

Abstract
The matricellular protein CCN1 (CYR61) regulates cell adhesion, migration, proliferation, survival, and differentiation through binding to integrin receptors and heparan sulfate proteoglycans. Here we show that Ccn1-null mice are impaired in cardiac valvulosepatal morphogenesis, resulting in severe atrioventricular septal defects (AVSD). Remarkably, haploinsufficiency for Ccn1 also results in delayed formation of the ventricular septum in the embryo and persistent ostium primum atrial septal defects (ASD) in ~20% of adults. Mechanistically, Ccn1 is not required for epithelial-to-mesenchymal transformation or cell proliferation and differentiation in the endocardial cushion tissue. However, Ccn1 deficiency leads to precocious apoptosis in the atrial junction of the cushion tissue and impaired gelatinase activities in the muscular component of the interventricular septum at E12.5, when fusion between the endocardial cushion tissue and the atrial and ventricular septa occurs, indicating that these defects may underlie the observed AVSD. Moreover, human CCN1 maps to 1p21-p31, the chromosomal location of an AVSD susceptibility gene. Together, these results provide evidence that deficiency in matrix signaling can lead to autosomal dominant AVSD, identify Ccn1+/− mice as a genetic model for ostium primum ASD, and implicate CCN1 as a candidate gene for AVSD in humans.
INTRODUCTION
Atrioventricular septal defect (AVSD) is a common family of genetic disorders, accounting for ~7.5% of the recognized congenital heart disease (CHD) in humans1. During cardiac development, the endocardial cushion tissue expands through formation of the cardiac jelly, a specialized extracellular matrix (ECM) between the endocardium and myocardium, into which endocardial cells that have undergone epithelial-to-mesenchymal transformation (EMT) invade. Subsequent growth and remodeling of the cushion tissue, coupled with its fusion with the developing atrial septum and muscular interventricular septum, lead to formation of the definitive atrioventricular (AV) septa and valves2, 3. Defects in this process are manifested in a spectrum of abnormalities with varying severities, ranging from partial forms of AVSD with ostium primum atrial septal defects (ASD) to complete forms with absence of the AV septa and lack of partitioning of the AV valve into separate mitral (left) and tricuspid (right) valves. While AVSD is frequently associated with Down’s syndrome (trisomy 21), non-syndromic AVSD also occur. Although non-syndromic AVSD may be a sporadic trait or the result of multifactorial inheritance, strong evidence also support the presence of susceptibility genes for AVSD that are autosomal dominant and incompletely penetrant4–6.
CCN1 (CYR61, cysteine-rich angiogenic inducer 61; NM_001554) is a secreted, cysteine-rich protein associated with the ECM. A dynamically expressed protein, CCN1 serves regulatory rather than structural roles and can be considered a matricellular protein7. CCN1 promotes cell adhesion, migration, proliferation, differentiation, and survival or death in a cell type-dependent manner8. CCN1 also induces angiogenesis in vivo and Ccn1-null mice suffer embryonic lethality, in part due to placental vascular insufficiency and compromised embryonic vessel integrity9, 10. Mechanistically, CCN1 binds and functions through specific integrin Whereas CCN1 promotes cell adhesion, migration, and proliferation in fibroblasts through integrins α6β1, αvβ5, and αvβ3, respectively, the pro-angiogenic activities of CCN1 in endothelial cells are mediated through αvβ3. Recently, the specific binding sites of CCN1 for these integrins and HSPGs have been identified11, 12.
Human CCN1 has been mapped to chromosome 1p22-p3113, the same region (1p21-p31) as an AVSD susceptibility gene identified in a large kindred4. In this study, we show that Ccn1-null mice exhibit AVSD with complete penetrance. Remarkably, haploinsufficiency for Ccn1 also results in ostium primum ASD in 20% of adults, resembling a common form of partial AVSD recognized clinically in humans. These results implicate CCN1 as a candidate gene for AVSD. Ccn1 deficiency does not affect endocardial cushion tissue formation or EMT, but results in accelerated apoptosis in the cushion tissue and reduced gelatinase activity in the developing ventricular septum and AV valves. These results show that the matricellular protein CCN1 is essential for heart septation, and underscore the importance of matrix signaling in valvuloseptal morphogenesis.
MATERIALS AND METHODS
Animals, histology and immunohistochemistry
Generation of Ccn1-deficient mice, knockin of a lacZ reporter gene, and detection of β-galactosidase activity were described previously10. For histology, formalin-fixed embryos or post-natal hearts were embedded in paraffin, and 7 μm sections were stained with hematoxylin and eosin. For immunohistochemistry, embryos fixed in 4% paraformaldehyde were sectioned and stained with antibodies against α-smooth-muscle actin antibody (clone 1A4; Sigma) or activated caspase-3 (5A1, Cell signaling), and detected with alkaline phosphatase (AP) chromogen (Zymed). For proliferation assay, sections were stained with polyclonal anti-Ki67 antibodies (Novocastra Lab) and Alexa Fluor 594-conjugated secondary antibody (Molecular Probes), and counterstained with 4′,6-diamidino-2-phenylindole dilactate (DAPI; Sigma). Images were acquired through a fluorescence microscope using a digital camera, and superimposition of Alexa Fluor 594 (red) and DAPI (blue) staining was done using the AxioVision (Zeiss) software.
In situ hybridization
Embryos were fixed in 4% paraformaldehyde and paraffin embedded; 20 μm sections were hybridized to digoxigenin-labeled Mox-1 anti-sense riboprobe (generous gift of Christopher Wright, Vanderbilt) and visualized with AP-conjugated anti-digoxigenin antibody (Roche) and AP-chromogen. Sense riboprobe of Mox-1 was used as control (data not shown).
AV cushion explant culture
AV cushion explants were cultured on collagen gels as described14. E9.5 (somite #21~26) AV cushion tissues were microdissected and cultured on type I collagen (BD Biosciences) gels, then fixed and stained with fluorescein phalloidin (Invitrogen) and counterstained with DAPI after 48 hrs.
TUNEL assay and zymography
Paraffin sections (10 μm) of 4% paraformaldehyde-fixed embryos were subjected to TUNEL assay using ApopTag Red In Situ Apoptosis Detection Kit (Chemicon) per vendor’s instructions. In situ zymography was performed as described15. H9c2 cardiomyocytes (ATCC) were treated with 4 μg/ml recombinant CCN1 protein16 in DMEM containing 0.1% BSA. Conditioned medium was collected after 1–3 days and subjected to gelatin gel zymography following standard protocol with precast 10% zymogram gel containing gelatin (Bio-Rad).
Quantitative real-time reverse transcriptase PCR (qRT-PCR)
cDNA reverse transcribed from total RNA isolated from E12.5 embryonic hearts was quantified using a MyiQ single-color real-time PCR detection system (Bio-Rad). PCR reaction in triplicate contains iQ SYBR Green Supermix (Bio-Rad), cDNA template and 200 nM primer pairs for either Ccn1 or cyclophilin (internal control). Sense (S) and antisense (AS) primers are: Ccn1-S, 5′-GCAGCAAGACCAAGAAATCC-3′; Ccn1-AS, 5′-TTCTGGTCTGCAGAGGTGTG-3′. Cyclophilin-S, 5′-GGCAAATGCTGGACCAAACAC-3′; cyclophilin-AS, 5′-TTCCTGGACCCAAAACGCTC-3′.
Statistical analysis
Values are expressed as mean ± standard derivations. Comparisons were made by Student’s t test; statistical significance was set at P<0.05.
