

Abstract
β-catenin, a pivotal component of the Wnt-signaling pathway, binds to and serves as a transcriptional coactivator for the T-Cell Factor/Lymphoid Enhancer Factor (TCF/LEF) family of transcriptional activator proteins, and also for the androgen receptor (AR), a nuclear receptor. Three components of the p160 nuclear receptor coactivator complex, including CARM1, p300/CBP, and GRIP1 (one of the p160 coactivators), bind to and cooperate with β-catenin to enhance transcriptional activation by TCF/LEF and AR. Here we report that another component of the p160 nuclear receptor coactivator complex, the coiled-coil coactivator (CoCoA), directly binds to and cooperates synergistically with β-catenin as a coactivator for AR and TCF/LEF. CoCoA uses different domains to bind GRIP1 and β-catenin, and it uses different domains to transmit the activating signal to the transcription machinery, depending on whether it is bound to GRIP1 or β-catenin. CoCoA associated specifically with the promoters of transiently transfected and endogenous target genes of TCF/LEF, and reduction of the endogenous CoCoA level decreased the ability of TCF/LEF and β-catenin to activate transcription of transient and endogenous target genes. Thus, CoCoA uses different combinations of functional domains to serve as a physiologically relevant component of the Wnt/β-catenin signaling pathway and the androgen signaling pathway.
The Wnt/β-catenin-signaling cascade plays important roles in developmental processes. Inappropriate activation of this pathway is associated with a variety of cancers such as colorectal cancer and hepatocellular carcinoma (1,2). Activation of this pathway by extracellular Wnt ligands results in increased intracellular levels of β-catenin, which consists of N- and C-terminal activation domains flanking twelve armadillo repeats and serves as a coactivator for various DNA-binding transcription factors. In the absence of stimulation by Wnt ligand, β-catenin levels are maintained at a low level through a specific degradation mechanism. Phosphorylation of β-catenin by glycogen synthase kinase GSK3β targets β-catenin for ubiquitylation and degradation via ubiquitin-mediated proteasomal degradation (2,3). Activation of the cell surface Frizzled receptor by binding of the Wnt ligand activates a signaling pathway which leads to inactivation of GSK3β by destabilizing its complex with axin and the adenomatous polyposis coli, or APC, tumor suppressor protein. The resulting reduced degradation of β-catenin leads to enhanced cellular levels of β-catenin, which allows its nuclear translocation and accumulation. In the nucleus β-catenin binds to and serves as a primary coactivator for the T-Cell Factor/Lymphoid Enhancer Factor (TCF/LEF) proteins (4,5). In so doing, β-catenin displaces the corepressors Groucho (6) and CtBP (7) and thus converts the TCF/LEF complex from a transcriptional repressor to a transcriptional activator.
β-catenin also serves as a coactivator for the androgen receptor (AR) (8,9), which is a member of the nuclear receptor (NR) family of hormone-regulated transcriptional activator proteins. Binding of hormone to AR results in a conformational change which allows AR to associate with specific target genes, either by direct binding of specific enhancer elements or through protein-protein interactions with other DNA-bound transcription factors (10,11). AR recruits a variety of coregulator proteins to the target gene promoter, and these coregulators mediate the activation or repression of transcription by modulation of chromatin conformation and recruitment and activation of RNA polymerase II and its associated transcription factors (11–13). The fact that more than 200 different putative coregulator proteins for NRs have been identified over the past decade (B. W. O’Malley and R. M. Evans, Nuclear Receptor Signaling Atlas, http://www.nursa.org/index.cfm) indicates that the process of transcriptional regulation is extremely complex, involving distinct contributions from many different coregulator complexes. For example, the TRAP/DRIP/Mediator complex helps to recruit and activate RNA polymerase II, and Swi/Snf is an ATP-dependent chromatin remodeling complex (12). The p160 coactivator complex also contributes to chromatin remodeling, but by a different mechanism that involves acetylation and methylation of histones (12,14,15). The p160 complex is anchored to the hormone-activated, DNA-bound NR by one of the three p160 coactivator proteins, which include SRC-1, GRIP1/TIF2, and pCIP/ACTR/AIB1/RAC3/TRAM1. The p160 protein is thus a primary coactivator, which serves as a scaffold to recruit a variety of secondary coactivators (16). These include the protein arginine methyltransferase CARM1 (17), the protein acetyltransferases p300 and CBP (18–20), and the coiled-coil coactivator CoCoA (21), which contributes to transcriptional activation by an unknown mechanism.
The mechanism by which β-catenin contributes to transcriptional activation, after binding to AR or TCF/LEF has recently begun to emerge. β-catenin can bind to various components of the p160 nuclear receptor coactivator complex, including GRIP1 (9,22), CARM1 (23), and p300/CBP (24–26); and β-catenin cooperates synergistically as a coactivator for AR and TCF/LEF with each of these NR coactivators. To further investigate the mechanism of β-catenin coactivator function and the collaboration between β-catenin and the p160 coactivator complex, we tested whether the recently identified coiled-coil coactivator CoCoA can bind to and cooperate with β-catenin. CoCoA is the product of the human calcoco1 gene and functions as a secondary coactivator for NRs. That is, it does not bind directly to NRs or act by itself as a NR coactivator; instead, it is apparently recruited to the promoter through its contact with the N-terminal bHLH-PAS domain of p160 coactivators, and the coactivator activity of CoCoA depends on the co-expression of a p160 coactivator (21). Here we demonstrate that CoCoA can bind to β-catenin and act as a secondary coactivator for the AR and TCF/LEF pathways through that interaction. We also found that CoCoA uses different domains for binding to and activating transcription in cooperation with β-catenin versus p160 coactivators.
MATERIALS AND METHODS
Plasmids
Plasmids pSG5.HA-β-catenin encoding chicken β-catenin with an N-terminal hemagglutinin (HA) epitope tag, pM-β-catenin encoding Gal4 DNA binding domain (DBD) fused to β-catenin, pSG5.HA-LEF1 encoding HA-tagged LEF1, pSG5.HA-GRIP1, pGEX-4T1-β-catenin encoding glutathione S-transferase (GST) fused to β-catenin (for bacterial expression), and luciferase reporter gene plasmids MMTV-LUC (for AR), pGL3OT (for LEF1), and GK1-LUC (for Gal4) were previously described (22). PCR amplified β-catenin cDNA fragments were inserted into XhoI and BglII sites of pSG5.HA vector. pcDNA3.1-CoCoA/V5-His, pGEX-5X1.CoCoA, pSG5.HA-CoCoA, pSG5.HA-CoCoA(1–149), pSG5.HA-CoCoA(1–190), pSG5.HA-CoCoA(1–500), pSG5.HA-CoCoA(140–274), pSG5.HA-CoCoA(150–500), pSG5.HA-CoCoA(274–510), pSG5.HA-CoCoA(150–691), pSG5.HA-CoCoA(470–691), pSG5.HA-CoCoA(501–691), pM.CoCoA(1–500), pM.CoCoA(150–691), and pM.GRIP1(5–479) were described previously (21). pSG5.HA-CoCoA-R, encodes wild type CoCoA but contains silent mutations in codons 29–31 to make it resistant to a CoCoA-specific small interfering RNA (siRNA); the Quick Change Site-Directed Mutagenesis kit (Stratagene) was used to generate the mutations with primers 5′-CATCCCCAACACCAAaGTcGAgTGTCACTACACTTTG-3′ (forward) and 5′-CAAAGTGTAGTGACAcTCgACtTTGGTGTTGGGGATG-3′ (reverse) (mutations are shown in lower case).
