

Abstract
Integrin-mediated cell adhesion to extracellular matrix proteins is known to promote cell survival, whereas detachment from the matrix can cause rapid apoptotic death in some cell types. Contrary to this paradigm, we show that fibroblast adhesion to the angiogenic matrix protein CCN1 (CYR61) induces apoptosis, whereas endothelial cell adhesion to CCN1 promotes cell survival. CCN1 induces fibroblast apoptosis through its adhesion receptors, integrin α6β1 and the heparan sulfate proteoglycan (HSPG) syndecan-4, triggering the transcription-independent p53 activation of Bax to render cytochrome c release and activation of caspase-9 and -3. Neither caspase-8 activity nor de novo transcription or translation is required for this process. These results show that cellular interaction with a specific matrix protein can either induce or suppress apoptosis in a cell type–specific manner and that integrin α6β1-HSPGs can function as receptors to induce p53-dependent apoptosis.
Introduction
Cellular interaction with the ECM, occurring largely through integrin receptors, is a principal regulator of cell survival. Several abundant ECM proteins are known to induce potent prosurvival signals in specific cell types, although others are neutral to cell survival (Ilic et al., 1998). Many types of normal cells are anchorage dependent; when detached from the ECM these cells undergo anoikis, a form of apoptotic death (Frisch and Screaton, 2001; Grossmann, 2002). This apoptotic mechanism plays a critical role in controlling homeostasis and the integrity of tissue architecture. Malignant tumor cells are often resistant to anoikis, thus promoting metastasis by allowing these cells to migrate through and proliferate in inappropriate environments (Igney and Krammer, 2002). During embryogenesis and tissue remodeling, however, many normal cells undergo programmed cell death under the control of apoptotic factors without detachment from the ECM (Rich et al., 1999; Baehrecke, 2002). In these instances, dynamic changes in the matrix environment may provide conditions that are permissive of, or conducive to, apoptotic processes. However, no specific ECM protein has been shown to directly trigger cell death through its cell-adhesive function to date.
The principal receptors for ECM molecules are integrins, which connect the ECM to the cytoskeleton and transduce signals induced by their extracellular ligands (Hynes, 2002; Miranti and Brugge, 2002). Engagement of several specific integrins has been shown to activate strong prosurvival signaling pathways; among these pathways FAK plays a critical role (Ilic et al., 1998; Parsons, 2003). In addition to FAK, some integrins also recruit Shc and integrin-linked kinase, and these three signaling molecules mediate ECM-dependent survival signals through activation of the phosphatidylinositol 3-kinase/Akt, c-Jun NH2-terminal kinase (JNK), and Erk pathways (Almeida et al., 2000; Frisch and Screaton, 2001). Detachment of cells from the ECM blocks these prosurvival pathways and induces cytoskeletal changes, resulting in the release and activation of proapoptotic molecules. Whereas cell death by anoikis is often associated with the activation of caspase-8 or p53 (Frisch and Screaton, 2001; Grossmann, 2002), unligated αvβ3 or β1 integrins, even in adherent cells, can also induce apoptosis by the direct recruitment and activation of caspase-8 (Stupack et al., 2001). In addition, unligated integrin α5β1 can trigger the release of the mitochondrial protein Bit1 into the cytoplasm, thereby activating a caspase-independent mechanism of cell death (Jan et al., 2004). Thus, integrin-mediated cell adhesion events play critical roles in the control of apoptosis.
CCN1 (CYR61) is a secreted matrix-associated heparin-binding protein that contains structural domains common to ECM proteins, including the von Willebrand factor type C repeat, the thrombospondin type 1 repeat, and the COOH terminus of muscins (Lau and Lam, 1999). Encoded by a growth factor–inducible immediate early gene, CCN1 regulates a broad spectrum of cellular activities, including cell adhesion, migration, proliferation, survival, and differentiation. Mechanistically, CCN1 acts through direct binding to at least five distinct integrins, which mediate CCN1 functions in a cell type– and context-dependent manner (Lau and Lam, 2005). For example, CCN1 promotes proangiogenic activities in activated endothelial cells through integrin αvβ3 (Leu et al., 2002) and supports fibroblast and smooth muscle cell adhesion through integrin α6β1 with heparan sulfate proteoglycans (HSPGs) as coreceptors (Chen et al., 2000; Grzeszkiewicz et al., 2002). Adhesion of primary human fibroblasts to immobilized CCN1 induces adhesive signaling, including the formation of filopodia and lamellipodia and activation of FAK, paxillin, Rac, and Erk1/2, culminating in the regulation of genes that control angiogenesis, inflammation, and matrix remodeling (Chen et al., 2001a,b). Consistent with these activities, Ccn1 expression in adulthood is associated with biological and pathological contexts in which angiogenesis and inflammation play important roles, such as wound healing, restenosis, atherosclerosis, and tumorigenesis (Grzeszkiewicz et al., 2002; for review see Menendez et al., 2003). Furthermore, CCN1 induces angiogenesis both in vitro and in vivo and is essential for successful vascular development, as evidenced by the embryonic lethality of Ccn1-null mice resulting from placental vascular insufficiency and loss of embryonic vessel integrity (Babic et al., 1998; Mo et al., 2002).
CCN1 protects activated endothelial cells from apoptosis by ligation to integrin αvβ3 and promotes survival in MCF7 breast cancer cells by up-regulation of X-linked inhibitor of apoptosis (Leu et al., 2002; Lin et al., 2004). However, Ccn1 expression has been associated with cell death in the hippocampal progenitor cell line H19 and in endometrial cancer cells (Kim et al., 2003; Chien et al., 2004), suggesting that CCN1 may regulate cell survival differently in distinct cell types. We show that CCN1 can promote the survival of activated endothelial cells but induces apoptosis in fibroblasts. Paradoxically, CCN1 induces fibroblast apoptosis as an adhesion substrate through its adhesion receptors, integrin α6β1 and the HSPG syndecan-4, despite activation of the prosurvival protein FAK. Ligation of CCN1 to the adhesion receptors in fibroblasts, neither of which has previously been implicated in apoptosis, results in the transcription-independent p53 activation of Bax and cytochrome c release, triggering the activation of caspase-9 and -3. These results show that cell adhesion to a specific matrix protein can either induce or suppress apoptosis in a cell type–dependent manner and that integrin α6β1 and syndecan-4 can function as apoptotic receptors to induce p53-dependent cell death.
Results
CCN1 induces apoptosis in fibroblasts as a cell adhesion substrate
To investigate whether CCN1 regulates cell survival in fibroblasts, Rat1a cells were adhered to surfaces coated with CCN1 or other ECM proteins and maintained in low serum for 24 h. Cells were subjected to the TUNEL assay to monitor apoptosis and counterstained with DAPI, which also marks apoptotic cells by their intensely staining condensed nuclei (Kennedy et al., 1997). As expected, cells adherent to fibronectin (FN), laminin (LN), or vitronectin (VN) were not apoptotic, even under serum deprivation (Fig. 1, A and B). Surprisingly, ∼30% of the cells adhered to CCN1 suffered apoptotic death. By contrast, cells did not succumb when attached to poly-l-lysine (PLL), indicating that a lack of ECM-dependent signals, per se, was not sufficient to induce apoptosis under these experimental conditions. Rather, CCN1 was actively inducing apoptosis. TUNEL-positive cells matched those with intense DAPI staining precisely, indicating that the same cells were scored as apoptotic using either assay. FACS analysis showed that the fraction of cells with sub-G1 DNA content increased from 9.5 to 31.4% after CCN1 treatment, further demonstrating that CCN1 triggered DNA degradation, a hallmark of apoptosis (unpublished data).
Figure 1.