RESULTS
Expression of Ccn1 during heart development
We have previously placed the lacZ gene under the control of the endogenous Ccn1 promoter, thereby allowing Ccn1 expression to be monitored by following β-galactosidase activity10. As judged by X-gal staining, Ccn1 is expressed in the E8.5 myocardium and endocardium of the bulbus cordis region in the primitive heart (Fig. 1A). Prominent cardiac expression of Ccn1 was seen at E10.5 (Fig. 1B), especially in the truncus arteriosus (arrowhead, Fig. 1C), which later divides to form the aorta and pulmonary trunk. Ccn1 expression was also seen around the AV canal at later stages and in the AV cushion tissue and septum (Fig. 1D,E). At E12.5, Ccn1 was expressed at the junctions between the muscular component of the ventricular septum and the membranous mesenchyme of the cushion tissue, and between the mesenchymal cap of the atrial septum and the AV cushion tissue (Fig. 1F). By E13.5, expression around the atrial junction had subsided, but was prominent in the developing AV valvular leaflets (Fig. 1G). Expression was high in the vessel wall of the aorta, although little or no expression was seen in the pulmonary and aortic valve leaflets (Fig. 1H). Thus, Ccn1 expression coincides with the temporal and spatial pattern of key events in atrioventricular valvuloseptal morphogenesis. The levels of Ccn1 mRNA expressed in E12.5 embryonic hearts were examined by quantitative real-time RT-PCR, confirming a true null phenotype in Ccn1−/ − embryos and haploinsufficiency in Ccn1+/− embryos (Fig. 1I)10.
Figure 1.
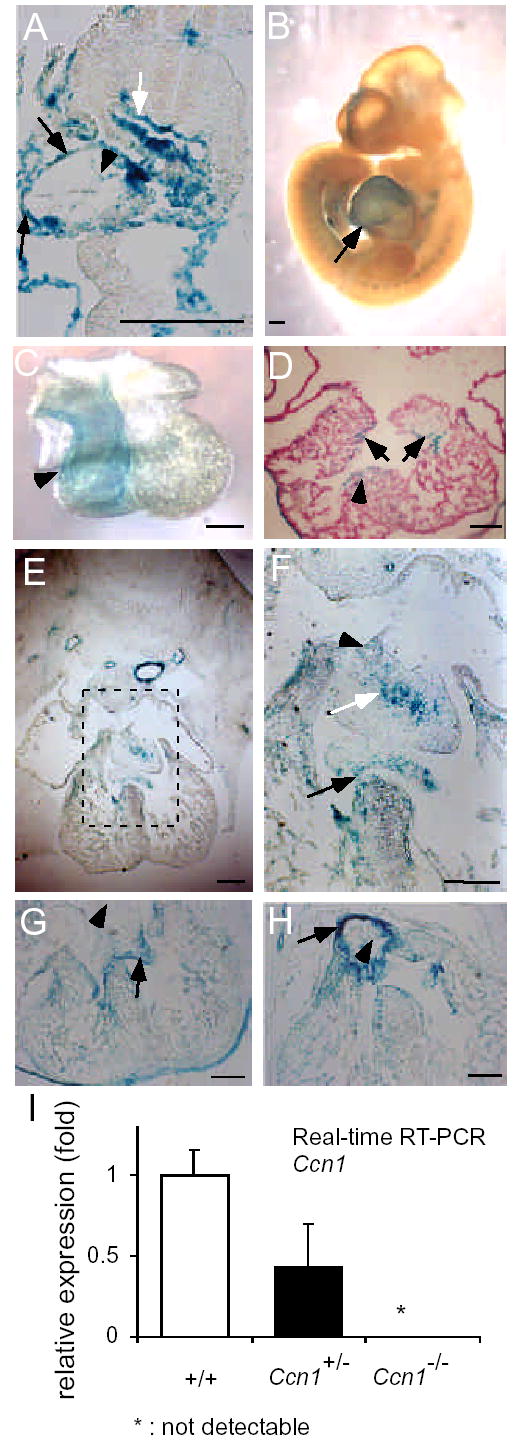
Expression of β-galactosidase driven by the Ccn1 promoter. (A) Intense X-gal staining in the myocardium (black arrow) of the bulbus cordis of the primitive heart of E8.5 Ccn1+/− embryos; less staining was seen at endocardium (black arrowhead). White arrow points to the foregut. (B) Whole-mount E10.5 embryo shows prominent cardiac staining (arrow). (C) X-gal staining was detected in the truncus arteriosus (arrowhead) at E10.5. (D) Expression in the developing atrioventricular cushion tissue (arrows) and ventricular septum (arrowhead) at E11.5. (E) X-gal staining in the E12.5 embryonic heart. (F) Higher magnification view of dashed box in E, showing staining in the mesenchymal cap of the atrial septum primum (black arrowhead), the cushion mesenchyme (white arrow), and at the junction between the ventricular septum and the cushion tissue (black arrow). (G) At E13.5, expression was observed at the valvular leaflets (arrow) and the ventricular septum junction, while expression around the atrial fusion sites had diminished (arrowhead). (H) Expression was weak in aortic leaflets (arrowhead), but remained intense in aortic wall at E13.5 (arrow in H). (I) The levels of Ccn1 mRNA in E12.5 hearts were quantified by qRT-PCR, normalized to cyclophilin mRNA as internal control. No detectable signal was observed in Ccn1−/− (n=3 per genotype, P<0.03 between +/+ and +/−). Bars: 200 μm.
Ccn1-null mice exhibit AVSD
Given the cardiac expression of Ccn1 described above, we examined the effects of Ccn1 deficiency in the heart. The atrial and ventricular septa were formed by E14.5 in WT embryos, as were the AV valves (Fig. 2A,B). Ccn1−/ − embryos, however, exhibited AVSD with ~100% penetrance, with 55% (10/18) showing complete AVSD and 45% (8/18) showing VSD (Fig. 2E-G). Severe malformation of the atrial and ventricular septa and defects in AV valve morphogenesis were evident. During AV septation and valve formation, the common AV canal is partitioned by the fusion of the inferior and superior endocardial cushion tissues into separate left and right components, which undergo further morphogenesis to form the mitral and tricuspid valves3. Fusion of the inferior and superior cushion tissue was impaired in affected Ccn1−/ − embryos as evidenced by gaps between the two endocardial cushions, resulting in a common AV valve orifice that is a hallmark of complete AVSD (Fig. 2F,G). Nearly all Ccn1-null embryos showed defects in maturation of the interventricular septum (IVS), which forms by the fusion of a muscular component that extends as an outgrowth of the ventricular wall, and a membranous component that forms from the AV cushion tissue. This fusion is defective in Ccn1-null embryos (Fig. 2E), consistent with the expression of Ccn1 in the junction between the muscular component of the septum and the membranous mesenchyme (black arrow, Fig.1F).
Figure 2.