Cell culture and transfections
CV-1, COS-7, and SW480 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum and penicillin and streptomycin. For reporter gene assays, CV-1 cells were transiently transfected in 12-well plates and assayed for luciferase activity as described previously (21).
Protein-protein interaction assay
GST pull-down assays were performed as described previously (21) using extracts from COS-7 cells transiently transfected with the indicated plasmids. The bound proteins were eluted from the beads and analyzed by immunoblot with anti-HA antibody (Roche Applied Science). Co-immunoprecipitation was performed as described previously (22) using rabbit antiserum against β-catenin (Santa Cruz Biotechnology) or rabbit antisera against CoCoA (21) for immunoprecipitation. Immunoblots were performed with anti-HA antibody, anti-β-catenin antibody or anti-V5 antibody (Invitrogen).
Reporter co-immunoprecipitation (Reporter Co-IP) and chromatin immunoprecipitation (ChIP) assays
Reporter Co-IP assays were performed as described previously (22). COS-7 cells in 100-mm dishes were transfected with reporter gene and expression plasmids as indicated. 24 hours after transfection, cells were treated with 20 mM LiCl to inhibit GSK3β and increase β-catenin levels, and soluble chromatin fractions were prepared 48 hours post-transfection. Immunoprecipitation was performed using 2 μg of anti-LEF1 antibody C-19 (Santa Cruz Biotechnology) or anti-β-catenin antibody, or 8 μl of an equal mixture of two rabbit antisera against CoCoA (21). ChIP assays were performed as described previously (21). Briefly, SW480 cells were cultured in 150-mm dishes and treated with 20 mM LiCl for 24 hours. Immunoprecipitation was performed as in Reporter Co-IP assays, and PCR amplification of the precipitated DNA was performed with primers spanning the cyclin D1 promoter: −795 to −502, 5′-CAAGGACCGACTGGTCAAG-3′ (forward) and 5′-ACACGTGTAAATTGCAAGAACTAA-3′ (reverse); +128 to +486, 5′-CGGGGCAGCAGAAGCGAGA-3′ (forward) and 5′- GTGAGTAGCAAAGAAACGTGG-3′ (reverse); −3892 to −3649, 5′-GGTCCTCCCCGCAGTCTTC-3′ (forward) and 5′-CTCTCCCCCGCAGTCAGG-3′ (reverse). The amount of each immunoprecipitated DNA sample was titrated to determine the linear range for the PCR reactions, and the results shown are within the linear range.
RNA interference and rescue assays
siRNA transfection into cultured mammalian cells was described previously (21). The following siRNA were used for transfection: Scramble#1 and siCoCoA#1 were described previously (21); Scramble#2: 5′-GUCCCUCGAUUACACUACCTT-3′ (sense) and 5′-GGUAGUGUAAUCGAGGGACTT-3′ (antisense); siCoCoA#2:5′-UGACCUGAUGCAGCUGAAGTT-3′ (sense) and 5′-CUUCAGCUGCAUCAGGUCATT-3′ (antisense); siβ-catenin: 5′-AGCUGAUAUUGAUGGACAGTT-3′ (sense) and 5′-CUGUCCAUCAAUAUCAGCUTT-3′ (antisense). COS-7 or CV-1 cells were transfected with siRNA duplex, reporter plasmid, and plasmids encoding the indicated proteins using Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions, and assayed for luciferase reporter activity. For RT-PCR, total RNA of transfected cells were extracted using Trizol reagent (Invitrogen), and analysis was performed using Access RT-PCR system (Promega). The following primers were used for RT-PCR: CoCoA, 5′-CACACCAGTGTCCAGTTCCAA-3′ (forward) and 5′-CTTCGTCAGCACTTTCTCACT-3′ (reverse); β-actin, 5′-CCTCGCCTTTGCCGATCC-3′ (forward) and 5′-GGATCTTCATGAGGTAGTCAGTC-3′ (reverse). siRNA rescue assays were performed by transfecting CV-1 cells with 40 pmol of siRNA using Lipofectamine 2000 according to the manufacturer’s instructions. 48 hours after siRNA transfection, reporter gene and expression plasmids were transfected using Targefect F1 (Targeting Systems) for 24 hours and cell lysates were then assayed for luciferase activity.
To assess the effect of reducing endogenous CoCoA level on the expression of an endogenous target gene of LEF1 and β-catenin, SW480 cells in 6-well plates were transfected with siRNA duplex using Lipofectamine 2000 following the manufacturer’s instructions. Total RNA was extracted with Trizol reagent (Invitrogen) and the reverse transcriptase reaction was performed with iScript cDNA Synthesis Kit (Bio-Rad Laboratories). Quantitative real-time PCR reactions were performed with Brilliant SYBR Green QPCR Master Mix (Stratagene) with the Mx3000P system (Stratagene) using the following primers: CoCoA, 5′-GACCTACATCCCCAACACCAA-3′ (forward) and 5′-CCAGGGTCACCAGTTCATCCA-3′ (reverse); Cyclin D1, 5′-GTGCTGCGGGCCATGCTGAAGG-3′ (forward) and 5′-TCGGGCCGGATGGAGTTGTCG-3′ (reverse); GAPDH, 5′-TCTGGTAAAGTGGATATTGTTG-3′ (forward) and 5′-GATGGTGATGGGATTTCC-3′ (reverse). Following PCR, a melting curve was obtained to confirm the purity of the amplification product. Relative expression levels of the target genes were obtained with the built in software of the Mx3000P system, using the standard curve method, and were normalized to the expression level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
RESULTS
CoCoA binds to β-catenin
Given that members of the p160 NR coactivator complex, including CARM1 (23), p300/CBP (24-26), and GRIP1 (9,22) have been shown to associate with β-catenin, we tested whether another component of the p160 NR coactivator complex, CoCoA, can interact with β-catenin. To test for binding in vitro, HA-tagged CoCoA (Fig. 1A, lane 1) or β-catenin (lane 4) was expressed in COS-7 cells by transient transfection, and the COS-7 cell extracts were incubated with GST-β-catenin or GST-CoCoA, respectively. CoCoA effectively bound to GST-β-catenin (lane 3) but not to GST (lane 2). Conversely, β-catenin in COS-7 cell extracts bound to GST-CoCoA (lane 6) but not to GST (lane 5).
FIG. 1.
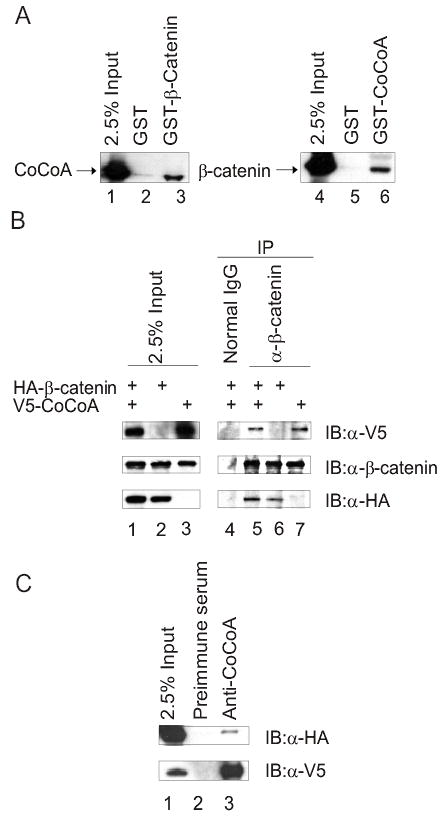
CoCoA interacts with β-catenin in vitro and in vivo. (A) Plasmids encoding HA-tagged CoCoA or β-catenin were transfected into COS-7 cells. Cell lysates were collected 48 h after transfection and incubated with glutathione-Sepharose beads containing GST (lanes 2 & 5), GST-β-catenin (lane 3), or GST-CoCoA (lane 6) for GST pull-down assays. Bound proteins were eluted with SDS sample buffer and analyzed by immunoblot with antibodies against the HA-epitope tag. A portion of the amount of cell lysate incubated with beads was loaded as control (2.5% Input, lanes 1 & 4). (B) Co-immunoprecipitation was performed with extracts of COS-7 cells transfected with 5βg each of plasmids encoding HA-tagged β-catenin and V5-tagged CoCoA, as indicated. After 48 h cell lysates were immunoprecipitated with anti-β-catenin antibody or normal rabbit IgG as control and analyzed by immunoblots using anti-V5 antibody (top panel), anti-β-catenin antibody (middle panel), or anti-HA antibody (bottom panel). (C) Co-immunoprecipitation was performed as in B, using anti-CoCoA antisera for immunoprecipitation and anti-HA antibody (top panel) or anti-V5 antibody (lower panel) for immunoblots.