CCN1 induces fibroblast apoptosis as an adhesion substrate. (A) Rat1a fibroblasts were adhered to glass coverslips coated with 10 μg/ml FN, 2.5 μg/ml VN, 50 μg/ml CCN1, 10 μg/ml LN, or 20 μg/ml PLL and cultured in medium containing 0.5% FBS for 24 h. After fixation, cells were subjected to TUNEL assay and counterstained with DAPI. Bar, 10 μm. (B) Rat1a cells were adhered to dishes coated with 20 μg/ml PLL, 2 μg/ml FN, 10 μg/ml LN, 0.4 μg/ml VN, or 10 μg/ml CCN1 and maintained in medium containing 0.5% FBS for 24 h. After fixation and staining with DAPI, cells were scored for apoptosis. (C) To test the effect of CCN1 as an adhesion substrate, HUVECs, HSF, or Rat1a cells were adhered to culture wells coated with 10 μg/ml CCN1 or 10 μg/ml LN as indicated and maintained for 24 h before being scored for apoptosis. To test the effect of CCN1 as a soluble factor, cells were adhered to tissue culture dishes overnight, washed, and incubated in serum-free medium with or without added soluble 10 μg/ml CCN1 for 24 h before being scored for apoptosis. (D) Rat1a cells were adhered on dishes coated with various ECM proteins as indicated and incubated further for 24 h with or without added 10 μg/ml CCN1 before being scored for apoptosis. (E) Rat1a cells were adhered to dishes coated with 10 μg/ml CCN1, 2 μg/ml FN, or 20 μg/ml PLL for 20 min. Cell lysates were prepared and resolved on 7.5% SDS-PAGE, followed by immunoblotting with antibodies against FAK, phospho-FAK Y397, or phospho-FAK Y576/577. (F) Rat1a cells were plated on coverslips coated with FN or CCN1 as in A and stained with antibodies against phospho-FAK Y397, phospho-paxillin Y118, or control IgG 20 min after plating. Arrowheads point to staining in focal complexes. Cells were counterstained with DAPI. Bar, 10 μm. (G) Cells were adhered to glass coverslips coated with FN or CCN1 as in A, and stained with antibodies against phospho-JNK T183/Y185 or control IgG 10 min after plating. Cells were counter stained with DAPI. Arrowheads point to phosphorylated JNK in focal complexes. Bar, 10 μm. Error bars represent ± SD from experiments done in triplicate.
CCN1 is known to protect activated endothelial cells from apoptosis (Leu et al., 2002), prompting us to compare its effects on different cell types. Because CCN1 also functions as a soluble protein (Babic et al., 1998), we assessed its activities as an immobilized adhesion substrate and as a soluble factor. Activated human umbilical vein endothelial cells (HUVECs), primary normal human skin fibroblasts (HSFs), and Rat1a cells were adhered to dishes coated with either CCN1 or LN and subjected to serum deprivation (Fig. 1 C). Alternatively, cells were adhered on tissue culture dishes overnight and allowed to secrete their own matrix before the addition of soluble CCN1. HUVECs were protected from apoptosis upon serum withdrawal in the presence of CCN1 but not LN, either as adhesion substrates or as soluble factors. By contrast, CCN1 induces apoptosis in primary HSFs and Rat1a cells, both as an adhesion substrate and in a soluble form. The induction of cell death in primary fibroblasts is remarkable because these cells are relatively resistant to apoptosis induced by many conditions, including serum withdrawal. Because the p21-deficient Rat1a fibroblasts are more prone to cell death (Kennedy et al., 1997) and showed an enhanced response to CCN1, we have used Rat1a fibroblasts for most of our subsequent experiments. However, key observations have been confirmed in primary HSFs as well (see Fig. 4 B and Fig. 6 D).
Figure 4.

Induction of fibroblast apoptosis by CCN1–integrin interaction. (A) Effects of integrin-binding defective CCN1 mutants in apoptosis in Rat1a fibroblasts. Cells adhered to 6-well tissue culture plates were either left untreated or treated with 10 μg/ml of soluble wild-type CCN1; 10 μg/ml of the mutants SM, DM, or TM; or 10 μg/ml D125A for 24 h, and apoptosis was assayed. (B) Integrin requirements of CCN1-induced apoptosis in HSF. Cells adhered to 6-well plates were either left untreated or pretreated with 50 μg/ml of antibodies against integrin α6 (GoH3), β1 (P5D2), α5 (P1D6), αvβ3 (LM609), or control IgG for 1 h. 10 μg/ml of soluble CCN1 was added where indicated and apoptosis was assayed 24 h thereafter. Error bars represent ± SD from experiments done in triplicate.
Figure 6.

CCN1 induces p53-dependent Bax activation. (A) Rat1a cells were transfected with either the pBabePuro vector or the same vector expressing GSE56. Cells were incubated with or without 10 μg/ml CCN1 for 6 h and immunostained and scored for activated Bax. (B) Cells were transfected with either the pBabePuro vector or the same vector expressing GSE56, or were pretreated with 200 μM of cyclic pifithrin for 1 h. Cells were incubated with or without 10 μg/ml CCN1 for 24 h and scored for apoptosis. (C) Primary human fibroblasts were either untreated or pretreated with 200 mM of cyclic pifithrin for 1 h and incubated with or without 10 mg/ml CCN1 for 24 h, as indicated, before scoring for apoptosis. (D) Parental 10.1 cells and cells transfected with pMV7 containing either temperature-sensitive p53 (ts-p53)or transcription transactivation inactive temperature-sensitive p53 (ts-p53 22–23) were transferred from nonpermissive (39°C) to permissive temperature (33°C) for 24 h to regain functional p53 protein. Cells were incubated with medium containing 0.5% FBS and treated with 10 mg/ml CCN1 for the next 6 h. Cells were fixed and scored for apoptosis. Error bars represent ± SD from experiments done in triplicate.
Acting as adhesion substrates, various ECM molecules display cell type–specific effects on cell survival. Cell adhesion to FN, a ligand of integrin α5β1, is known to induce potent prosurvival signals in endothelial cells and fibroblasts, whereas adhesion to other proteins such as collagen or LN appears neutral to cell survival in fibroblasts (Ilic et al., 1998). Thus, we tested the activity of CCN1 in cells adhered to specific ECM molecules. Fibroblasts adherent to FN, VN, or LN were susceptible to CCN1 cytotoxicity (Fig. 1 D), indicating that the apoptotic activity of CCN1 can override any prosurvival signals resulting from cell adhesion to these ECM proteins.
Fibroblast adhesion to CCN1 is mediated through integrin α6β1-HSPGs, resulting in α6β1-containing focal adhesion complexes and the activation of FAK, paxillin, Rac, and Erk1/2 (Chen et al., 2001a). Like primary HSFs, Rat1a cells also adhere to and spread on CCN1, leading to adhesive signaling including tyrosyl phosphorylation of FAK (Fig. 1, A and E). Specifically, FAK was phosphorylated at Y397, a site known to be autophosphorylated upon integrin signaling and that serves as a docking site for phosphatidylinositol 3-kinase, as well as at Y576 and Y577, which are sites that enhance FAK kinase activity when phosphorylated (Parsons, 2003). Furthermore, similar to cells adhered to FN, virtually 100% of cells adhered to CCN1 had phosphorylated FAK, leading to the phosphorylation of paxillin at Y118, a specific substrate of FAK (Schaller and Parsons, 1995; Fig. 1 F). FAK can also activate JNK, and phosphorylated JNK is localized in focal adhesions of fibroblasts cultured on prosurvival matrix (Almeida et al., 2000). We found that fibroblasts adhered to both FN and CCN1 showed the same pattern of rapid and transient phosphorylation of JNK, peaking between 5 and 15 min after adhesion (unpublished data). In fibroblasts adhered to both FN and CCN1, phosphorylated JNK was localized to the focal complexes (Fig. 1 G). These results show that Rat1a cell adhesion to CCN1 induces signaling through FAK, even though apoptosis ensues under these conditions. Thus, the phosphorylation of FAK, either by FN or CCN1, is not sufficient to circumvent CCN1-induced apoptosis.
Induction of apoptosis by CCN1 is dose dependent, observable at 1.0 μg/ml (25 nM) CCN1, and maximal at 20 μg/ml when ∼90% of cells were apoptotic (Fig. 2 A). This active concentration range is consistent with that of other integrin-mediated CCN1 activities (Lau and Lam, 2005). Neither cycloheximide nor 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB) was able to block CCN1-induced apoptosis, indicating that this process does not require de novo translation or transcription (Fig. 2 B). The inclusion of 2% serum in the culture medium, which is sufficient to sustain cell proliferation for Rat1a cells (Conzen et al., 2000), did not eliminate CCN1-induced cell death (Fig. 2 C). Moreover, the addition of 100 ng/ml EGF or 10 ng/ml of basic FGF failed to confer protection from CCN1 cytotoxicity (Fig. 2 C). Therefore, CCN1 can actively induce cell death even in the presence of mitogenic serum growth factors.