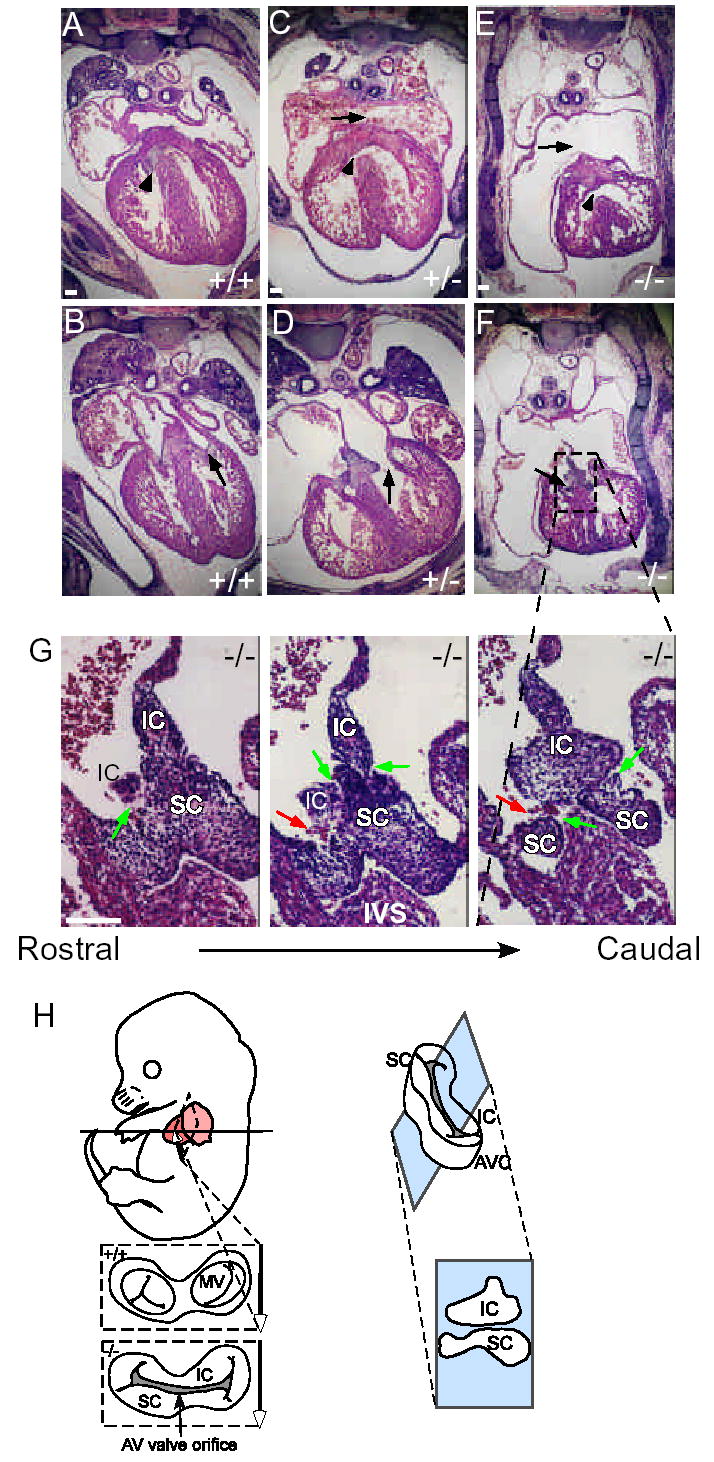
Atrioventricular septal defects in Ccn1-deficient embryos. Transverse sections of E14.5 embryonic hearts were H & E stained. (A,B) WT embryos showed well developed four-chambered hearts, with complete closure of the ventricular septum (arrowhead, A) and extended AV valvular structure (arrow, B). (C,D) Ccn1+/− embryos displayed VSD (arrowheads in C), ASD (arrow, C), and dysplastic atrioventricular valves (arrow, D). (E,F) Ccn1−/− hearts exhibited VSD (arrowhead, E), ASD (arrow, E), and AV valve orifice (arrow, F). (G) High magnification views of dashed box in F and its adjacent sections reveal evidence of AV valve orifice, including gaps between superior (SC) and inferior (IC) endocardial cushions of AV canal (green arrows) with blood cells in between the two AV cushions (red arrows). IVS: interventricular septum. (H) Left, diagram illustrates the partitioning of the AV canal (AVC) into tricuspid (TV) and mitral (MV) valves, viewed from the atrial perspective. A hallmark of complete AVSD is the presence of a common AV valve orifice resulting from impaired fusion between the inferior (IC) and superior (SC) cushion tissue. Right, the plane of section in sections shown in (G), showing how the IC and SC are viewed. Bars: 100 μm.
Haploinsufficiency in Ccn1 leads to atrial septal defects
Surprisingly, Ccn1+/− embryos also display AVSD. This finding is significant because autosomal dominant inheritance has been observed in non-syndromic AVSD in humans4, 6. Histological analysis showed that ~67% of Ccn1+/− embryos (18/27) exhibited cardiac defects of varying severity at E14.5, including complete AVSD (4/27), VSD (11/27), or dysplastic mitral valves (3/27)(Fig. 2C,D). Although VSD phenotypes in Ccn1+/− hearts were conspicuous at E13.5 and E14.5, they were not observed after E15.5 (n=35), indicating that IVS formation is delayed in Ccn1+/− embryos but not persistently impaired. However, ~20% of adult Ccn1+/− mice (5/25) show persistent ASD (Fig. 3). Although cardiac septal defects might form as secondary effects of placental abnormalities17, these cardiac defects are not likely related to the placenta since the Ccn1+/− placenta develop normally10.
Figure 3.
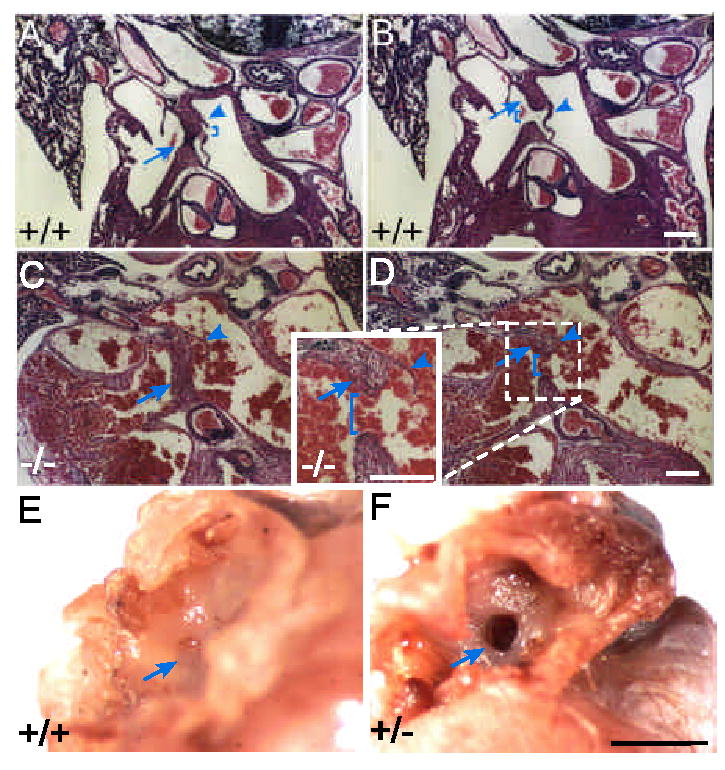
Haploinsufficiency in Ccn1 results in ASD. (A,B) WT neonatal hearts were sectioned and H & E stained. The septum primum (arrowheads) and septum secundum (arrows) were properly formed, separating the two atrial chambers. The ostium secundum (bracket in A) and fossa ovalis (bracket in B) are seen. (C,D) In Ccn1+/− pups, fusion of the septum primum (arrowheads) with the cushion tissue was unable to occur, leaving a detached septum primum and foramen ovale (brackets in D). Formation of the septum secundum (arrows) appeared normal. (E,F) In hearts of 10 month old mice, the fossa ovalis (arrow, E) was closed in the WT but the embryonic foramen ovale remained patent (arrow, F) in the Ccn1+/− mouse. White bars: 100 μm, black bar: 1 mm.