To test binding in vivo by co-immunoprecipitation, HA-tagged β-catenin and V5-tagged CoCoA were expressed in COS-7 cells (Fig. 1B, lane 1–3). Anti-β-catenin antibody effectively precipitated CoCoA (lane 5), even in the absence of exogenously expressed β-catenin (lane 7). This was presumably due to the presence of endogenous β-catenin (middle panels); note that the total β-catenin level was the same regardless of whether the β-catenin expression vector was transfected or not (lanes 1–3, middle panels), indicating a high level of endogenous β-catenin expression. Normal IgG failed to bring down either CoCoA or β-catenin (lane 4). Co-immunoprecipitation of the two proteins was also observed using anti-CoCoA antiserum (Fig. 1C). Thus, the β-catenin-CoCoA complex was observed in vitro and in vivo.
CoCoA and β-catenin synergistically enhance transcriptional activation in AR and TCF/LEF-mediated pathways
β-catenin can bind directly to AR and TCF/LEF proteins and function as a primary coactivator (3,9). In light of the physical interaction between β-catenin and NR coactivator CoCoA (Fig. 1), we tested whether they can function synergistically as coactivators for AR or TCF/LEF proteins in transient transfection assays. β-catenin alone, but not CoCoA, enhanced the ability of AR to activate a transiently transfected luciferase reporter gene controlled by a mouse mammary tumor virus (MMTV) promoter (which contains AR-responsive enhancer elements) in dihydrotestosterone (DHT)-treated CV-1 cells (Fig. 2A, assays 3–4). β-catenin and CoCoA synergistically enhanced AR-mediated transcriptional activation (assay 5). When the AR-responsive elements in the MMTV promoter of the reporter gene were replaced by an estrogen-responsive enhancer element, the reporter gene was not activated by AR or by AR with β-catenin and CoCoA (data not shown).
FIG. 2.
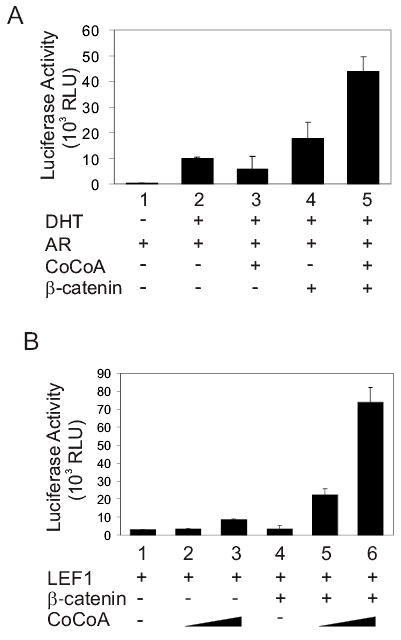
CoCoA and β-catenin synergistically enhance transcriptional activation by AR and LEF1. (A) CV-1 cells were transfected in 12-well plates with MMTV-LUC reporter plasmid (200 ng), pSV40-AR (50 ng), pSG5.HA-CoCoA (200 ng), and pSG5.HA-β-catenin (200 ng) as indicated, and grown in medium containing 20 nM DHT. Cell extracts were assayed for luciferase activity. (B) CV-1 cells were transfected with pGL3OT reporter plasmid (200 ng), pSG5.HA-LEF1 (10 ng), pSG5.HA-β-catenin (200 ng), and pSG5.HA-CoCoA (200 or 400 ng) as indicated. Results shown are from a single experiment and representative of three independent experiments.
Similarly, CoCoA and β-catenin also synergistically enhanced LEF1-mediated expression of luciferase reporter plasmid pGL3OT, which contains TCF/LEF-responsive elements (Fig. 2B). In contrast, the mutant reporter plasmid pGL3OF, containing mutant TCF/LEF responsive elements, was not activated by LEF1 alone or LEF1 with β-catenin and CoCoA (data not shown). Over-expression of CoCoA alone enhanced LEF1-mediated transcriptional activation of the wild type LEF1 reporter gene pGL3OT, presumably due to cooperation with endogenous β-catenin. However, the dramatic coactivator synergy between exogenously expressed CoCoA and β-catenin (Fig. 2B, assays 5–6) coupled with the failure of CoCoA to bind LEF1 in GST pull-down assays (data not shown), suggest that CoCoA functions as a secondary coactivator for AR and TCF/LEF-mediated transcriptional activation. I.e. CoCoA is apparently recruited to the promoter through its interaction with the primary coactivator β-catenin, not through a direct association with DNA-bound LEF1.
CoCoA is specifically targeted to transient and endogenous TCF/LEF-responsive enhancer elements
Reporter Co-IP assays were used to test whether CoCoA is specifically recruited to TCF/LEF-responsive enhancer elements in vivo. COS-7 cells were transiently transfected with reporter plasmid pGL3OT along with plasmids encoding LEF1, β-catenin, and CoCoA. pGL3-SV40, which contains the same vector backbone as pGL3OT but lacks LEF1 binding sites, was also included as an internal control. Chromatin preparations from the transfected cells were immunoprecipitated with antibodies against LEF1, β-catenin, or CoCoA and protein A/G Sepharose beads. The precipitated DNA was analyzed by PCR with primers recognizing the same backbone sequences in pGL3OT and pGL3-SV40; these primers produce amplification products of 318 base pairs from the pGL3OT plasmid and 412 base pairs from the pGL3-SV40 plasmid (Fig. 3A). PCR products amplified from the chromatin preparation before immunoprecipitation shows that the transfected COS-7 cells contain similar amounts of pGL3OT and pGL3-SV40 plasmids (Fig. 3A, lane 1). In the PCR products generated from DNA precipitated by antibodies against LEF1 (lane 2), β-catenin (lane 3), and CoCoA (lane 4), the 318-base pair product was enriched relative to the 412-base-pair product, indicating that LEF1, β-catenin, and CoCoA associated preferentially with the pGL3OT plasmid containing the TCF/LEF-responsive elements. Beads with no antibody did not precipitate the plasmids (lane 5).
FIG. 3.