Figure 2.

Apoptotic activities of CCN proteins in Rat1a fibroblasts. (A) Cells were grown in 6-well plates and treated with the indicated concentrations of soluble CCN1 for 24 h, followed by fixation and scoring for apoptosis. (B) Cells were pretreated for 1 h with 25 μM cycloheximide and 40 μM DRB before further incubation for 6 h with or without 10 μg/ml CCN1. Cells were fixed and scored for apoptosis. (C) Cells were grown in tissue culture dishes in 10% serum, washed, and maintained in medium with 0% FBS, 2% FBS, 100 ng/ml EGF, or 10 ng/ml of basic FGF, in the presence or absence of 10 mg/ml CCN1 for 24 h before scoring for apoptosis. (D) Cells were adhered to tissue culture dishes or dishes coated with CCN1, CCN2, or PLL (10 mg/ml each) and maintained in medium containing 0.5% FBS with or without soluble CCN1 or CCN2 for 24 h before apoptosis assay. Error bars represent ± SD from experiments done in triplicate.
The CCN family of proteins includes six homologous members (Lau and Lam, 1999). Both CCN1 and CCN2 (connective tissue growth factor) are encoded by growth factor–inducible immediate early genes, induce angiogenesis in vitro and in vivo, and have similar activities in several cell types (Lau and Lam, 2005). CCN2 also supports endothelial cell adhesion through αvβ3, protects the cells from apoptosis, and induces adhesive signaling in fibroblasts similar to CCN1 (Babic et al., 1999; Chen et al., 2001a). We found that CCN2 also induces cell death, both as an adhesion substrate in Rat1a fibroblasts (Fig. 2 D) and when added as a soluble factor (unpublished data). Thus, both CCN1 and CCN2 are able to promote endothelial cell survival while inducing apoptosis in fibroblasts.
Apoptotic activity of CCN1 is mediated through integrin α6β1 and syndecan-4
Because CCN1 induces apoptosis as an adhesion substrate, we investigated the role of its adhesion receptors, integrin α6β1 and HSPGs (Chen et al., 2000), although neither has been previously implicated in apoptosis. The presence of soluble heparin in the culture medium blocked CCN1-induced apoptosis completely (Fig. 3 A), suggesting that soluble heparin may saturate the heparin binding sites of CCN1 and prevent it from interacting with cell surface HSPGs. Consistent with this interpretation, treatment of fibroblasts with NaClO3, which inhibits 3-phosphoadenosine 5′-phosphosulfate synthesis and blocks sulfation of proteoglycans, abrogated CCN1-induced apoptosis (Fig. 3 A). The inhibitory effect of NaClO3 was reversed by the inclusion in the culture medium of 10 mM Na2SO4, which overrides the sulfation block exerted by NaClO3 (Rapraeger et al., 1991), thus confirming that the inhibitory effect of NaClO3 was attributable to impaired sulfation of HSPGs. Among the HSPGs expressed in fibroblasts, syndecan-4 is uniquely colocalized with integrins in focal adhesions, where it activates PKC in support of cell adhesion and spreading (Couchman et al., 2001; Simons and Horowitz, 2001). We found that syndecan-4, but not other syndecans, is localized to focal adhesion complexes in fibroblasts adhered to CCN1 (unpublished data), suggesting that it might act as an HSPG coreceptor with α6β1. Preincubation of fibroblasts with anti–syndecan-4 antibodies completely abolished CCN1-induced apoptosis, whereas control IgG had no effect (Fig. 3 B). These results support the involvement of a cell surface HSPG, and implicate syndecan-4 as a coreceptor that plays a critical role in CCN1-induced apoptosis.
Figure 3.

CCN1 induces apoptosis through integrin α6β1 and HSPGs. (A) Cells were pretreated with 1 mg/ml heparin for 1 h in serum-free medium or with 20 mM Na2SO4 and/or 100 mM NaClO3 for 24 h in media containing 10% FBS, after which cells were washed and subjected to further incubation with or without 10 μg/ml CCN1 in serum-free medium containing the pretreatment level of Na2SO4 and/or NaClO3. (B) Cells were pretreated with 100 μg/ml of control rabbit IgG or 100 μg/ml anti–syndecan-4 antibody for 1 h in serum-free medium before incubation with or without CCN1. (C) Cells were pretreated with the peptides T1 (4 mM), T1-mut (4 mM), H2 (5 mM), or T4 (5 mM) for 1 h before further incubation with or without 10 mg/ml CCN1. (D) Cells were pretreated with 40 μg/ml GoH3, an mAb against integrin α6, or 40 μg/ml of control mouse IgG for 1 h before incubation with or without CCN1. (E) Cells were pretreated for 1 h with GRGDSP and GRGESP peptides (0.2 mM) before further incubation with or without CCN1. Error bars represent ± SD from experiments done in triplicate.
To test the possibility that integrin α6β1 may also be involved in CCN1-induced apoptosis, we took advantage of two recently described CCN1 peptides, T1 and H2, which contain α6β1-binding sites and are able to block α6β1-mediated CCN1 functions (Leu et al., 2003, 2004). Whereas the addition of synthetic T1 or H2 peptide alone to the culture medium had no effect on cell survival, either peptide was able to abrogate CCN1-induced apoptosis (Fig. 3 C). The control peptides T1-mut, a mutated T1 peptide with a two-residue substitution that rendered it unable to bind α6β1 (Leu et al., 2003), and T4, a CCN1 peptide with irrelevant sequence, had no effect. These results indicate that CCN1-induced apoptosis requires its binding to α6β1, for which the T1 and H2 peptides act as competitive inhibitors. Furthermore, pretreatment of cells with an anti–α6 integrin monoclonal antibody (GoH3) completely annihilated the apoptotic activity of CCN1, whereas control IgG had no effect (Fig. 3 D). These results show that α6β1, in addition to syndecan-4, is required for mediating CCN1-induced apoptosis.
Aside from interaction with the α6β1-HSPG coreceptors, CCN1 induces fibroblast migration and enhances DNA synthesis through αvβ5 and αvβ3, respectively (Grzeszkiewicz et al., 2001). To test the role of αv integrins, cells were treated with a peptide containing the canonical αv integrin–binding sequence RGD, which did not protect Rat1a cells from CCN1-induced apoptosis (Fig. 3 E). The GRGDSP peptide induced apoptosis on its own, whereas the control peptide GRGESP had no effect. This apoptotic effect is expected because RGD-containing peptides can activate caspase-3 directly (Buckley et al., 1999). However, the apoptotic activities of GRGDSP peptide and CCN1 were additive, indicating that they work through largely nonoverlapping pathways (Fig. 3 E).
The aforementioned findings indicate the requirement for α6β1-HSPGs, but not αv-containing integrins, in CCN1-induced apoptosis. To further substantiate these findings, we evaluated the importance of direct interaction between CCN1 and these receptors using CCN1 mutants that are defective in binding αvβ3 or α6β1-HSPGs specifically. Biochemical and functional studies identified three sites involved in binding α6β1 and HSPGs in CCN1, namely T1, H1, and H2 (Leu et al., 2003, 2004), whereas the mutation D125A disrupts an αv integrin binding site, V2 (Chen et al., 2004; Leu et al., 2004). The full-length CCN1 mutant SM, which disrupts T1 alone, had relatively minor effects, whereas the mutant DM, which alters both H1 and H2, severely damaged α6β1-HSPG–mediated CCN1 activities. Disruption of all three sites in the mutant TM completely abolished α6β1-HSPG–mediated functions (Leu et al., 2004). Consistent with these findings, the mutants DM and TM were completely defective for induction of apoptosis, whereas SM showed only modest impairment of apoptotic activity (Fig. 4 A). Notably, all three mutants have intact αvβ3 binding sites and are fully active in αvβ3-mediated functions (Leu et al., 2004), indicating that interaction with αvβ3 alone does not induce apoptosis. Furthermore, the mutant D125A, which disrupts binding to αvβ3 and impairs αvβ3-dependent CCN1 activities (Chen et al., 2004), was able to induce apoptosis similar to wild type (Fig. 4 A). Thus, binding to αvβ3 is not critical to the induction of Rat1a cell apoptosis by CCN1. To determine the receptor requirement for CCN1-induced apoptosis in HSFs, we examined the inhibitory effects of monoclonal antibodies that are available against the human integrins. Monoclonal antibodies against integrins α6 (GoH3) and β1 (P5D2) strongly inhibited CCN1-induced apoptosis, whereas antibodies against integrin α5 (P1D6) or αvβ3 (LM609) had no effect (Fig. 4 B). Thus, CCN1-induced apoptosis is also dependent on integrin α6β1, but not αvβ3, in HSFs.