Septation of the common atrial chamber begins at E10.5 with the formation of the septum primum, which approaches the AV cushion tissue from the atrial chamber roof. The opening between the two atrial chambers, the ostium primum, is obliterated upon fusion of the septum primum with the AV cushion and is replaced by the ostium secundum, which forms by apoptosis in the septum primum. The septum secundum then forms, with the foramen ovale serving as the opening between the atria3. Postnatal fusion of the septum primum and septum secundum closes the openings of both septa and ensures complete separation of the left and right atria after birth when pulmonary circulation is established (Fig. 3A,B). In affected Ccn1+/− neonates, the septum primum failed to fuse completely with the cushion tissue and the ostium primum remained (Fig. 3C,D). Consistent with a role in this fusion event, Ccn1 was expressed in the AV cushion where fusion with the mesenchymal cap of the septum primum occurs (Fig. 1F). Although formation of the septum secundum was successful, postnatal fusion of the two septa was precluded by the defective septum primum. Consequently, blood cells were trapped between the atria as the hemodynamic flow was in disarray (Fig. 3C,D), and the foraman ovale remained patent in affected adult Ccn1+/− mice (Fig. 3E,F).
EMT and differentiation of cushion mesenchyme
Central to valvuloseptal morphogenesis is the AV cushion tissue, which forms when endocardial cells undergo EMT and invade into the cardiac jelly2, 18. We therefore examined whether EMT occurs in AV cushion tissue explants isolated from E9.5 Ccn1−/ − embryos. Endocardial cells that successfully undergo EMT are capable of migrating into collagen gel on which the explant is cultured (Fig. 4A-D). The percentages of cells migrating into collagen gel in wild-type and Ccn1−/ − cushion explants were indistinguishable (Fig. 4E), indicating that Ccn1 is not essential for EMT. We next investigated the differentiation of the mesenchymal cells within the cushion tissue using molecular markers. Smooth muscle α-actin is expressed as mesenchymal cells gain characteristics of smooth muscle-like myofibroblasts19, and Mox-1 is a marker of more differentiated mesenchymal cells in the cushion20. Expression of both smooth muscle α-actin and Mox-1 was similar in wild-type and Ccn1−/ − embryonic hearts (Fig. 4F), indicating that EMT and further differentiation of cushion tissue mesenchymal cells were successful in Ccn1−/ − embryos.
Figure 4.
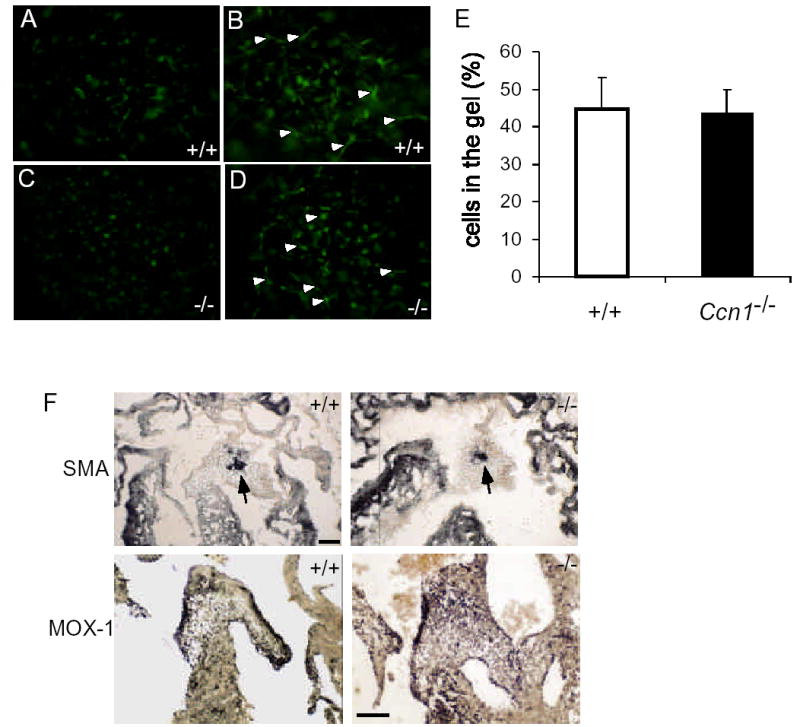
Epithelial-mesenchymal-transformation in AV cushion explant culture. E9.5 AV cushions were isolated from Ccn1+/+ (A,B) and Ccn1−/− (C,D) embryos and cultured on collagen gel for 48 hrs. (A,C) Undifferentiated cells remaining on the gel surface. (B,D) Differentiated cells (arrowheads) migrated into the collagen gel. (E) Percentages of cells migrated into the collagen gel over the total number of cells in AV explants from Ccn1+/+ (n=6) and Ccn1−/− (n=5) embryos (P>0.80). (F) Immunostaining of smooth muscle α-actin (SMA, arrows) in E13.5 cushions showed similar patterns in Ccn1+/+ and Ccn1−/− embryonic hearts. Mox-1 expression was detected in E14.5 cushions and developing valves in both wild-type and Ccn1−/− embryos by in situ hybridization. Bars: 100 μm.
Cell proliferation
Valvuloseptal morphogenesis begins with the expansion of the cushion tissue and fusion with the AV septa, followed by extensive remodeling and shrinkage to form the mature AV septa and valves. Coordinated with these morphogenetic events are highly regulated cell proliferation and programmed cell death in the cushion tissue21, and BMP-4 deficiency causes septal defects due to reduced cushion mesenchymal cell proliferation22. As judged by Ki67 staining, the percentages of proliferating cells in WT and Ccn1-null AV cushion were similar as the cushion tissue actively expands at E11.5 and E12.5 (Fig. 5). Thus, cell proliferation in the AV cushion is not affected by Ccn1 deficiency.
Figure 5.
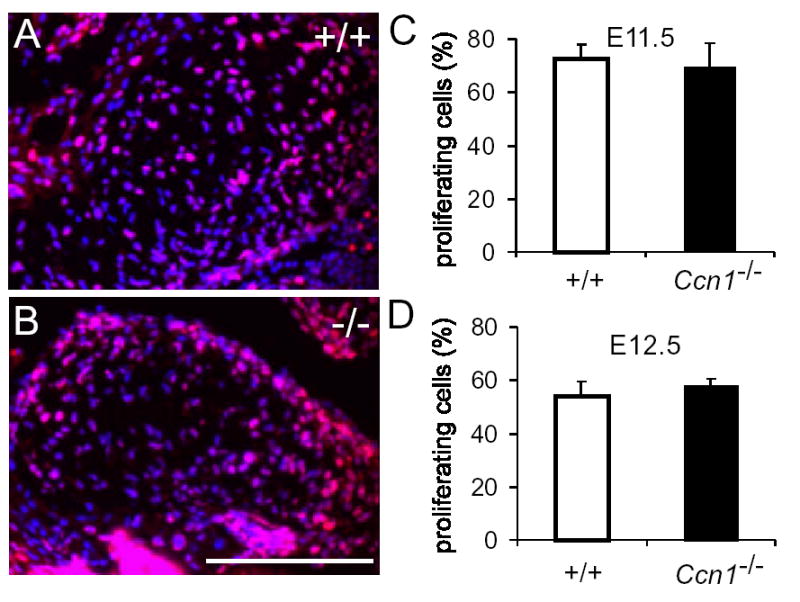
Cell proliferation in the AV cushion tissue. E11.5 (shown in A, B) and E12.5 AV cushion tissues of wild-type (+/+) and Ccn1−/− embryos were immunostained for Ki67 (red) and countered stained DAPI (blue). Results were quantified, showing equivalent proliferation rates in wild-type and Ccn1−/− embryos in E11.5 (C, n=6 each; P>0.59) and E12.5 embryos (D, n=5 each, P>0.34).