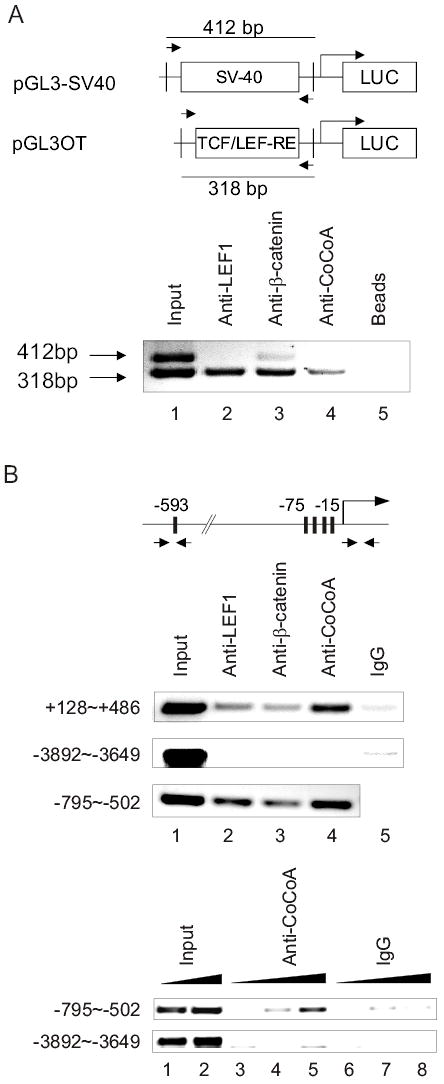
CoCoA is specifically recruited to transient and endogenous TCF/LEF-responsive elements in vivo. (A) COS-7 cells in 100-mm dishes were transfected with pGL3OT reporter plasmid (3 μg), pGL3-SV40 control plasmid (3 μg), pSG5.HA-LEF1 (1 μg), pSG5.HA-β-catenin (1 μg), and pSG5.HA-CoCoA (1 μg). 24 h after transfection, cells were treated with 20 mM LiCl for stabilization of β-catenin. After 24 h reporter-CoIP assays were performed with the transfected cell extracts using the indicated antibodies. PCR analysis of precipitated DNA was performed using primers (opposing arrows in diagrams) that span the backbone sequence of pGL3 basic vector but produce amplification products of different sizes for the reporter and control plasmids. Results shown are representative of three independent experiments. (B) ChIP assay was performed with the endogenous CyclinD1 promoter in SW480 cells. SW480 cells in 150-mm dishes were treated with 20 mM LiCl for 24 h. Chromatin-immunoprecipitation was performed with the indicated antibodies, and the precipitated DNA was analyzed by PCR using primers (arrows) that span or are close to the two TCF/LEF binding regions (−795 to −502 and +128 to +486) (28), or control primers which span the upstream region of the promoter (−3892 to −3649) which lacks TCF/LEF binding sites. Upper panel, PCR analysis was performed with 1 μl of a 1:10 dilution of precipitated DNA and a 1:20 dilution for input. Lower panel, PCR analysis was performed with 1 μl of 1:50, 1:10 or 1:1 dilution of precipitated DNA and a 1:50 or 1:20 dilution for input. Results shown are representative of two independent experiments.
To test whether CoCoA is specifically recruited to a verified endogenous target gene of TCF/LEF and β-catenin, we employed the colon adenocarcinoma cell line SW480. These cells contain elevated levels of wild type β-catenin due to a mutation in the APC protein, which regulates β-catenin degradation, and they therefore have elevated expression of cyclin D1 mRNA and protein. The critical role of LEF1 and β-catenin in the elevated expression of the cyclin D1 gene has been demonstrated in SW480 cells by over-expression of the cytoplasmic region of N-cadherin, which competes with LEF1 for binding to β-catenin (27); by expression of a dominant negative form of TCF-4E (28); and by use of siRNA against β-catenin (29). ChIP assays on SW480 cells demonstrated that LEF1, β-catenin, and CoCoA associated specifically with both sets of TCF/LEF enhancer elements in the cyclin D1 promoter, but not with a region 3 kb upstream which lacked TCF/LEF binding sites (Fig. 3B). These results demonstrated the specific recruitment of LEF1, β-catenin, and CoCoA to TCF/LEF-responsive elements in vivo, supporting the roles of β-catenin and CoCoA as coactivators which are recruited to TCF/LEF-responsive elements through their interaction with LEF1.
Endogenous CoCoA is required for the coactivator function of and transcriptional activation by β-catenin
To further test the role of CoCoA as a mediator of transcriptional activation by β-catenin, small interfering RNA (siRNA) targeting CoCoA was transfected into COS-7 cells. Transfection of siRNA targeting CoCoA, but not control siRNA with scrambled sequences or siRNA targeting β-catenin, specifically reduced the level of endogenous CoCoA mRNA; but the β-actin mRNA level was not affected (Fig. 4A, upper panels). Similarly, β-catenin mRNA level was reduced by siRNA against β-catenin but not by the two scrambled sequence siRNAs (middle panel). Activation of the transiently transfected, TCF/LEF-controlled pGL3OT luciferase reporter gene was reduced by 70% by siRNA targeting CoCoA and siRNA targeting β-catenin. In contrast, lucifease activity was not reduced by similar amounts of one of the control siRNAs and was reduced only 30% by the second control siRNA (Fig. 4A, lower panel).
FIG. 4.
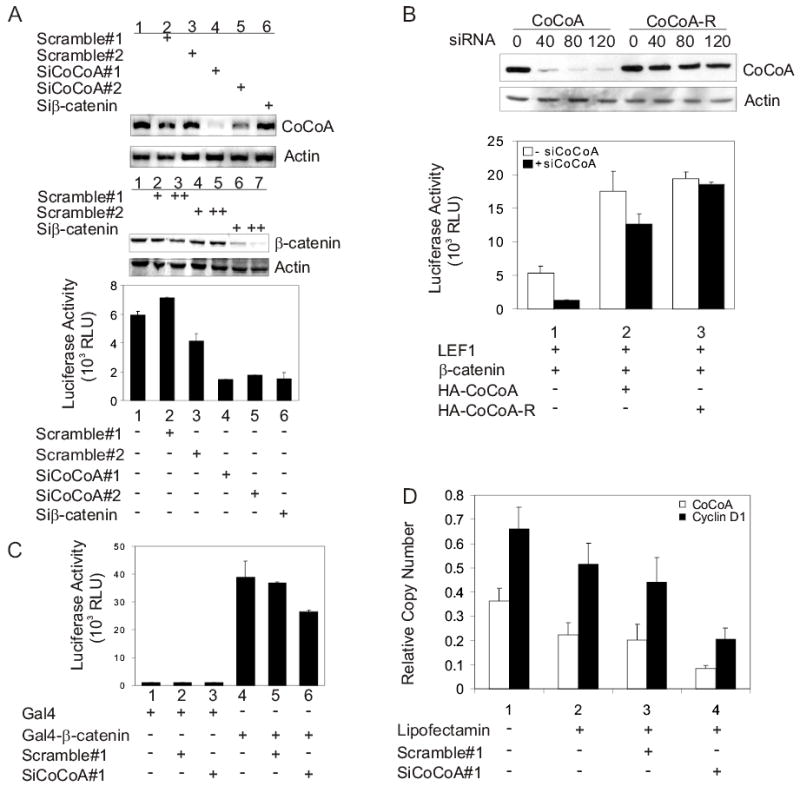
Requirement of endogenous CoCoA for β-catenin and LEF1 function. (A) COS-7 cells were transfected in 12-well plates with pGL3OT reporter plasmid (200 ng), pSG5.HA-LEF1 (100 ng), and either siRNA targeting CoCoA, siRNA with scrambled sequence, or siRNA targeting β-catenin (+=40 pmole, ++=60 pmole). Cell lysates were collected 48 h after transfection and used for luciferase assays (lower panel), for immunoblots detected with anti-β-catenin and anti-actin antibodies (middle panels), or for preparation of total RNA for RT-PCR analysis using primers for CoCoA or β-actin cDNA (upper panels). (B) For immunoblots (upper panels), COS-7 cells were transfected in 12-well plates with 1 μg of either pSG5.HA-CoCoA or pSG5.HA-CoCoA-R and the indicated amount (pmole) of siRNA targeting CoCoA. Expression of HA-CoCoA was analyzed with anti-HA antibodies, and endogenous β-actin was detected with anti-actin antibodies. For transient reporter plasmid assays, CV-1 cells were transfected in 12-well plates with or without siRNA targeting CoCoA (40 pmole) using Lipofectamine 2000. 48 h after siRNA transfection, cells were transfected using Targefect F1 with pGL3OT (200 ng), pSG5.HA-LEF1 (1 ng), pSG5.HA-β-catenin (10 ng), and either pSG5.HA-CoCoA or pSG5HA-CoCoA-R (200 ng) as indicated. Cell lysates were prepared 24 h after plasmid transfection for measurement of luciferase activity. Results shown are representative of two independent experiments. (C) COS-7 cells were transfected in 12-well plates with GK1-LUC reporter plasmid (200 ng), plasmids encoding Gal4 DBD or Gal4DBD-β-catenin (150 ng), and 20 pmole of either siRNA targeting CoCoA or siRNA with scrambled sequence as indicated. Luciferase activity was measured 48 h after transfection. Results shown are representative of four independent experiments. (D) SW480 cells were transfected with 60 pmole of either siRNA targeting CoCoA or siRNA with scrambled sequence as indicated. Total RNA was collected and cDNA was produced for quantitative real-time PCR analysis. Results shown are normalized to GAPDH mRNA levels and are the mean and standard deviation from 4 QPCR reactions for a single transfection experiment, which is representative of six independent experiments. For the six independent experiments, comparing cells treated with siCoCoA versus scrambled sequence siRNA, p=0.004 for CoCoA mRNA and p=0.01 for Cyclin D1 mRNA. Supplementary Fig. S1 shows the results and statistical analysis of all six experiments. In addition Supplementary Fig. S1 shows that the Correlation Coefficient R2 is 0.91 for the 6 experiments when the levels of CoCoA mRNA and Cyclin D1 mRNA are compared in the cells which were treated with siRNA against CoCoA; this indicates that the siRNA against CoCoA causes a proportional decrease in the levels of CoCoA mRNA and Cyclin D1 mRNA.