CCN1 induces apoptosis through the intrinsic mitochondrial pathway
The induction of cell death through the ligation of an ECM molecule to α6β1-HSPGs establishes a novel signaling pathway for apoptosis. To define the intracellular events involved, we first analyzed the role of caspases. Two principal apoptotic pathways involving caspases have been characterized: the extrinsic pathway, which is induced by direct ligand–receptor interaction, resulting in the recruitment and activation of caspase-8; and the intrinsic pathway, which is induced by various stress stimuli that activate proapoptotic molecules targeting the mitochondria, rendering the release of cytochrome c and activation of caspase-9 and -3 (Li et al., 1997). Cell-permeable, irreversible inhibitors of caspase-9 (z-LEHD-fmk) and -3 (z-DEVD-fmk), and of all caspases (z-VAD-fmk), effectively abrogated CCN1-induced apoptosis (Fig. 5 A). By contrast, caspase-8 inhibitor (z-IETD-fmk) had no effect, whereas the same concentration of this inhibitor completely blocked FasL-induced apoptosis in HSFs (unpublished data). Immunoblot analysis showed that both caspase-9 and -3 were cleaved into their active forms upon CCN1 treatment (Fig. 5 B). These results suggest that CCN1 induces apoptosis via the intrinsic pathway, in which disruption of mitochondrial integrity leads to cytochrome c release from the mitochondria to form a complex with Apaf and procaspase-9 to assemble the apoptosome (Nicholson and Thornberry, 2003). This process activates caspase-9, which in turn cleaves and activates caspase-3. To test this possibility, the subcellular localization of cytochrome c was detected by indirect immunofluorescence labeling using anti–cytochrome c antibodies. Whereas cytochrome c staining largely followed the mitochondria, the staining became diffuse in CCN1-treated apoptotic cells, indicating cytochrome c release (Fig. 5 C).
Figure 5.
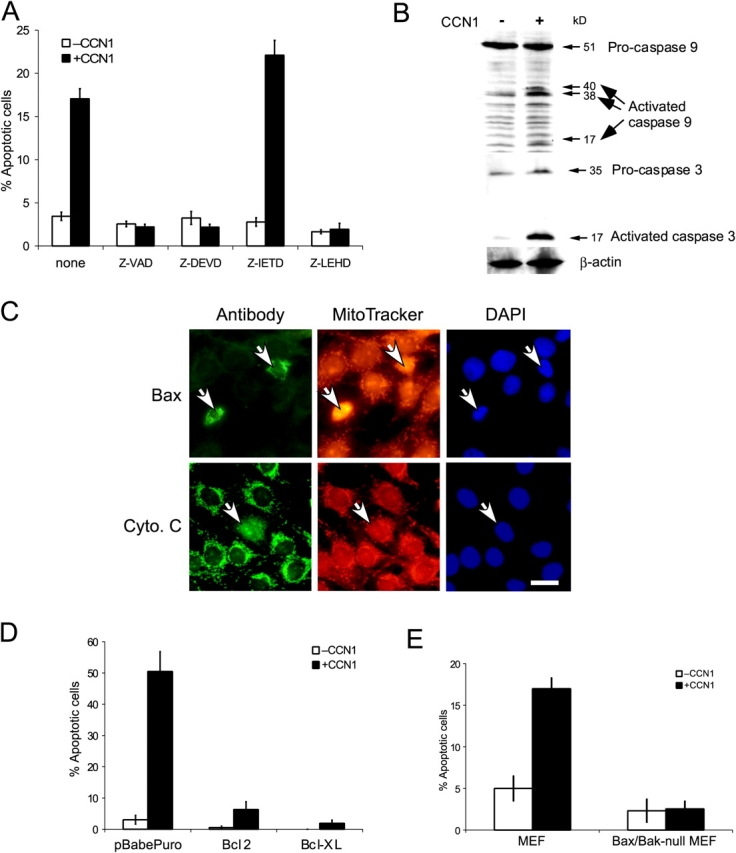
CCN1 activates the intrinsic apoptotic pathway. (A) Rat1a cells were pretreated for 1 h with 50 μM Z-VAD (pan-caspase), Z-DEVD (caspase-3), Z-IETD (caspase-8), and Z-LEHD (caspase-9) inhibitors. Cells were treated with 10 μg/ml CCN1 for 6 h, fixed, and scored for apoptosis. (B) Cells cultured as in A were treated with CCN1 for 6 h and lysed in Laemmli buffer. Proteins were resolved on 12% SDS-PAGE, followed by immunoblotting with antibodies against caspase-3, caspase-9, or β-actin. (C) Rat1a cells were adhered on glass coverslips overnight, washed, and maintained in serum-free medium. Cells were treated with 10 μg/ml CCN1 for 6 h, stained with MitoTracker for 5 min, fixed, immunostained for either Bax or cytochrome c, and counterstained with DAPI. Arrows point to apoptotic cells. Bar, 10 μm. (D) Fibroblasts transfected with pBabePuro empty vector or the same vector driving the expression of Bcl-2 or Bcl-XL were incubated with or without 10 μg/ml CCN1 for 24 h, fixed, and scored for apoptosis. (E) Immortalized wild-type or Bax/Bak-null MEFs were incubated with or without 10 μg/ml CCN1 for 6 h, fixed, and scored for apoptosis. Error bars represent ± SD from experiments done in triplicate.
The balance of pro- and antiapoptotic Bcl family members regulates mitochondrial integrity and activation of the apoptotic cascade (Cory and Adams, 2002). Transfection of Rat1a cells with expression vectors for Bcl-2 or Bcl-XL abrogated CCN1-induced apoptosis, whereas transfection of the empty vector had no effect (Fig. 5 D). These results suggest the regulation of CCN1-induced apoptosis by proapoptotic Bcl family members, among which the Bax/Bak subfamily plays prominent roles. Upon activation, both proteins can homooligomerize and localize to the outer mitochondrial membrane to facilitate cytochrome c release (Cory and Adams, 2002). Because Bax can act downstream of integrins (Gilmore et al., 2000), we examined Bax activation using antibodies specific for the oligomer form of Bax. Consistent with its involvement in CCN1-induced apoptosis, we found that Bax oligomerized and colocalized with the mitochondria in apoptotic cells (Fig. 5 C). Furthermore, Bax/Bak double-null mouse embryonic fibroblasts (MEFs; Wei et al., 2001), but not the corresponding wild-type MEFs, were resistant to CCN1-induced apoptosis (Fig. 5 E). Together, these results show that Bax is activated upon CCN1 treatment and Bax/Bak are indispensable for CCN1-induced apoptosis in fibroblasts.
CCN1-induced apoptosis requires p53-dependent Bax activation
p53 is known to induce apoptosis via Bax and Bak, either through up-regulation of their expression or through protein–protein interaction to trigger their oligomerization and mitochondrial localization (Haupt et al., 2003). To investigate the potential role of p53 in CCN1-induced apoptosis, we tested the effects of the genetic suppressor element GSE56, which has been widely used to inhibit p53 function (Ossovskaya et al., 1996). Expression of GSE56 completely abolished activation of Bax upon CCN1 treatment (Fig. 6 A). Furthermore, either expression of GSE56 or treatment of cells with the p53 inhibitor cyclic pifithrin (Pietrancosta et al., 2005) completely abolished CCN1-induced apoptosis in Rat1a cells (Fig. 6 B). Likewise, cyclic pifithrin also blocked CCN1-induced apoptosis in HSFs (Fig. 6 C). Thus, CCN1-induced apoptosis requires p53 function, which mediates the activation of Bax.