Precocious apoptosis in the Ccn1-deficient cushion tissue
CCN1 can induce apoptosis in fibroblasts but promote endothelial cell survival in vitro16, 23. Consistent with its pro-survival function in endothelial cells, vascular cells of large vessels in Ccn1-null mice suffer apoptosis10. As previously reported, very few apoptotic cells were detected in the WT AV cushion tissue as it began to fuse with the AV septa at E12.5 (Fig. 6A,B)24. By contrast, a large number of TUNEL- or activated caspase-3-positive apoptotic cells were detected in the Ccn1−/ − cushion tissue proximal to the atrial septum (Fig. 6A-E, K-N) where Ccn1 is expressed (Fig. 1F). These results suggest that CCN1 is required for cushion tissue cell survival at this embryonic stage, and its absence results in apoptosis. This early onset of apoptosis in the E12.5 cushion tissue may be detrimental to atrial septum fusion, leading to the ASD phenotype. By E13.5, the atrial septum primum fusion has occurred, and expression of Ccn1 was diminished in the atrial fusion site of the AV cushion (Fig. 1G), correlating with a dramatic increase in apoptosis in the WT cushion tissue (Fig. 6F,G), consistent with previous reports24. Thus, while programmed cell death occurs in the AV cushion upon fusion with the atrial septum21, precocious onset of apoptosis in Ccn1-null mice may prevent the fusion event from occurring successfully. Although Ccn1 deficiency also leads to VSD, no abnormal cell death was detected at the fusion site between the ventricular septum and the cushion tissue (Fig. 6).
Figure 6.
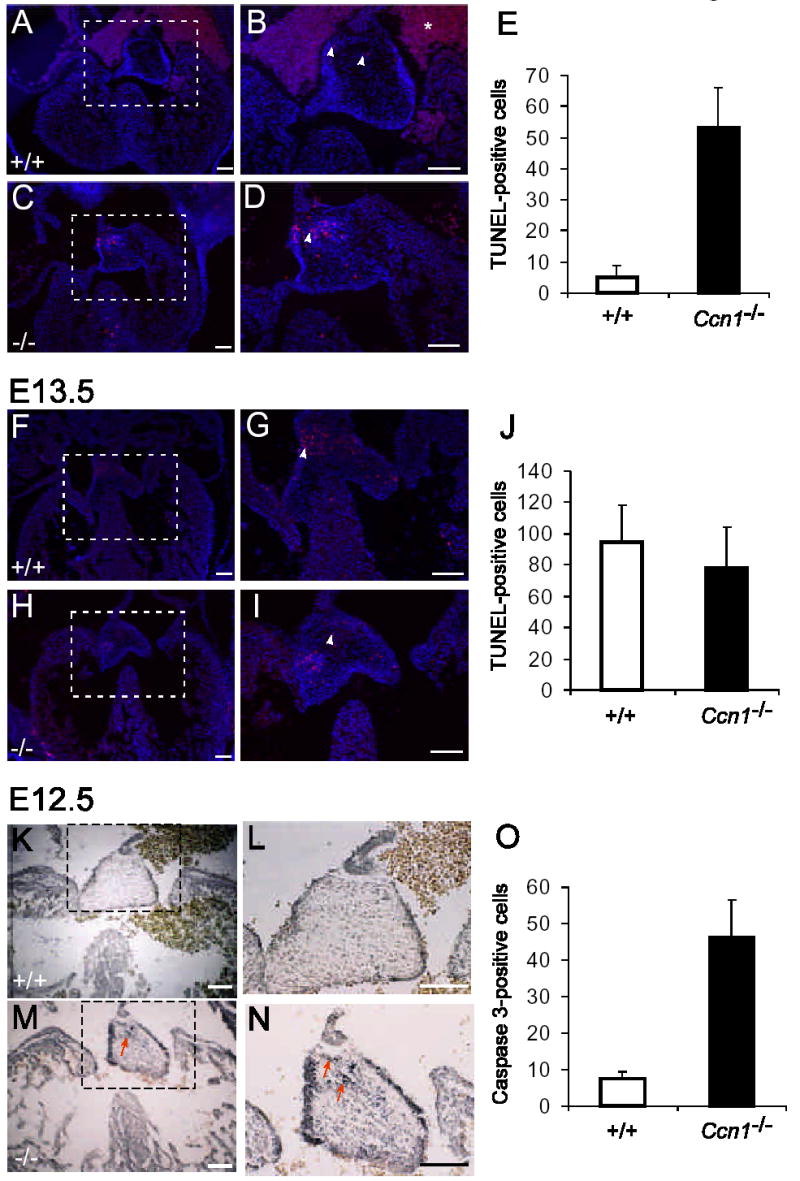
Apoptosis in the AV cushion. Apoptotic cells were identified in sections of embryonic hearts using TUNEL assay and counterstained with DAPI (A-J, arrowheads), or by immunohistochemical staining for activated caspase-3 (K-O, arrows). Higher magnifications of dashed boxes in A, C, F, H, K, and M are shown in B, D, G, I, L, and N, respectively. Precocious apoptosis was observed in E12.5 Ccn1−/− AV cushion (C,D), compared to the Ccn1+/+ cushion (A,B) by either TUNEL assay (E; P<0.001) or immunohistochemical staining of activated caspase-3 (K-O; P<0.001). At E13.5, similar numbers of TUNEL-positive cells were observed in AV cushions and valvular leaflets of both WT (F, G) and Ccn1−/− (H, I) hearts (J; P>0.30). Apoptotic cells were counted from transverse sections of AV cushions of Ccn1+/+ and Ccn1−/− hearts (each group n=5), and the average numbers of TUNEL-positive cells in 10 μm sections are shown in E, J, and O. Error bars are standard deviations. The star in B indicates background staining from red blood cells. Bars = 100 μm.
Gelatinase deficiency in the Ccn1-null heart
Matrix metalloproteinases (MMPs) and their regulators are thought to be involved in aspects of cardiac development as regulators of growth factors activity and ECM composition25, 26. Several MMPs have been identified as key targets of CCN1-regulated signaling pathways in fibroblasts27, prompting us to examine MMP activity in embryos. Gelatinases (MMP2 and MMP9) degrade type IV collagen in basement membrane and are of particular importance in tissue remodeling. Gelatinase activity was detected by in situ gelatin zymography in the E12.5 WT heart (Fig. 7A,D). This activity was blocked by the inhibitor 1, 10-phenanthroline (PNT), confirming that it is due to a metalloprotease (Fig. 7B,E). By contrast, gelatinase activity was greatly diminished in the Ccn1−/ − heart, particularly in the muscular component of the IVS and the valvular leaflets where defects are observed (Fig. 7C,F). To test whether CCN1 is able to directly regulate the expression of gelatinases, which are synthesized as zymogens and are proteolytically cleaved to the active forms, we treated H9c2 cardiomyocytes with purified CCN1 protein. Indeed, CCN1 treatment dramatically increased both secreted pro- and activated-MMP-2 (Fig. 7G). Thus, CCN1 is an inducer of MMP2 in cardiomyocytes, and a deficiency in gelatinase activities in Ccn1-null mice may compromise the matrix remodeling process that is crucial for fusion of the IVS and AV cushion tissue.