To confirm the specificity of the siRNA, we used a plasmid expressing an si-RNA-resistant mutant of CoCoA (CoCoA-R) to perform a rescue experiment on cells treated with the CoCoA-specific siRNA. The CoCoA-R expression plasmid contains three silent point mutations in the portion of the coding region of CoCoA that is targeted by the CoCoA-specific siRNA. Immunoblots indicated that expression of CoCoA-R in transient transfections was not affected by the CoCoA-specific siRNA, while the expression of wild type CoCoA was severely reduced (Fig. 4B, upper panels). In the rescue experiment, the CoCoA-specific siRNA reduced the ability of transiently expressed LEF1 and β-catenin to activate the pGL3OT reporter plasmid by 75% (Fig. 4B, lower panel, assays 1). In the absence of the siRNA, CoCoA wild type and CoCoA-R mutant expression plasmids increased the reporter gene activity 3–4 fold (Fig. 4B, assays 2–3, white bars). The CoCoA-specific siRNA reduced by 30% the reporter gene activity in the cells transfected with the wild type CoCoA expression plasmid but had no effect on the cells transfected with the CoCoA-R expression plasmid (assays 2–3, black bars). The enhanced rescue achieved by over-expression of CoCoA-R compared with wild type CoCoA confirms that the reduction in the expression of the pGL3OT reporter gene caused by the CoCoA-specific siRNA was specifically due to the reduction of cellular CoCoA levels.
To test further the role of CoCoA in mediating transcriptional activation by β-catenin, we tested the effect of the CoCoA-specific siRNA on the transcriptional activation function of β-catenin fused to Gal4 DBD. Activation of a reporter plasmid containing Gal4 response elements by Gal4DBD-β-catenin was reduced by about 30% by siRNA targeting CoCoA but not by the scrambled-sequence siRNA (Fig. 4C). Thus, endogenous CoCoA is important for mediating the activity of the β-catenin activation domains, whether β-catenin is tethered to the promoter by the Gal4 DBD or by binding as a coactivator to TCF/LEF transcription factors.
To investigate the role of endogenous CoCoA on the expression of a specific endogenous target gene that is activated by LEF1 and β-catenin, we tested the effect of CoCoA-directed siRNA on the expression of the cyclin D1 gene in SW480 cells. In 6 independent experiments siRNA against CoCoA caused a substantial reduction in the cyclin D1 mRNA level, compared with a control siRNA with a scrambled sequence (Fig. 4D shows results of one typical experiment and Supplementary Fig. S1 shows the results and statistical analysis of all six experiments.). These effects are gene-specific, since the results are normalized to GAPDH mRNA levels. Thus, although several other coactivators (CBP/p300, CARM1, and GRIP1) are known to act along with CoCoA as downstream mediators of β-catenin in transcriptional activation (9,22–26), simply reducing endogenous CoCoA levels causes a substantial decrease in the coactivator function of endogenous β-catenin on the endogenous cyclin D1 gene.
Supplementary Fig. S1.
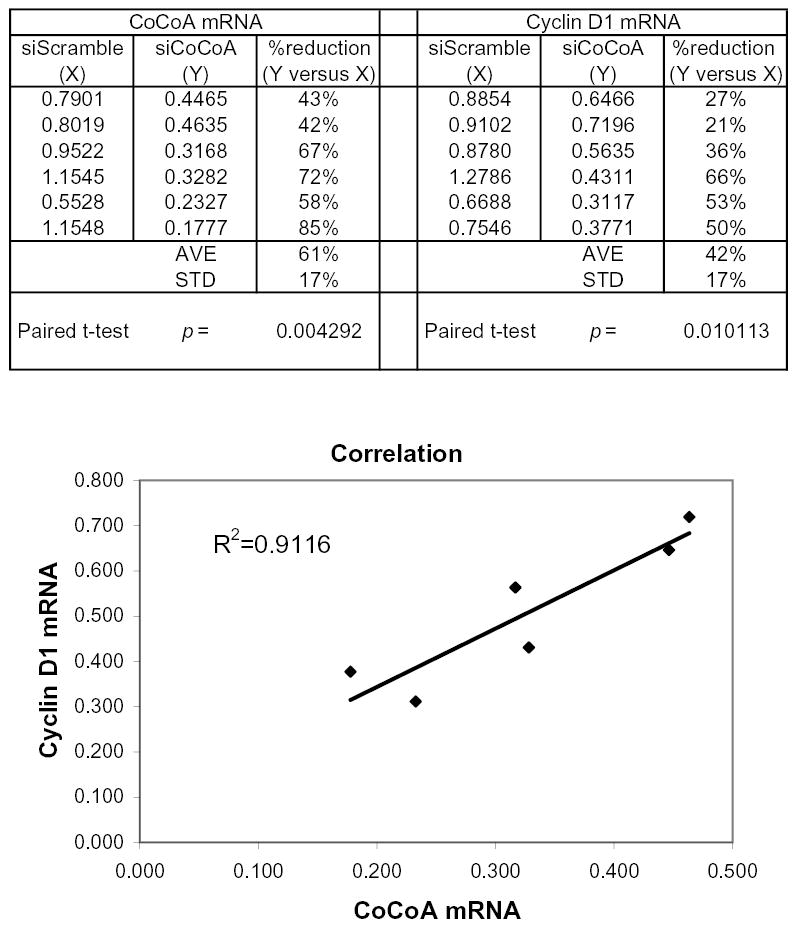
Statistical analysis of endogenous CoCoA and Cyclin D1 mRNA levels. SW480 cells were transfected with 60 pmole of either siRNA targeting CoCoA or siRNA with scrambled sequence. Total RNA was collected and cDNA was produced for quantitative real-time PCR analysis. Each number shown in columns X and Y for the CoCoA or Cyclin D1 mRNA level represents the mean from 4 QPCR reactions, which was then normalized in two steps: first, to the GAPDH mRNA level determined from the same cDNA sample; and second, to the control level of CoCoA or Cyclin D1 mRNA, respectively, determined for the cells (from the same experiment) which were not treated with siRNA. In other words, if the control CoCoA or Cyclin D1 mRNA level (in the absence of any transfected siRNA) in each experiment is set equal to 1.0, then the numbers in columns X and Y represent the relative levels to which CoCoA and Cyclin D1 mRNA were reduced by the transfection with scrambled (X) or specific (Y) siRNAs. For the 6 independent experiments shown, comparison of cells treated with siCoCoA versus scrambled siRNA by a paired, two-tailed T-test yielded the following p values: p = 0.004 for CoCoA mRNA and p = 0.01 for Cyclin D1 mRNA. The accompanying figure shows the Y value for Cyclin D1 mRNA plotted against the Y value for CoCoA mRNA for each of the 6 experiments. The Correlation Coefficient R2 for the resulting plot is 0.91, indicating a strong correlation between the CoCoA mRNA level and the Cyclin D1 mRNA level. I.e., the siRNA against CoCoA caused proportionate decreases in the levels of both CoCoA mRNA and Cyclin D1 mRNA.