To establish the role of p53 further, we tested the responsiveness of p53-deficient cells. p53-null 10.1 mouse fibroblasts (Livingstone et al., 1992) were left untreated or were infected with retroviruses driving the expression of a temperature-sensitive p53 (ts-p53; Wagner et al., 1994) or of the temperature-sensitive, transcription transactivation–defective mutant ts-p53 22–23 (Lin et al., 1994). Stable cell populations were selected and propagated at the nonpermissive temperature (39°C) because prolonged exposure to the permissive temperature (33°C) for p53 leads to p21 induction and cell cycle arrest (Buschmann et al., 2001). After propagation, cells were shifted to 33°C and subjected to CCN1 treatment in low serum medium. The parental p53-null 10.1 cell line was completely nonresponsive to CCN1-induced apoptosis, whereas 10.1 cells expressing ts-p53 or ts-p53 22–23 were highly sensitive to CCN1 exposure, showing 20–25% cell death (Fig. 6 D). These results clearly show that CCN1-induced apoptosis requires p53 but not its transcription transactivation activity, which is consistent with this apoptotic process being independent of de novo transcription and translation (Fig. 2 B).
Discussion
Numerous studies have demonstrated that integrin-mediated interaction with the ECM can induce prosurvival signals, whereas detachment from matrix proteins results in rapid apoptotic death in many cell types (Frisch and Screaton, 2001; Grossmann, 2002). As cell adhesion substrates, specific ECM proteins are known to promote, or are indifferent to, cell survival with cell type specificity, and none has been shown to induce apoptosis to date (Ilic et al., 1998). In this study we show that, unexpectedly, cell adhesion to the matrix protein CCN1 induces apoptosis in fibroblasts but promotes survival in activated endothelial cells. Thus, a new category of cell adhesion events that induce apoptosis is beginning to emerge, suggesting that cellular interaction with the ECM can help to program both cell survival and death in a cell type–specific manner. Furthermore, CCN1-induced apoptosis in fibroblasts is p53 dependent and is mediated through its cell adhesion receptors, α6β1 and the HSPG syndecan-4, thus linking these receptors to apoptotic pathways for the first time.
Most of the activities of CCN proteins identified to date in isolated cell systems can be attributed to their direct interaction with integrin receptors, which function with HSPGs as coreceptors in some contexts (Lau and Lam, 2005). It is well documented that CCN1 supports fibroblast adhesion through integrin α6β1 and HSPGs, and the CCN1 binding sites for these receptors have been identified (Leu et al., 2003, 2004). Remarkably, these adhesion receptors also mediate CCN1-induced fibroblast apoptosis (Figs. 3 and 4). Analyses of CCN1 mutants indicate that the CCN1 binding sites H1 and H2, which interact with both α6β1 and HSPGs, are essential for apoptotic activity. By contrast, the T1 site, which binds α6β1, or the V2 site, which binds integrin αvβ3, is not essential (Fig. 4). These results suggest that concomitant binding of CCN1 to both α6β1 and HSPGs, possibly through closely juxtaposed binding sites, may be necessary for induction of apoptosis. Such a requirement might explain why other α6β1 ligands, such as LN, do not induce apoptosis (Fig. 1). Furthermore, our studies suggest syndecan-4 as the HSPG that acts with α6β1 to induce apoptosis (Fig. 3 B), thus implicating α6β1 and syndecan-4 as apoptotic receptors.
Cell type–specific differences have been observed regarding the regulation of apoptosis by cell adhesion events. Epithelial and endothelial cells undergo rapid cell death by anoikis when detached from the ECM, whereas fibroblasts are resistant to such a challenge for a prolonged period of time (Meredith et al., 1993). The differential effects of CCN1 on endothelial cells as opposed to fibroblastic cells can be ascribed to distinct integrin utilization in these cell types. CCN1 confers proangiogenic and antiapoptotic functions in activated endothelial cells through ligation to integrin αvβ3, whose ability to transduce cell survival signals is well documented (Eliceiri and Cheresh, 2000; Leu et al., 2002). Furthermore, vascular cells in Ccn1-null mice suffer a high level of apoptosis, showing that CCN1 is critical for sustaining vascular cell survival in vivo (Mo et al., 2002). By contrast, CCN1 induces apoptosis in fibroblasts through its adhesion receptors, α6β1 and syndecan-4, thereby selectively promoting or suppressing apoptosis in different cell types through the engagement of distinct integrins.
Fibroblast adhesion to CCN1 induces adhesive signaling including the activation of FAK (Fig. 1 E), which is generally associated with a prosurvival outcome (Frisch and Screaton, 2001). Although ECM molecules such as FN are highly efficient in promoting the survival of fibroblasts through activation of FAK (Ilic et al., 1998), cells adhered to FN or to their endogenous matrices are nevertheless susceptible to CCN1-induced apoptosis (Fig. 1). The presence of 2% serum or mitogenic growth factors (Fig. 2 C) also did not prevent CCN1-induced apoptosis. Thus, CCN1 can induce cell death despite the prosurvival signals conferred by either matrix molecules or growth factors and must activate signaling pathways that override cell adhesion–dependent prosurvival signals. These findings suggest that CCN1 can induce apoptosis under physiological conditions, such as during wound healing (Chen et al., 2001b), where cells may be attached to prosurvival ECM proteins and exposed to growth factors and cytokines.
The loss of proper integrin–ligand interactions can lead to apoptosis. Anoikis, or apoptosis resulting from detachment from the ECM, has been associated with activation of caspase-8 or p53 (Frisch and Screaton, 2001; Grossmann, 2002), although a caspase-independent mechanism of anoikis also occurs (Jan et al., 2004). In adherent cells, unligated integrins can also recruit and activate caspase-8, leading to “integrin-mediated death” (Stupack et al., 2001). CCN1-induced apoptosis is independent of caspase-8 and is instead mediated through the mitochondrial pathway (Fig. 5). In this context, p53 is required for the activation of Bax, which in turn leads to cytochrome c release and activation of caspase-9 and -3 (Figs. 5 and 6). p53 is known to mediate apoptosis through both transcription-dependent and -independent mechanisms (Haupt et al., 2003; Slee et al., 2004). As a transcription factor, p53 activates proapoptotic genes encoding Bax, the BH3-only proteins PUMA and Noxa, AIP-1, Apaf-1, and PERP. It can also repress antiapoptotic genes encoding Bcl2 and immunosuppressive acidic proteins. Because preincubation of cells with cycloheximide and DRB did not block CCN1-induced apoptosis (Fig. 2 B), and transactivation-defective p53 was able to restore CCN1-induced apoptosis in p53-null cells, we conclude that p53 acts through a transcription-independent mechanism in this context. Our results showing the transcription-independent, p53-mediated activation of Bax in CCN1-mediated apoptosis (Fig. 6) are consistent with recent findings that direct interaction of p53 with Bax/Bak, or with Bcl family members to displace BH3-only proteins, can lead to Bax activation and apoptosis independent of de novo protein synthesis (Mihara et al., 2003; Chipuk et al., 2004).
Both CCN1 and its homologous family member CCN2 are angiogenic in vitro and in vivo, support cell adhesion and promote survival in endothelial cells, and induce adhesion signaling in HSFs in a similar manner (Chen et al., 2001a; Lau and Lam, 2005). Like CCN1, CCN2 also induces fibroblast apoptosis, suggesting that the control of cell survival and death may be a general function of CCN proteins (Fig. 2 D). The finding that CCN1 and CCN2 are novel proapoptotic matrix molecules has implications on their biological roles in vivo. CCN1 and CCN2 are both encoded by growth factor–inducible immediate early genes, and their expression in the adult is associated with biological and pathological events that involve apoptosis, such as in wound healing, arthritis, and tumorigenesis (Lau and Lam, 1999; for review see Menendez et al., 2003; Planque and Perbal, 2003). It is tempting to speculate that their dynamic expression in the ECM may regulate cell survival and death during tissue remodeling, including the resolution of granulation tissue during wound healing.