Figure 7.
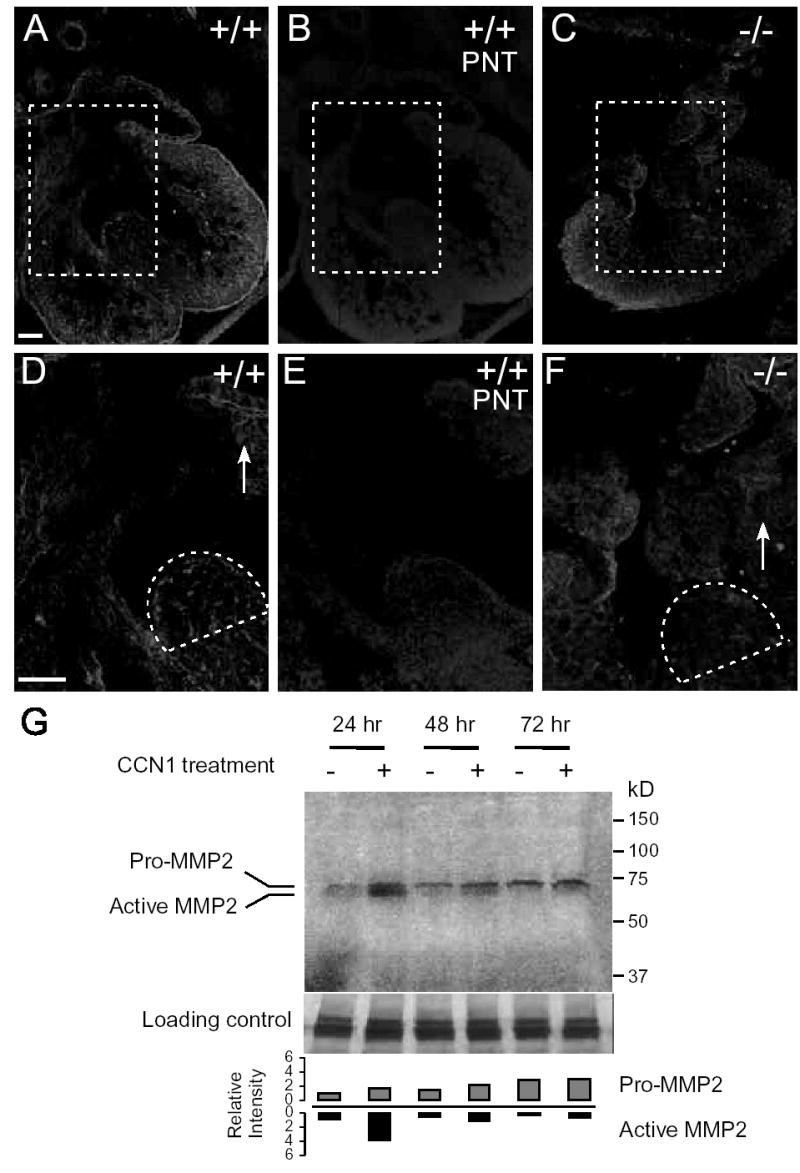
Gelatinase zymography. Gelatinase activity was visualized by in situ zymography as bright fluorescence in E12.5 hearts (A-F). D, E and F are the higher magnification views of dashed boxes in A, B and C, respectively. Enzyme activities can be seen in the hearts of wild-type E12.5 embryos (A,D), including the tip of the ventricular septum (dashed area in D) and the valvular leaflets (arrow). (B,E) The MMP inhibitor 1,10-phenanthroline (PNT) blocked this activity. (C,F) Ccn1−/− hearts show deficiency in gelatinase activity at the ventricular septum (dashed) and the valvular leaflets (arrow). (G) Conditioned media of H9c2 cardiomyocytes cultured with or without purified CCN1 were collected 24, 48 or 72 hrs after treatment and analyzed by gelatin zymogram, showing induction of active MMP-2 by CCN1 at 24 hrs. The intensity of bands corresponding to either the pro- or active MMP-2 were quantified with ImageJ (NIH). The same samples were electrophoresed in acrylamide gel and Coomassie blue-stained, showing equal protein in each sample (lower panel). Four independent experiments were performed with similar results.
DISCUSSION
ECM signaling plays a critical role in developmental processes. As demonstrated in this study, the matricellular protein CCN1 is indispensable for cardiovascular development, and nullizygosity in Ccn1 results in severe AVSD and embryonic death. Although heterozygous Ccn1 mice are largely viable, 20% of them display ostium primum ASD. Furthermore, CCN1 maps to the same human chromosomal location (1p21-p31) as that of a susceptibility gene for non-syndromic AVSD, AVSD1, mutations in which also leads to predominantly ostium primum ASD4, 13. These results provide the first evidence that haploinsufficiency in an ECM protein can lead to autosomal dominant ASD, and implicate CCN1 as a candidate gene for AVSD in humans.
CHDs are the most common group of birth defects in humans, encountered in ~1% of live births and 10% of stillborns1. Consistent with the complex nature of cardiac development, nullizygosity of a multitude of genes in mice result in heart defects28. Whereas human CHDs exhibit predominantly autosomal dominance rather than homozygous recessive modes of transmission, only a very few genes, encoding cardiac-specific transcription factors, have been identified to cause autosomal dominant CHD due to haploinsufficiency in mice. Heterozygous mutations in these transcription factor genes, including Tbx1 (DiGeorge syndrome)29, Tbx5 (Holt-Oram syndrome)30, and Nkx2-5 (conduction defects, tetralogy of Fallot)31, also cause CHDs in humans. Since CCN1 is a matricellular protein that acts through binding to integrins and HSPGs, Ccn1 mutant mice provide a unique animal model for addressing the role of matrix signaling in heart development. Aside from Ccn1 and AVSD1, another genetic locus known to be associated with non-syndromic AVSD is CRELD1, which encodes a predicted matricellular cell adhesion molecule based on its sequence6, 32. Although the activities of CRELD1 have not yet been examined, these findings are consistent with the notion that matrix signaling is crucial for AV septation and valvular formation.
The AV cushion tissue is central to valvuloseptal morphogenesis, and malformations in the cushion tissue, such as those caused by deficiency in versican, hyaluronan synthase 2 or the TGF-β type III receptor, result in AVSD28, 33. Although Ccn1 is not required for the formation of the AV cushion, EMT of endocardial cells, or proliferation or further differentiation of cushion mesenchymal cells, Ccn1 deficiency leads to precocious apoptosis in the AV cushion (Fig. 4–6). Programmed cell death plays a key role in cardiac development and is often found at causative factor in the AVSD phenotype of transforming growth factor-β2 (TGF-β2)-deficient mice34. The early onset of apoptosis in the AV cushion in Ccn1-deficient mice is likely an impediment to successful fusion between the cushion tissue and the atrial septum, and may lead to defects in tissue remodeling. This finding also indicates that CCN1 is required for cushion tissue mesenchymal cell survival. CCN1 is known to promote the survival of endothelial cells in culture through the engagement of integrin αvβ3 16, 23, consistent with aberrant vascular cell apoptosis in Ccn1-null embryos10. CCN1 may serve a similar pro-survival function for the endocardium-derived cushion mesenchymal cells, and thus Ccn1 deficiency results in apoptosis.