Interacting domains of β-catenin and CoCoA
To transmit the activating signal from the enhancer element-bound transcriptional activator protein to the transcription machinery, coactivators must have one or more signal input domains which interact with upstream components of the signaling pathway and one or more signal output domains which interact with downstream components of the pathway (16). Since CoCoA is downstream from β-catenin in the TCF/LEF transcriptional activation signaling pathway, we defined the signal input domain(s) of CoCoA which interact with β-catenin. GST-β-catenin was incubated with CoCoA fragments over-expressed in COS-7 cells. Surprisingly, the central coiled-coil region of CoCoA (amino acids 150–500), which is important for binding to p160 coactivators (21), was neither necessary nor sufficient for binding to β-catenin (Fig. 5A). Instead, both the N-terminal (amino acids 1–190) and C-terminal (amino acids 501–691) regions of CoCoA were capable of binding to β-catenin. Thus, CoCoA has two independent β-catenin binding sites.
FIG. 5.
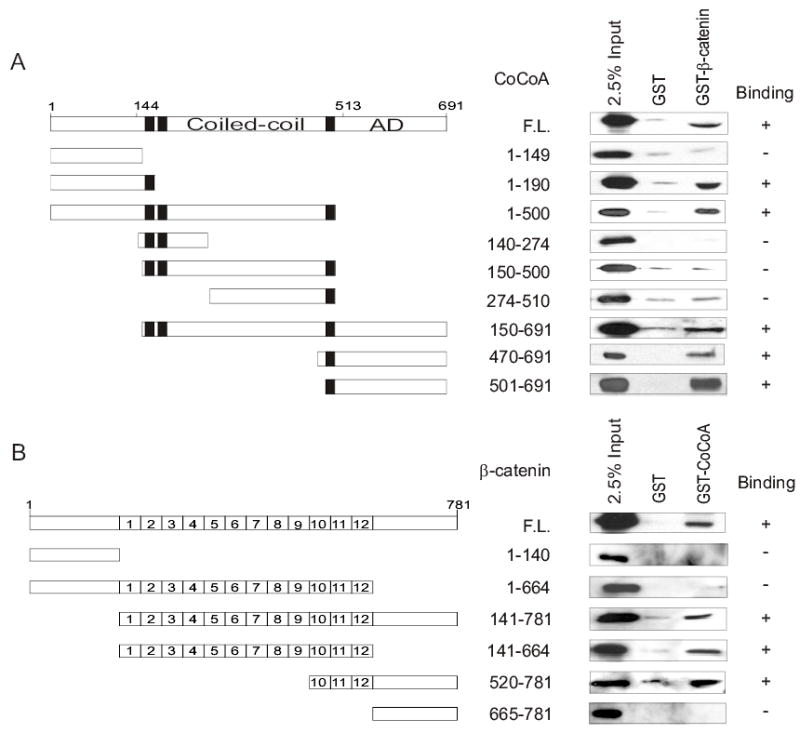
Domain requirements for β-catenin and CoCoA interaction. (A) GST pull-down assays were performed as in Fig. 1, using COS-7 cell extracts containing HA-tagged CoCoA (full length or fragments) and GST or GST-β-catenin bound to beads. The bound proteins were analyzed by immunoblot with anti-HA antibody. (B) GST pull-down assays were performed as in Fig.1, using COS-7 cell extracts containing HA-tagged β-catenin (full length or fragments) and GST or GST-CoCoA bound to beads. The bound proteins were analyzed by immunoblot with anti-HA antibody.
To define the region(s) of β-catenin that bind CoCoA, GST-CoCoA was incubated with β-catenin fragments over-expressed in COS-7 cells. In addition to full length β-catenin, CoCoA bound to β-catenin fragments consisting of the 12 armadillo repeats or the last three armadillo repeats still attached to the C-terminal region of β-catenin (Fig. 5B). This indicates that armadillo repeats 10–12 of β-catenin are important for binding CoCoA. However, since a fragment consisting only of armadillo repeats 10–12 failed to bind CoCoA (data not shown), adjacent N-terminal or C-terminal regions flanking armadillo repeats 10–12 are also required. It is interesting to note that β-catenin(1–664), which contains all 12 armadillo repeats, but lacks the C-terminus, failed to bind CoCoA, while full length β-catenin and a fragment consisting only of the 12 repeats did bind. This suggests that the N-terminal region of β-catenin may have a negative-regulatory effect on the interaction, while the presence of the C-terminal region overcomes this negative regulation.
Different domains of CoCoA are required to mediate transcriptional activation by β-catenin versus GRIP1
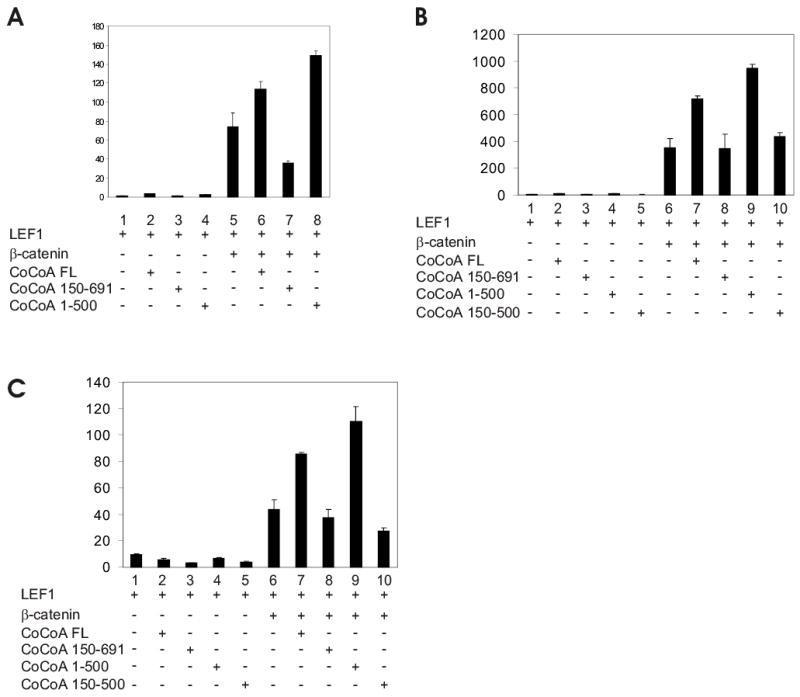
When CoCoA cooperates synergistically with the p160 coactivator GRIP1 to enhance ER-mediated transcriptional activation, the central coiled-coil domain of CoCoA is required for binding to GRIP1, and the C-terminal activation domain of CoCoA is needed as a signal output domain (21). As shown above (Fig. 5), different domains of CoCoA are required for binding to β-catenin than to GRIP1. Therefore, we also tested whether different domains of CoCoA are required for its coactivator function in cooperation with β-catenin and GRIP1. When CoCoA cooperates with β-catenin to enhance LEF1-mediated transcriptional activation, full-length CoCoA and a mutant lacking the C-terminal end were effective coactivators, but a mutant lacking the N-terminal end was inactive (Fig. 6A). These mutant forms of CoCoA were expressed at levels similar to those of wild type CoCoA (Fig. 5). In contrast to our findings for CoCoA domain requirements with β-catenin, the C-terminal region of CoCoA is required for its cooperation with GRIP1 to enhance ER-mediated transcriptional activation (21). Thus, the C-terminal region, but not the N-terminal region of CoCoA is required for its coactivator function with GRIP1 (21) (and data not shown), while the N-terminal but not the C-terminal region of CoCoA is needed for cooperation with β-catenin (Fig. 6A).