Integrin-mediated signaling resulting from engagement of ECM proteins is known to regulate diverse biological processes, and its role in promoting cell survival is well established (Pullan et al., 1996; Colognato et al., 2002). It has been observed that in human mammary epithelial cells where p53 function was suppressed, exposure to reconstituted ECM can induce growth arrest followed by apoptosis in an integrin α3/β1-dependent fashion (Seewaldt et al., 2001). These observations suggest that p53 may suppress, rather than mediate, apoptotic signals from the ECM. Nevertheless, these results reinforce our findings that matrix proteins can transduce apoptotic signals via integrins and suggest that the induction of apoptosis by the ECM may be more general than originally thought. Thus, in addition to the two well characterized categories of cell adhesion events—one that promotes cell survival and one that is neutral to this process—evidence of another category that induces apoptosis is beginning to emerge.
Materials and methods
Cell culture
Rat1a, 10.1 p53-null fibroblasts (Livingstone et al., 1992), and MEFs from either wild-type or Bax/Bak double-null animals (Wei et al., 2001) were grown at 37°C with 10% CO2 in DME (Invitrogen) containing 10% FBS (Intergen). Normal HSFs were obtained from the American Type Culture Collection (CRL-2076) and maintained in Iscove's modified DME (Invitrogen) with 10% FBS at 37°C with 5% CO2 and used before passage 8. Primary HUVECs were maintained at 37°C with 5% CO2 in Medium 200 containing 2% serum and endothelial growth supplements (Cascade Biologics, Inc.). Cells were used between passages 16–20 to ensure activation of integrin αVβ3 (Leu et al., 2002). Rat1a cells stably expressing Bcl-2 and Bcl-XL were prepared by infection with retroviruses that drive their expression (Gottlob et al., 2001), and cells harboring the empty vector (pBabePuro) were used as controls. The 10.1 p53-null fibroblasts were stably transfected with a pMV7-derived vector driving expression of a temperature-sensitive p53 (Wagner et al., 1994) or a temperature-sensitive, transcription transactivation–defective p53 mutant (Lin et al., 1994), and grown at 39°C. Growth at 33°C is permissive for expression of p53 activity in these cells.
Proteins, antibodies, peptides, and reagents
Wild-type and mutant CCN1 were produced using the baculovirus expression system and purified as previously described (Chen et al., 2004; Leu et al., 2004). Human FN and mouse LN were purchased from BD Biosciences. Recombinant human EGF and basic FGF were obtained from Invitrogen. DRB, human VN, monoclonal anti–β-actin antibody (AC-15), and rabbit and mouse IgGs were purchased from Sigma-Aldrich. Function-blocking mAbs against integrins, including NKI-GoH3 (anti-α6), P5D2 (anti-β1), P1D6 (anti-α5), and LM609 (anti-αVβ3) were purchased from CHEMICON International, Inc. Function-blocking antibodies against syndecan-4 and TRITC-conjugated mAb against phospho-JNK T183/Y185 were obtained from Santa Cruz Biotechnology, Inc. Monoclonal anti–cytochrome c (6H2.B4) and anti-Bax (6A7) antibody were obtained from BD Biosciences. Rabbit polyclonal caspase-3, -9, FAK, phospho-FAK Y576/577, and phospho-paxillin Y118 antibodies were purchased from Cell Signaling Technology, and antibodies against phospho-FAK Y397 were obtained from Calbiochem. HRP-conjugated anti–mouse and anti–rabbit secondary antibodies were purchased from GE Healthcare. Alexa Fluor 488–conjugated anti–mouse and anti–rabbit secondary antibodies were obtained from Invitrogen. Synthetic GRGDSP and GRGESP peptides were purchased from Life Technologies, Inc. The synthetic peptides T1 (GQKCIVQTTSWSQCSKS), T1-mut (GQKCIVQTTSAAQCSKS), T4 (RLVKETRICEVRPCGQPVYSSLK), and H2 (FTYAGCSSVKKYRPKY) were prepared by Research Genetics (Leu et al., 2003, 2004). The pan-caspase inhibitor Q-VD-Oph was purchased from Valeant Pharmaceuticals; the pan-caspase inhibitor z-VAD-fmk, caspase-3 inhibitor z-DEVD-fmk, caspase-8 inhibitor z-IETD-fmk, caspase-9 inhibitor z-LEHD-fmk, cyclic pifithrin-α, and cycloheximide were purchased from Calbiochem. The mitochondrion-selective probe MitoTracker Orange was obtained from Invitrogen.
Apoptosis assays
To examine cell death resulting from cell adhesion, cells were plated in medium supplemented with 0.5% FBS on 35-mm Petri dishes precoated overnight with various proteins. After 24 h of incubation, cells were fixed with a 10% formaldehyde solution overnight, washed with PBS, and stained by 1 μg/ml DAPI in 1× PBS. Apoptotic cells were quantified by DAPI staining as described previously (Kennedy et al., 1997). A total of five random fields were counted per sample, with a minimum of 50 cells per field. All experiments were repeated at least twice in triplicate. In experiments where apoptotic factors were added in a soluble form, cells were plated at a low density (50,000 cells per well in a 6-well plate) overnight, replaced with serum-free medium (unless otherwise indicated), and treated with test molecules for 24 h. In experiments using inhibitors with cytotoxicity (e.g., cycloheximide, DRB, and caspase inhibitors), cells were plated at high density (500,000 cells per well in a 6-well plate) and assayed 6 h after treatment. For the TUNEL assay, fibroblasts were plated on glass coverslips coated with test proteins and cultured in basal medium containing 0.5% FBS for 24 h. Apoptosis was assayed using the ApopTag Red in situ Apoptosis Detection Kit as instructed by the manufacturer (CHEMICON International, Inc.), and cells were counterstained with DAPI. Cells were then observed using standard UV, rhodamine, or FITC filters by fluorescence microscopy using an inverted microscope (model DM IRB/E; Leica). Images were obtained with a digital camera (model DC-330; Dage-MTI, Inc.) and Image-Pro Plus software (Media Cybernetics, Inc.). All image processing in this paper was performed using Canvas 7 (ACD Systems, Inc.).
Immunoblot analysis
To detect FAK activation, Rat1a cells were serum-starved overnight, detached with trypsin-free cell detachment solution (2.5 mM EDTA and 0.1% wt/vol glucose in 1× PBS), and plated in serum-free medium on 35-mm dishes coated with test proteins. For immunoblotting, equal amounts of proteins from cell lysates were separated on 12% (caspases) or 7.5% (FAK) SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blotted with primary antibodies, detected by secondary antibodies, and developed by chemiluminescence (GE Healthcare).
Indirect immunofluorescence
To localize cytochrome c and Bax, Rat1a cells were adhered on glass coverslips overnight and treated with serum-free medium with or without CCN1 for 6 h. To label the mitochondria, 100 nM of MitoTracker was added for 5 min before fixation with 3% PFA. Cytochrome c immunostaining was performed as described previously, with minor modifications (Kennedy et al., 1997). In brief, cells were permeabilized by incubation in 0.2% saponin, blocked with 10% normal goat serum, and stained with 1 μg/ml of monoclonal anti–cytochrome c antibody and 1 μg/ml of Alexa Fluor 488–conjugated secondary antibody. For Bax localization, PFA-fixed cells were permeabilized with 1% Triton X-100, blocked with 1% BSA, and stained with polyclonal anti-Bax antibodies. Cells were observed by fluorescence microscopy using an Optiphot microscope (Nikon). Photographs were taken with a 35-mm camera (model FX-35WA; Nikon) and scanned with a Perfection scanner (Epson) using TWAIN 5 software (Epson) to import images to Canvas.