MMPs and their inhibitors have been shown to mediate various aspects of cardiac development, including heart tube formation, cardiac neural crest migration, and EMT of the endocardial cushions25. The Ccn1-null hearts displayed a significant reduction in gelatinase activities in the interventricular septum, left ventricular wall, and the AV valves, whereas residual activity was detected in the right ventricular wall (Fig. 7). CCN1 regulates the expression of genes involved in matrix remodeling in fibroblasts27, and upregulates MMP-2 levels and enhances its activation in cardiomyocytes (Fig. 7G). Although deficiency in gelatinase activities may contribute to cardiac defects, it is by itself insufficient to cause cardiac abnormalities. No heart defects have been reported in mouse models with null mutations in MMP-2 (gelatinase A), MMP-9 (gelatinase B), or both35–37. However, loss of MMP-2 and/or -9 protects against the tissue fibrosis seen following myocardial infarction in adults, demonstrating that these gelatinases do have significant effects on myocardiocytes in vivo38.
Ccn2 (CTGF, connective tissue growth factor) is a closely related homologue of Ccn1, and both genes are highly expressed in the myocardium and AV cushion during heart development, and play a role in adaptation of the heart to cardiovascular stress39, 40. Although Ccn2-null mice show apparently normal heart development through birth41, there may be significant functional redundancies between these genes that allow Ccn1 to compensate for the loss of Ccn2 in the cardiovascular system, since CCN1 and CCN2 have similar activities in vitro42. Hence, while specific functions of CCN1 are clearly indispensable, the potential roles of CCN2 and possible overlapping functions between CCN1 and CCN2 in cardiac development remain to be established.
How Ccn1 is integrated into the regulatory circuits that control heart development is currently unknown, although consensus binding sites for the cardiac transcription factors Nkx2.5 and GATA-4 are found on its promoter43. Mechanistically, CCN1 can function to regulate cell survival/death and the expression of genes involved in matrix remodeling. In addition, CCN1 may potentially modulate the activities of other regulatory pathways through interactions with growth or morphogenic factors or their receptors44, 45. Since haploinsufficiency in Ccn1 leads to ostium primum ASD resembling that observed in man, the possibility that mutations in CCN1 may lead to clinical AVSD clearly warrants further investigation.
Acknowledgments
We thank Grady Chang and Theresa Lo for technical assistance, and Dr. Karen Lyons for a critical reading of the manuscript.
Footnotes
DISCLOSURE
None
SOURCES OF FUNDING
This work was supported by NIH grant CA46565.
References
- 1.Ferencz C. Armonk, N.Y.: Futura Publishing; 1997. Genetic and environmental risk factors of major cardiovascular malformations: the Baltimore-Washington infant study 1981–1989. [Google Scholar]
- 2.Eisenberg LM, Markwald RR. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res. 1995;77:1–6. doi: 10.1161/01.res.77.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Anderson RH, Webb S, Brown NA, Lamers W, Moorman A. Development of the heart: (2) Septation of the atriums and ventricles. Heart. 2003;89:949–58. doi: 10.1136/heart.89.8.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheffield VC, Pierpont ME, Nishimura D, Beck JS, Burns TL, Berg MA, Stone EM, Patil SR, Lauer RM. Identification of a complex congenital heart defect susceptibility locus by using DNA pooling and shared segment analysis. Hum Mol Genet. 1997;6:117–21. doi: 10.1093/hmg/6.1.117. [DOI] [PubMed] [Google Scholar]
- 5.Robinson SW, Morris CD, Goldmuntz E, Reller MD, Jones MA, Steiner RD, Maslen CL. Missense mutations in CRELD1 are associated with cardiac atrioventricular septal defects. Am J Hum Genet. 2003;72:1047–52. doi: 10.1086/374319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zatyka M, Priestley M, Ladusans EJ, Fryer AE, Mason J, Latif F, Maher ER. Analysis of CRELD1 as a candidate 3p25 atrioventicular septal defect locus (AVSD2) Clin Genet. 2005;67:526–8. doi: 10.1111/j.1399-0004.2005.00435.x. [DOI] [PubMed] [Google Scholar]
- 7.Bornstein P. Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J Cell Biol. 1995;130:503–6. doi: 10.1083/jcb.130.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lau LF, Lam SCT. Integrin-mediated CCN functions. In: Perbal B, Takigawa M, editors. CCN proteins: a new family of cell growth and differentiation regulators. London: Imperial College Press; 2005. pp. 61–79. [Google Scholar]
- 9.Babic AM, Kireeva ML, Kolesnikova TV, Lau LF. CYR61, product of a growth factor-inducible immediate-early gene, promotes angiogenesis and tumor growth. Proc Natl Acad Sci U S A. 1998;95:6355–60. doi: 10.1073/pnas.95.11.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mo FE, Muntean AG, Chen CC, Stolz DB, Watkins SC, Lau LF. CYR61 (CCN1) Is Essential for Placental Development and Vascular Integrity. Mol Cell Biol. 2002 December;22(24):8709–20. doi: 10.1128/MCB.22.24.8709-8720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leu S-J, Chen N, Chen C-C, Todorovic V, Bai T, Juric V, Liu Y, Yan G, Lam SCT, Lau LF. Targeted mutagenesis of the matricellular protein CCN1 (CYR61): selective inactivation of integrin α6β1-heparan sulfate proteoglycan coreceptor-mediated cellular activities. J Biol Chem. 2004;279:44177–87. doi: 10.1074/jbc.M407850200. [DOI] [PubMed] [Google Scholar]
- 12.Chen N, Leu S-J, Todorovic V, Lam SCT, Lau LF. Identification of a novel integrin αvβ3 binding site in CCN1 (CYR61) critical for pro-angiogenic activities in vascular endothelial cells. J Biol Chem. 2004;279:44166–76. doi: 10.1074/jbc.M406813200. [DOI] [PubMed] [Google Scholar]
- 13.Jay P, Berge-Lefranc JL, Marsollier C, Mejean C, Taviaux S, Berta P. The human growth factor-inducible immediate early gene, CYR61, maps to chromosome 1p. Oncogene. 1997;14:1753–7. doi: 10.1038/sj.onc.1200986. [DOI] [PubMed] [Google Scholar]
- 14.Dor Y, Camenisch TD, Itin A, Fishman GI, McDonald JA, Carmeliet P, Keshet E. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development. 2001;128:1531–8. doi: 10.1242/dev.128.9.1531. [DOI] [PubMed] [Google Scholar]
- 15.Oh LY, Larsen PH, Krekoski CA, Edwards DR, Donovan F, Werb Z, Yong VW. Matrix metalloproteinase-9/gelatinase B is required for process outgrowth by oligodendrocytes. J Neurosci. 1999;19:8464–75. doi: 10.1523/JNEUROSCI.19-19-08464.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leu S-J, Lam SCT, Lau LF. Proangiogenic activities of CYR61 (CCN1) mediated through integrins αvβ3 and α6β1 in human umbilical vein endothelial cells. J Biol Chem. 2002;277:46248–55. doi: 10.1074/jbc.M209288200. [DOI] [PubMed] [Google Scholar]