FIG. 6.
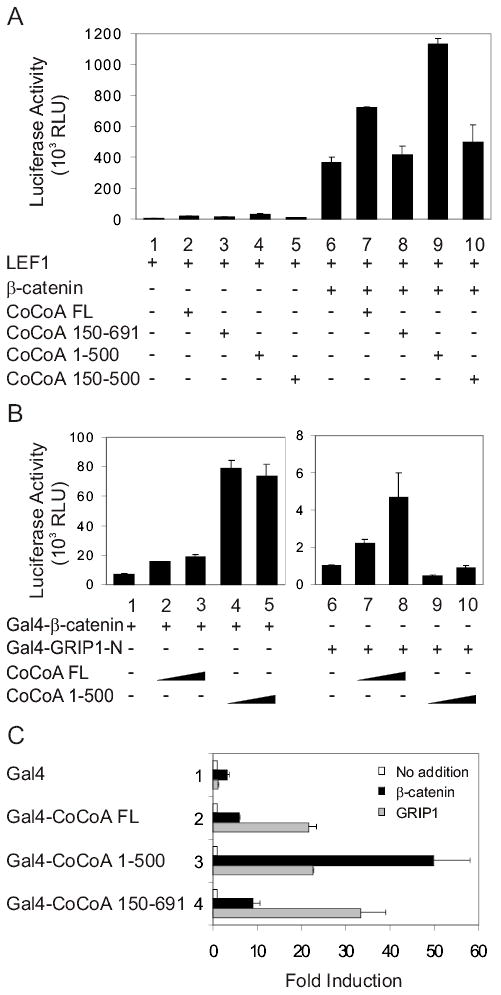
Domain requirements for CoCoA coactivator function with β-catenin or with GRIP1. (A) CV-1 cells were transfected in 12-well plates with pGL3OT reporter plasmid (200 ng), pSG5.HA-LEF1 (1 ng), pSG5.HA-β-catenin (50 ng), and pSG5.HA-CoCoA full-length or fragments (200 ng) as indicated. Luciferase activities shown are from a single experiment and are representative of seven independent experiments. Supplementary Fig. S2 shows results from 3 additional experiments. (B) CV-1 cells were transfected with pGK1-LUC reporter plasmid (200 ng), pM.β-catenin (100 ng) encoding Gal4DBD-β-catenin, pM.GRIP1N (100 ng) encoding Gal4DBD-GRIP1(5–479), and pSG5.HA-CoCoA full-length or fragment (200 or 400 ng) as indicated. Luciferase activities shown are representative of two independent experiments. (C) CV-1 cells were transfected with GK1-LUC reporter plasmid (200 ng), pM plasmids encoding Gal4DBD or Gal4DBD fused with CoCoA full-length or fragments (100 ng), and either pSG5.HA-GRIP1 or pSG5.HA-β-catenin (250 ng) as indicated. Luciferase activities are shown as fold increase above the activity observed for the corresponding Gal4DBD or Gal4DBD-CoCoA fusion protein expressed without GRIP1 or β-catenin. The results shown are representative of two independent experiments.
To test domain requirements of CoCoA’s coactivator function in a different setting, we used a mammalian-one hybrid format. Full-length CoCoA enhanced the ability of both Gal4DBD-β-catenin and Gal4DBD fused to the GRIP1 N-terminal domain (amino acids 5–479, the region that binds CoCoA) to activate transcription of a reporter gene containing Gal4 response elements (Fig. 6B, assays 1–3 & 6–8). In contrast, a CoCoA mutant lacking the C-terminus strongly enhanced the activity of Gal4DBD-β-catenin, but had little or no effect on the activity of Gal4DBD-GRIP1N (Fig. 6B, assays 4–5 & 9–10). These results confirm the importance of the C-terminal region of CoCoA for mediating transcriptional activation by GRIP1 but not by β-catenin.
A third test format provided additional support for this conclusion. Reporter gene activation by Gal4DBD-CoCoA lacking the CoCoA C-terminal region was enhanced more strongly by β-catenin than by GRIP1 (Fig. 6C, assays 3). In contrast, Gal4DBD-CoCoA lacking the N-terminal region of CoCoA was more active with GRIP1 than with β-catenin (assays 4). Thus, while CoCoA can function as secondary coactivator through either β-catenin or GRIP1, the specific domains required for its cooperation with each of these primary coactivators are different.
DISCUSSION
CoCoA functions as a secondary coactivator for LEF1
The Wnt/β-catenin signaling pathway plays important roles in developmental processes such as cell fate determination and axis formation (30). The pathway regulates expression of target genes such as cyclin D1, c-Myc, and BMP4 (2). Misregulation of this pathway leads to developmental defects and formation or progression of certain cancers. For example, β-catenin homozygous knockout mice are embryonic lethal (31). In addition, mice with a transgene expressing a constitutively active form of β-catenin developed intestinal adenomas (32,33). Approximately 90% of colorectal cancers and a smaller fraction of hepatocellular carcinomas were shown to have activating mutations in this pathway (2). Thus, it is important to understand the molecular basis for regulation of this pathway.
β-catenin plays an important role near the distal end of the Wnt-signaling pathway as a primary coactivator for the TCF/LEF transcription factors. The transcriptional activation signal is transmitted from the TCF/LEF proteins to the transcription machinery through primary coactivator β-catenin and its secondary coactivators GRIP1 (9,22), CARM1 (23), CBP/p300 (24–26), and the Swi/Snf ATPase subunit Brg1 (34). Here we report CoCoA as a novel participant in β-catenin-mediated transcription. CoCoA binds to β-catenin (Fig. 1) and cooperates synergistically with β-catenin to enhance the ability of LEF1 to activate transcription of a transiently transfected reporter gene (Fig. 2B). Because CoCoA does not directly interact with LEF1 (data not shown), and because the coactivator function of CoCoA depends strongly on β-catenin (Fig. 2B), CoCoA is a secondary coactivator for LEF1. The role of CoCoA as a coactivator for TCF/LEF transcriptional activator proteins is further supported by our finding that CoCoA, along with LEF-1 and β-catenin, is specifically recruited to transiently transfected and endogenous promoters which are activated by LEF1 (Fig. 3).