To quantify Bax activation, Rat1a cells were adhered on 2-well permanox slides (Nalge Nunc), pretreated with 100 μM of pan-caspase inhibitor, and treated with test molecules as described in Apoptosis assays. Cells were fixed and stained with 0.5 μg/ml mAb against Bax and 1 μg/ml of TRITC-conjugated anti–mouse antibody (Jackson ImmunoResearch Laboratories). Cells were rinsed and coverslips were mounted onto slides using the aqueous mounting solution Crystal/Mount (Biomeda). To stain for FAK or paxillin, cells were plated on glass coverslips coated with either 10 μg/ml FN or 50 μg/ml CCN1 for 20 min, fixed, and permeabilized as for Bax immunostaining before staining with the cognate antibodies and 1 μg/ml of Alexa Fluor 488–conjugated secondary antibody. Cells were observed by fluorescence microscopy using a microscope (Axioplan 2; Carl Zeiss MicroImaging, Inc.). Images were obtained with a digital camera (Axiocam HR) and Axiovision software (Carl Zeiss MicroImaging, Inc.). To stain for phosphorylated JNK, cells were plated for 10 min on FN- or CCN1-coated coverslips as for Bax immunostaining, stained with 4 μg/ml of TRITC-conjugated antibody against phospho-JNK T183/Y185 or 4 μg/ml of TRITC-conjugated mouse IgG (Jackson ImmunoResearch Laboratories), and counter stained with DAPI.
Acknowledgments
This work was supported by grants from the National Institutes of Health to L.F. Lau (CA46565) and N. Hay (AG016927) and a predoctoral fellowship from the American Heart Association to V. Todorović.
Abbreviations used in this paper: DRB, 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole; FN, fibronectin; HSF, human skin fibroblast; HSPG, heparan sulfate proteoglycan; HUVEC, human umbilical vein endothelial cell; JNK, c-Jun NH2-terminal kinase; LN, laminin; MEF, mouse embryonic fibroblast; PLL, poly-l-lysine; VN, vitronectin.
References
- Almeida, E.A., D. Ilic, Q. Han, C.R. Hauck, F. Jin, H. Kawakatsu, D.D. Schlaepfer, and C.H. Damsky. 2000. Matrix survival signaling: from fibronectin via focal adhesion kinase to c-Jun NH2-terminal kinase. J. Cell Biol. 149:741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babic, A.M., M.L. Kireeva, T.V. Kolesnikova, and L.F. Lau. 1998. CYR61, a product of a growth factor-inducible immediate early gene, promotes angiogenesis and tumor growth. Proc. Natl. Acad. Sci. USA. 95:6355–6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babic, A.M., C.-C. Chen, and L.F. Lau. 1999. Fisp12/mouse connective tissue growth factor mediates endothelial cell adhesion and migration through integrin αvβ3, promotes endothelial cell survival, and induces angiogenesis in vivo. Mol. Cell. Biol. 19:2958–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehrecke, E.H. 2002. How death shapes life during development. Nat. Rev. Mol. Cell Biol. 3:779–787. [DOI] [PubMed] [Google Scholar]
- Buckley, C.D., D. Pilling, N.V. Henriquez, G. Parsonage, K. Threlfall, D. Scheel-Toellner, D.L. Simmons, A.N. Akbar, J.M. Lord, and M. Salmon. 1999. RGD peptides induce apoptosis by direct caspase-3 activation. Nature. 397:534–539. [DOI] [PubMed] [Google Scholar]
- Buschmann, T., O. Potapova, A. Bar-Shira, V.N. Ivanov, S.Y. Fuchs, S. Henderson, V.A. Fried, T. Minamoto, D. Alarcon-Vargas, M.R. Pincus, et al. 2001. Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Mol. Cell. Biol. 21:2743–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C.-C., N. Chen, and L.F. Lau. 2001. a. The angiogenic factors Cyr61 and CTGF induce adhesive signaling in primary human skin fibroblasts. J. Biol. Chem. 276:10443–10452. [DOI] [PubMed] [Google Scholar]
- Chen, C.-C., F.-E. Mo, and L.F. Lau. 2001. b. The angiogenic inducer Cyr61 induces a genetic program for wound healing in human skin fibroblasts. J. Biol. Chem. 276:47329–47337. [DOI] [PubMed] [Google Scholar]
- Chen, N., C.C. Chen, and L.F. Lau. 2000. Adhesion of human skin fibroblasts to Cyr61 is mediated through integrin α6β1 and cell surface heparan sulfate proteoglycans. J. Biol. Chem. 275:24953–24961. [DOI] [PubMed] [Google Scholar]
- Chen, N., S.-J. Leu, V. Todorović, S.C.T. Lam, and L.F. Lau. 2004. Identification of a novel integrin αvβ3 binding site in CCN1 (CYR61) critical for pro-angiogenic activities in vascular endothelial cells. J. Biol. Chem. 279:44166–44176. [DOI] [PubMed] [Google Scholar]
- Chien, W., T. Kumagai, C.W. Miller, J.C. Desmond, J.M. Frank, J.W. Said, and H.P. Koeffler. 2004. Cyr61 suppresses growth of human endometrial cancer cells. J. Biol. Chem. 279:53087–53096. [DOI] [PubMed] [Google Scholar]
- Chipuk, J.E., T. Kuwana, L. Bouchier-Hayes, N.M. Droin, D.D. Newmeyer, M. Schuler, and D.R. Green. 2004. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 303:1010–1014. [DOI] [PubMed] [Google Scholar]
- Colognato, H., W. Baron, V. Avellana-Adalid, J.B. Relvas, A. Baron-Van Evercooren, E. Georges-Labouesse, and C. French-Constant. 2002. CNS integrins switch growth factor signalling to promote target-dependent survival. Nat. Cell Biol. 4:833–841. [DOI] [PubMed] [Google Scholar]
- Conzen, S.D., K. Gottlob, E.S. Kandel, P. Khanduri, A.J. Wagner, M. O'Leary, and N. Hay. 2000. Induction of cell cycle progression and acceleration of apoptosis are two separable functions of c-Myc: transrepression correlates with acceleration of apoptosis. Mol. Cell. Biol. 20:6008–6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory, S., and J.M. Adams. 2002. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer. 2:647–656. [DOI] [PubMed] [Google Scholar]
- Couchman, J.R., L. Chen, and A. Woods. 2001. Syndecans and cell adhesion. Int. Rev. Cytol. 207:113–150. [DOI] [PubMed] [Google Scholar]
- Eliceiri, B.P., and D.A. Cheresh. 2000. Role of alpha v integrins during angiogenesis. Cancer J. 6:S245–S249. [PubMed] [Google Scholar]
- Frisch, S.M., and R.A. Screaton. 2001. Anoikis mechanisms. Curr. Opin. Cell Biol. 13:555–562. [DOI] [PubMed] [Google Scholar]
- Gilmore, A.P., A.D. Metcalfe, L.H. Romer, and C.H. Streuli. 2000. Integrin-mediated survival signals regulate the apoptotic function of Bax through its conformation and subcellular localization. J. Cell Biol. 149:431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlob, K., N. Majewski, S. Kennedy, E. Kandel, R.B. Robey, and N. Hay. 2001. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 15:1406–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann, J. 2002. Molecular mechanisms of “detachment-induced apoptosis—Anoikis”. Apoptosis. 7:247–260. [DOI] [PubMed] [Google Scholar]
- Grzeszkiewicz, T.M., D.J. Kirschling, N. Chen, and L.F. Lau. 2001. CYR61 stimulates human skin fibroblast migration through integrin αvβ5 and enhances mitogenesis through integrin αvβ3, independent of its carboxyl-terminal domain. J. Biol. Chem. 276:21943–21950. [DOI] [PubMed] [Google Scholar]
- Grzeszkiewicz, T.M., V. Lindner, N. Chen, S.C.T. Lam, and L.F. Lau. 2002. The angiogenic factor CYR61 supports vascular smooth muscle cell adhesion and stimulates chemotaxis through integrin α6β1 and cell surface heparan sulfate proteoglycans. Endocrinology. 143:1441–1450. [DOI] [PubMed] [Google Scholar]
- Haupt, S., M. Berger, Z. Goldberg, and Y. Haupt. 2003. Apoptosis- the p53 network. J. Cell Sci. 116:4077–4085. [DOI] [PubMed] [Google Scholar]
- Hynes, R.O. 2002. Integrins: bidirectional, allosteric signaling machines. Cell. 110:673–687. [DOI] [PubMed] [Google Scholar]
- Igney, F.H., and P.H. Krammer. 2002. Death and anti-death: tumour resistance to apoptosis. Nat. Rev. Cancer. 2:277–288. [DOI] [PubMed] [Google Scholar]
- Ilic, D., E.A. Almeida, D.D. Schlaepfer, P. Dazin, S. Aizawa, and C.H. Damsky. 1998. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J. Cell Biol. 143:547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan, Y., M. Matter, J.T. Pai, Y.L. Chen, J. Pilch, M. Komatsu, E. Ong, M. Fukuda, and E. Ruoslahti. 2004. A mitochondrial protein, Bit1, mediates apoptosis regulated by integrins and Groucho/TLE corepressors. Cell. 116:751–762. [DOI] [PubMed] [Google Scholar]
- Kennedy, S.G., A.J. Wagner, S.D. Conzen, J. Jordan, A. Bellacosa, P.N. Tsichlis, and N. Hay. 1997. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 11:701–713. [DOI] [PubMed] [Google Scholar]
- Kim, K.H., Y.K. Min, J.H. Baik, L.F. Lau, B. Chaqour, and K.C. Chung. 2003. Expression of angiogenic factor Cyr61 during neuronal cell death via the activation of c-Jun N-terminal kinase and serum response factor. J. Biol. Chem. 278:13847–13854. [DOI] [PubMed] [Google Scholar]
- Lau, L.F., and S.C. Lam. 1999. The CCN family of angiogenic regulators: the integrin connection. Exp. Cell Res. 248:44–57. [DOI] [PubMed] [Google Scholar]
- Lau, L.F., and S.C.T. Lam. 2005. Integrin-mediated CCN functions. CCN Proteins: A New Family of Cell Growth and Differentiation Regulators. B. Perbal and M. Takigawa, editors. Imperial College Press, London. 61–79.