- 17.Adams RH, Porras A, Alonso G, Jones M, Vintersten K, Panelli S, Valladares A, Perez L, Klein R, Nebreda AR. Essential role of p38alpha MAP kinase in placental but not embryonic cardiovascular development. Mol Cell. 2000;6:109–16. [PubMed] [Google Scholar]
- 18.Wessels A, Markwald R. Cardiac morphogenesis and dysmorphogenesis. I. Normal development. Methods Mol Biol. 2000;136:239–59. doi: 10.1385/1-59259-065-9:239. [DOI] [PubMed] [Google Scholar]
- 19.Nakajima Y, Mironov V, Yamagishi T, Nakamura H, Markwald RR. Expression of smooth muscle alpha-actin in mesenchymal cells during formation of avian endocardial cushion tissue: a role for transforming growth factor beta3. Dev Dyn. 1997;209:296–309. doi: 10.1002/(SICI)1097-0177(199707)209:3<296::AID-AJA5>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 20.Candia AF, Wright CV. Differential localization of Mox-1 and Mox-2 proteins indicates distinct roles during development. Int J Dev Biol. 1996;40:1179–84. [PubMed] [Google Scholar]
- 21.Zhao Z, Rivkees SA. Programmed cell death in the developing heart: regulation by BMP4 and FGF2. Dev Dyn. 2000;217:388–400. doi: 10.1002/(SICI)1097-0177(200004)217:4<388::AID-DVDY6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 22.Jiao K, Kulessa H, Tompkins K, Zhou Y, Batts L, Baldwin HS, Hogan BL. An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. 2003;17:2362–7. doi: 10.1101/gad.1124803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Todorovic V, Chen C-C, Hay N, Lau LF. The matrix protein CCN1 (CYR61) induces apoptosis in fibroblasts. J Cell Biol. 2005;171:559–68. doi: 10.1083/jcb.200504015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdelwahid E, Pelliniemi LJ, Jokinen E. Cell death and differentiation in the development of the endocardial cushion of the embryonic heart. Microsc Res Tech. 2002;58:395–403. doi: 10.1002/jemt.10159. [DOI] [PubMed] [Google Scholar]
- 25.Song W, Jackson K, McGuire PG. Degradation of type IV collagen by matrix metalloproteinases is an important step in the epithelial-mesenchymal transformation of the endocardial cushions. Dev Biol. 2000;227:606–17. doi: 10.1006/dbio.2000.9919. [DOI] [PubMed] [Google Scholar]
- 26.Brauer PR, Cai DH. Expression of tissue inhibitor of metalloproteinases (TIMPs) during early cardiac development. Mech Dev. 2002;113:175–9. doi: 10.1016/s0925-4773(02)00016-3. [DOI] [PubMed] [Google Scholar]
- 27.Chen C-C, Mo F-E, Lau LF. The angiogenic inducer Cyr61 induces a genetic program for wound healing in human skin fibroblasts. J Biol Chem. 2001;276:47329–37. doi: 10.1074/jbc.M107666200. [DOI] [PubMed] [Google Scholar]
- 28.Srivastava D, Olson EN. A genetic blueprint for cardiac development. Nature. 2000;407:221–6. doi: 10.1038/35025190. [DOI] [PubMed] [Google Scholar]
- 29.Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27:286–91. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- 30.Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE, Seidman JG. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell. 2001;106:709–21. doi: 10.1016/s0092-8674(01)00493-7. [DOI] [PubMed] [Google Scholar]
- 31.Biben C, Weber R, Kesteven S, Stanley E, McDonald L, Elliott DA, Barnett L, Koentgen F, Robb L, Feneley M, Harvey RP. Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2-5. Circ Res. 2000;87:888–95. doi: 10.1161/01.res.87.10.888. [DOI] [PubMed] [Google Scholar]
- 32.Rupp PA, Fouad GT, Egelston CA, Reifsteck CA, Olson SB, Knosp WM, Glanville RW, Thornburg KL, Robinson SW, Maslen CL. Identification, genomic organization and mRNA expression of CRELD1, the founding member of a unique family of matricellular proteins. Gene. 2002;293:47–57. doi: 10.1016/s0378-1119(02)00696-0. [DOI] [PubMed] [Google Scholar]
- 33.Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A, Jr, Kubalak S, Klewer SE, McDonald JA. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest. 2000;106:349–60. doi: 10.1172/JCI10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartram U, Molin DG, Wisse LJ, Mohamad A, Sanford LP, Doetschman T, Speer CP, Poelmann RE, Gittenberger-de Groot AC. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-beta(2)-knockout mice. Circulation. 2001;103:2745–52. doi: 10.1161/01.cir.103.22.2745. [DOI] [PubMed] [Google Scholar]
- 35.Itoh T, Ikeda T, Gomi H, Nakao S, Suzuki T, Itohara S. Unaltered secretion of beta-amyloid precursor protein in gelatinase A (matrix metalloproteinase 2)-deficient mice. J Biol Chem. 1997;272:22389–92. doi: 10.1074/jbc.272.36.22389. [DOI] [PubMed] [Google Scholar]
- 36.Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–22. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perez SE, Cano DA, Dao-Pick T, Rougier JP, Werb Z, Hebrok M. Matrix metalloproteinases 2 and 9 are dispensable for pancreatic islet formation and function in vivo. Diabetes. 2005;54:694–701. doi: 10.2337/diabetes.54.3.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Panek AN, Bader M. Matrix reloaded: the matrix metalloproteinase paradox. Hypertension. 2006;47:640–1. doi: 10.1161/01.HYP.0000208603.97395.44. [DOI] [PubMed] [Google Scholar]
- 39.Chuva de Sousa Lopes SM, Feijen A, Korving J, Korchynskyi O, Larsson J, Karlsson S, ten Dijke P, Lyons KM, Goldschmeding R, Doevendans P, Mummery CL. Connective tissue growth factor expression and Smad signaling during mouse heart development and myocardial infarction. Dev Dyn. 2004;231:542–50. doi: 10.1002/dvdy.20162. [DOI] [PubMed] [Google Scholar]
- 40.Hilfiker-Kleiner D, Kaminski K, Kaminska A, Fuchs M, Klein G, Podewski E, Grote K, Kiian I, Wollert KC, Hilfiker A, Drexler H. Regulation of proangiogenic factor CCN1 in cardiac muscle: impact of ischemia, pressure overload, and neurohumoral activation. Circulation. 2004;109:2227–33. doi: 10.1161/01.CIR.0000127952.90508.9D. [DOI] [PubMed] [Google Scholar]
- 41.Ivkovic S, Yoon BS, Popoff SN, Safadi FF, Libuda DE, Stephenson RC, Daluiski A, Lyons KM. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development. 2003;130:2779–91. doi: 10.1242/dev.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen C-C, Chen N, Lau LF. The angiogenic factors Cyr61 and CTGF induce adhesive signaling in primary human skin fibroblasts. J Biol Chem. 2001;276:10443–52. doi: 10.1074/jbc.M008087200. [DOI] [PubMed] [Google Scholar]
- 43.Latinkic BV, O'Brien TP, Lau LF. Promoter function and structure of the growth factor-inducible immediate early gene cyr61. Nucleic Acids Res. 1991;19:3261–7. doi: 10.1093/nar/19.12.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abreu JG, Ketpura NI, Reversade B, De Robertis EM. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat Cell Biol. 2002;4:599–604. doi: 10.1038/ncb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Latinkic BV, Mercurio S, Bennett B, Hirst EM, Xu Q, Lau LF, Mohun TJ, Smith JC. Xenopus Cyr61 regulates gastrulation movements and modulates Wnt signalling. Development. 2003;130:2429–41. doi: 10.1242/dev.00449. [DOI] [PubMed] [Google Scholar]