: Structure, biological source, and properties. Methods Enzymol. 1989;179:167–214. doi: 10.1016/0076-6879(89)79122-9. [DOI] [PubMed] [Google Scholar]
- Ångstrom J, Teneberg S, Milh MA, Larsson T, Leonardsson I, et al. The lactosylceramide binding specificity of Helicobacter pylori. Glycobiology. 1998;8:297–309. doi: 10.1093/glycob/8.4.297. [DOI] [PubMed] [Google Scholar]
- Scatchard G. The attractions of proteins for small molecules and ions. Ann N Y Acad Sci. 1949;51:660–672. [Google Scholar]
- Bäckström A, Lundberg C, Kersulyte D, Berg DE, Borén T, et al. Metastability of bab adhesin genes in Helicobacter pylori confers dynamics in Lewis b antigen binding. Proc Natl Acad Sci U S A. 2004;101:16923–16928. doi: 10.1073/pnas.0404817101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solnick JV, Hansen LM, Salama NR, Boonjakuakul JK, Syvanen M. Modification of Helicobacter pylori outer membrane protein expression during experimental infection of rhesus macaques. Proc Natl Acad Sci U S A. 2004;101:2106–2111. doi: 10.1073/pnas.0308573100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Mitoma J, Nakamura N, Katsuyama T, Nakamura N, et al. Induction of peripheral lymp node addressin in human gastric mucosa infected by Helicobacter pylori. Proc Natl Acad Sci U S A. 2004;101:17807–17812. doi: 10.1073/pnas.0407503101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvarieux M, Demmer RT, Rundek T, Boden-Albala B, Jacobs DR, Jr., et al. Periodontal microbiota and carotid intima-media thickness: The Oral Infections and Vascular Disease Epidemiology Study (INVEST) Circulation. 2005;111:576–582. doi: 10.1161/01.CIR.0000154582.37101.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein SE. The multiple mechanisms by which infection may contribute to atherosclerosis development and course. Circulation Research. 2002;90:2–4. [PubMed] [Google Scholar]
- Gillum RF. Infection with Helicobacter pylori, coronary heart disease, cardiovascular risk factors, and systemic inflammation: The third national health and nutrition survey. J Natl Med Assoc. 2004;96:1470–1476. [PMC free article] [PubMed] [Google Scholar]
- Taylor NS, Hasubski AT, Fox JG, Lee A. Haemagglutination profiles of Helicobacter species that cause gastritis in man and animals. J Med Microbiol. 1992;37:299–303. doi: 10.1099/00222615-37-5-299. [DOI] [PubMed] [Google Scholar]
- Roche N, Angstrom J, Hurtig M, Larsson T, Borén T, et al. Helicobacter pylori and complex gangliosides. Infect Immun. 2004;72:1519–1529. doi: 10.1128/IAI.72.3.1519-1529.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson KW, Pinkner JS, Rose T, Magnusson G, Hultgren SJ, et al. Structural basis of the interaction of the pyelonephritic E. coli adhesin to its human kidney receptor. Cell. 2001;105:733–743. doi: 10.1016/s0092-8674(01)00388-9. [DOI] [PubMed] [Google Scholar]
- Strömberg N, Nyholm PG, Pascher I, Normark S. Saccharide orientation at the cell surface affects glycolipid receptor function. Proc Natl Acad Sci U S A. 1991;88:9340–9344. doi: 10.1073/pnas.88.20.9340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas WE, Trintchina E, Forero M, Vogel V, Sokurenko EV. Bacterial adhesion to target cells enhanced by shear force. Cell. 2002;109:913–923. doi: 10.1016/s0092-8674(02)00796-1. [DOI] [PubMed] [Google Scholar]