- Leu, S.-J., S.C.T. Lam, and L.F. Lau. 2002. Proangiogenic activities of CYR61 (CCN1) mediated through integrins αvβ3 and α6β1 in human umbilical vein endothelial cells. J. Biol. Chem. 277:46248–46255. [DOI] [PubMed] [Google Scholar]
- Leu, S.-J., Y. Liu, N. Chen, C.-C. Chen, S.C. Lam, and L.F. Lau. 2003. Identification of a novel integrin α6β1 binding site in the angiogenic inducer CCN1 (CYR61). J. Biol. Chem. 278:33801–33808. [DOI] [PubMed] [Google Scholar]
- Leu, S.-J., N. Chen, C.-C. Chen, V. Todorović, T. Bai, V. Juric, Y. Liu, G. Yan, S.C.T. Lam, and L.F. Lau. 2004. Targeted mutagenesis of the angiogenic protein CCN1 (CYR61): selective inactivation of integrin α6β1-heparan sulfate proteoglycan coreceptor-mediated cellular activities. J. Biol. Chem. 279:44177–44187. [DOI] [PubMed] [Google Scholar]
- Li, P., D. Nijhawan, I. Budihardjo, S.M. Srinivasula, M. Ahmad, E.S. Alnemri, and X. Wang. 1997. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 91:479–489. [DOI] [PubMed] [Google Scholar]
- Lin, J., J. Chen, B. Elenbaas, and A.J. Levine. 1994. Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev. 8:1235–1246. [DOI] [PubMed] [Google Scholar]
- Lin, M.T., C.C. Chang, S.T. Chen, H.L. Chang, J.L. Su, Y.P. Chau, and M.L. Kuo. 2004. Cyr61 expression confers resistance to apoptosis in breast cancer MCF-7 cells by a mechanism of NF-kappaB-dependent XIAP up-regulation. J. Biol. Chem. 279:24015–24023. [DOI] [PubMed] [Google Scholar]
- Livingstone, L.R., A. White, J. Sprouse, E. Livanos, T. Jacks, and T.D. Tlsty. 1992. Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell. 70:923–935. [DOI] [PubMed] [Google Scholar]
- Menendez, J.A., I. Mehmi, D.W. Griggs, and R. Lupu. 2003. The angiogenic factor CYR61 in breast cancer: molecular pathology and therapeutic perspectives. Endocr. Relat. Cancer. 10:141–152. [DOI] [PubMed] [Google Scholar]
- Meredith, J.E., Jr., B. Fazeli, and M.A. Schwartz. 1993. The extracellular matrix as a cell survival factor. Mol. Biol. Cell. 4:953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara, M., S. Erster, A. Zaika, O. Petrenko, T. Chittenden, P. Pancoska, and U.M. Moll. 2003. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell. 11:577–590. [DOI] [PubMed] [Google Scholar]
- Miranti, C.K., and J.S. Brugge. 2002. Sensing the environment: a historical perspective on integrin signal transduction. Nat. Cell Biol. 4:E83–E90. [DOI] [PubMed] [Google Scholar]
- Mo, F.-E., A.G. Muntean, C.-C. Chen, D.B. Stolz, S.C. Watkins, and L.F. Lau. 2002. CYR61 (CCN1) is essential for placental development and vascular integrity. Mol. Cell. Biol. 22:8709–8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson, D.W., and N.A. Thornberry. 2003. Apoptosis. Life and death decisions. Science. 299:214–215. [DOI] [PubMed] [Google Scholar]
- Ossovskaya, V.S., I.A. Mazo, M.V. Chernov, O.B. Chernova, Z. Strezoska, R. Kondratov, G.R. Stark, P.M. Chumakov, and A.V. Gudkov. 1996. Use of genetic suppressor elements to dissect distinct biological effects of separate p53 domains. Proc. Natl. Acad. Sci. USA. 93:10309–10314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons, J.T. 2003. Focal adhesion kinase: the first ten years. J. Cell Sci. 116:1409–1416. [DOI] [PubMed] [Google Scholar]
- Pietrancosta, N., F. Maina, R. Dono, A. Moumen, C. Garino, Y. Laras, S. Burlet, G. Quelever, and J.L. Kraus. 2005. Novel cyclized Pifithrin-alpha p53 inactivators: synthesis and biological studies. Bioorg. Med. Chem. Lett. 15:1561–1564. [DOI] [PubMed] [Google Scholar]
- Planque, N., and B. Perbal. 2003. A structural approach to the role of CCN (CYR61/CTGF/NOV) proteins in tumourigenesis. Cancer Cell Int. 3:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullan, S., J. Wilson, A. Metcalfe, G.M. Edwards, N. Goberdhan, J. Tilly, J.A. Hickman, C. Dive, and C.H. Streuli. 1996. Requirement of basement membrane for the suppression of programmed cell death in mammary epithelium. J. Cell Sci. 109:631–642. [DOI] [PubMed] [Google Scholar]
- Rapraeger, A.C., A. Krufka, and B.B. Olwin. 1991. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science. 252:1705–1708. [DOI] [PubMed] [Google Scholar]
- Rich, T., C.J. Watson, and A. Wyllie. 1999. Apoptosis: the germs of death. Nat. Cell Biol. 1:E69–E71. [DOI] [PubMed] [Google Scholar]
- Schaller, M.D., and J.T. Parsons. 1995. pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol. Cell. Biol. 15:2635–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seewaldt, V.L., K. Mrozek, R. Sigle, E.C. Dietze, K. Heine, D.M. Hockenbery, K.B. Hobbs, and L.E. Caldwell. 2001. Suppression of p53 function in normal human mammary epithelial cells increases sensitivity to extracellular matrix–induced apoptosis. J. Cell Biol. 155:471–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons, M., and A. Horowitz. 2001. Syndecan-4-mediated signalling. Cell. Signal. 13:855–862. [DOI] [PubMed] [Google Scholar]
- Slee, E.A., D.J. O'Connor, and X. Lu. 2004. To die or not to die: how does p53 decide? Oncogene. 23:2809–2818. [DOI] [PubMed] [Google Scholar]
- Stupack, D.G., X.S. Puente, S. Boutsaboualoy, C.M. Storgard, and D.A. Cheresh. 2001. Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. J. Cell Biol. 155:459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, A.J., J.M. Kokontis, and N. Hay. 1994. Myc-mediated apoptosis requires wild-type p53 in a manner independent of cell cycle arrest and the ability of p53 to induce p21waf1/cip1. Genes Dev. 8:2817–2830. [DOI] [PubMed] [Google Scholar]
- Wei, M.C., W.X. Zong, E.H. Cheng, T. Lindsten, V. Panoutsakopoulou, A.J. Ross, K.A. Roth, G.R. MacGregor, C.B. Thompson, and S.J. Korsmeyer. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 292:727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]