- Thankavel K, Madison B, Ikeda T, Malaviya R, Shah AH, et al. Localization of a domain in the FimH adhesin of Escherichia coli type 1 fimbriae capable of receptor recognition and use of a domain-specific antibody to confer protection against experimental urinary tract infection. J Clin Invest. 1997;100:1123–1136. doi: 10.1172/JCI119623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkholm B, Sjölund M, Falk PG, Berg OG, Engstrand L, et al. Mutation frequency and biological cost of antibiotic resistance in Helicobacter pylori. Proc Natl Acad Sci U S A. 2001;98:14607–14612. doi: 10.1073/pnas.241517298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, Kraft C, Taylor NS, Correa P, Fox JG, et al. Recombination and mutation during long-term gastric colonization by Helicobacter pylori: Estimates of clock rates, recombination size, and minimal age. Proc Natl Acad Sci U S A. 2001;98:15056–15061. doi: 10.1073/pnas.251396098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JR, Fuster MM, Whisenant T, Esko JD. Expression patterns of alpha 2,3-sialyltransferases and alpha 1,3-fucosyltransferases determine the mode of sialyl Lewis X inhibition by disaccharide decoys. J Biol Chem. 2003;278:23352–23359. doi: 10.1074/jbc.M303093200. [DOI] [PubMed] [Google Scholar]
- Amado M, Carneiro F, Seixas M, Clausen H, Sobrinho-Simoes M. Dimeric sialyl-Le(x) expression in gastric carcinoma correlates with venous invasion and poor outcome. Gastroenterology. 1998;114:462–470. doi: 10.1016/s0016-5085(98)70529-3. [DOI] [PubMed] [Google Scholar]
- Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, et al. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279:373–377. doi: 10.1126/science.279.5349.373. [DOI] [PubMed] [Google Scholar]
- Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- Hacker J, Schmidt G, Hughes C, Knapp S, Marget M, et al. Cloning and characterization of genes involved in production of mannose-resistant, neuraminidase-susceptible (X) fimbriae from a uropathogenic O6:K15:H31 Escherichia coli strain. Infect Immun. 1985;47:434–440. doi: 10.1128/iai.47.2.434-440.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borén T, Wadström T, Normark S, Gordon JI, Falk P. Helicobacter pylori protocols. Totowa (New Jersey): Humana Press; 1997. 274 [Google Scholar]
- Yamaoka Y, Kodama T, Kita M, Imanishi J, Kashima K, et al. Relationship of vacA genotypes of Helicobacter pylori to cagA status, cytotoxin production, and clinical outcome. Helicobacter. 1998;3:241–253. doi: 10.1046/j.1523-5378.1998.08056.x. [DOI] [PubMed] [Google Scholar]
- Karlsson KA, Strömberg N. Overlay and solid-phase analysis of glycolipid receptors for bacteria and viruses. Methods Enzymol. 1987;138:220–232. doi: 10.1016/0076-6879(87)38019-x. [DOI] [PubMed] [Google Scholar]
- Samuelsson BE, Pimlott W, Karlsson KA. Mass spectrometry of mixtures of intact glycosphingolipids. Methods Enzymol. 1990;193:623–646. doi: 10.1016/0076-6879(90)93442-n. [DOI] [PubMed] [Google Scholar]
- Koerner TA, Jr., Prestegard JH, Demou PC, Yu RK. High-resolution proton NMR studies of gangliosides. 1. Use of homonuclear two-dimensional spin-echo J-correlated spectroscopy for determination of residue composition and anomeric configurations. Biochemistry. 1983;22:2676–2687. doi: 10.1021/bi00280a014. [DOI] [PubMed] [Google Scholar]
- Yang HJ, Hakomori SI. A sphingolipid having a novel type of ceramide and lacto-N-fucopentaose 3. J Biol Chem. 1971;246:1192–1200. [PubMed] [Google Scholar]
- Stellner K, Saito H, Hakomori SI. Determination of aminosugar linkages in glycolipids by methylation. Aminosugar linkages of ceramide pentasaccharides of rabbit erythrocytes and of Forssman antigen. Arch Biochem Biophys. 1973;155:464–472. doi: 10.1016/0003-9861(73)90138-0. [DOI] [PubMed] [Google Scholar]