The identification of armadillo repeats 10–12 as the CoCoA binding site (Fig. 5B) defines this region of β-catenin as a signal output domain of β-catenin. It is interesting to note that both the N-terminal region of β-catenin and the region from armadillo repeat 10 to the C-terminus can bind to CBP and p300 (25,26). It is not clear whether the C-terminal region of β-catenin can bind to both CBP/p300 and CoCoA at the same time or uses them as alternative or sequential downstream targets. The regions of β-catenin which bind to CARM1 and GRIP1 have not been determined.
The physiological relevance of p300 and CBP as downstream targets for β-catenin was demonstrated by reducing their cellular level or inhibiting their activity with adenoviral protein E1A (25,26). Here, the physiological relevance of CoCoA for mediating the action of LEF-1 and β-catenin on transiently transfected and endogenous target genes was demonstrated by reducing the endogenous levels of CoCoA with siRNA, and by subsequently rescuing LEF-1 and β-catenin activity from the siRNA effect with an siRNA-resistant mutant of CoCoA (Fig. 4). In addition, endogenous CoCoA was specifically associated with the TCF/LEF enhancer elements of the endogenous cyclin D1 promoter in SW480 cells (Fig. 3B). In contrast, the roles of GRIP1 and CARM1 as secondary coactivators for LEF1 and β-catenin were primarily demonstrated through their abilities to bind to β-catenin and stimulate transcriptional activation by β-catenin and LEF1 in transient reporter gene assays (9,22,23).
Differential domain requirements for CoCoA as a coactivator
Coactivators constitute a multi-branched signal transduction pathway that emanates from the DNA-bound transcriptional activator protein and results in the recruitment or activation of multiple target proteins that are components of the chromatin and transcription machinery. Each subunit of the coactivator signaling pathway presumably serves as a conduit for one branch of the signal and thus must have signal input domains which bind to one or more upstream components of the pathway and signal output domains which interact with downstream components (16).
Here we show that different regions of CoCoA are used as signal input and output domains when CoCoA cooperates with β-catenin than when it acts with GRIP1. When GRIP1 and CoCoA cooperate as primary and secondary coactivators, respectively (Fig. 7B), the central coiled-coil region of CoCoA acts as the signal input domain, which binds to and receives the activating signal from GRIP1 (21). The C-terminal activation domain of CoCoA is essential for its coactivator function and apparently serves as a signal output domain, transmitting the activating signal to an unknown downstream component (21).
FIG. 7.
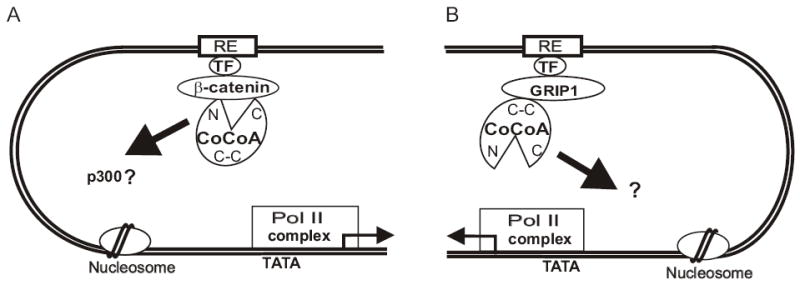
Differential use of domains by CoCoA with β-catenin and GRIP1. (A) When CoCoA serves as a secondary coactivator for β-catenin, the N-terminal and C-terminal regions of CoCoA (N and C in the diagram) bind to β-catenin and serve as signal input domains. The N-terminal domain may also serve as a signal output domain to transmit the activating signal (thick arrow) to an unknown target (possibly p300/CBP which bind to this domain) in the transcription machinery, but the C-terminal domain is dispensable. Other coactivators, such as GRIP1, CARM1, and p300/CBP, can also participate in this complex. (B) When CoCoA serves as a secondary coactivator for GRIP1, the coiled-coil domain of CoCoA binds to GRIP1 and serves as signal input domain. The C-terminal activation domain serves as a signal output domain, transmitting the signal to an unknown downstream target. Other coactivators, such as CARM1 and p300/CBP, can also participate in this complex. RE, response element or enhancer element; TF, DNA-binding transcription factor; C-C, coiled-coil domain; pol II complex, RNA polymerase II plus basal transcription factors; TATA, TATA box in the basal promoter; arrow emanating from the pol II complex, transcription start site and direction.
In contrast, when β-catenin and CoCoA cooperate as primary and secondary coactivators, respectively (Fig. 7A), the N-terminal and C-terminal regions of CoCoA can bind to β-catenin and thus represent two potential signal input domains (Fig. 5A). The C-terminal region of CoCoA, which is essential for its coactivator function with GRIP1, is dispensable for CoCoA’s coactivator function with β-catenin (Fig. 6A). The fact that the CoCoA mutant lacking the C-terminus still cooperates with β-catenin as a coactivator validates the N-terminal domain as a signal input domain. In addition, this finding indicates that the C-terminus of CoCoA is not required as a signal output domain when CoCoA cooperates with β-catenin; instead, the ability of the C-terminal domain to bind β-catenin suggests that it may serve as a redundant signal input domain. The CoCoA domain that serves as a signal output domain when CoCoA binds to β-catenin remains to be determined. However, our preliminary data indicate that the N-terminal region of CoCoA can bind p300 (our unpublished data), which suggests that the N-terminal region may provide both signal input and output functions when CoCoA collaborates with β-catenin.
Thus, CoCoA utilizes different signal input and output domains in response to activation signals sent from different primary coactivators (Fig. 7). Similar conclusions have been made for other coactivators. For example GRIP1 uses its LXXLL motifs to bind NRs (12), but uses its N-terminal bHLH-PAS domain to bind to other classes of DNA-binding transcriptional activators, such as MEF-2C (35). In addition, GRIP1 uses one set of signal output domains when it acts as a coactivator for steroid hormone-dependent transcriptional activation on some promoters (36), and uses a different set of signal output domains when it functions as a corepressor for steroid hormone-dependent repression on other promoters (37).
Implication of CoCoA as a general transcription coactivator in multiple pathways
The fact that CoCoA, GRIP1, CARM1, p300/CBP, and β-catenin can form heterodimers in a variety of combinations suggests the possibility that they could form a multi-subunit coactivator complex which associates with AR or LEF-1 on their target gene promoters. However, the potential activity of this group or complex of coactivators is not limited to the NR and TCF/LEF classes of transcriptional activators. Each of these five coactivators is known to bind directly to one or more DNA-binding transcriptional activator proteins. GRIP1 binds to NRs, AP-1, NFκB, HNF-1, MEF-2C, TEF4, and the aryl hydrocarbon receptor, among others (12,35,38–42). CBP and p300 interact with a huge variety of transcriptional activators (43). CARM1 binds to p53 and MEF-2C (44,45). β-catenin can bind to AR and LEF1 (4,5,8). CoCoA binds directly to the aryl hydrocarbon receptor and ARNT, which serves as a DNA-binding heterodimer partner for the aryl hydrocarbon receptor and a number of other transcriptional activators (46). Thus, complexes composed of these five coactivators or subsets of them could potentially mediate transcriptional activation for an enormous variety of DNA-binding transcriptional activator proteins. Obviously, different subunits would serve as primary and secondary coactivators in different situations; and as shown here for CoCoA, different domains of each coactivator subunit will presumably serve as signal input and output domains in different situations.
If the same cofactor is used for mediating transcriptional regulation in multiple coactivator complexes or by multiple DNA-binding transcription factors, and if the cofactor is present in limiting amounts, then there will be competition for this limiting cofactor among the various transcriptional regulators that require its function. In essence, this means that when one of the competing transcriptional regulators is activated, it will sequester the limiting cofactor and thus have a negative impact on signaling by the other transcriptional regulators that use the same limiting cofactor (47). For example, p300 and CBP serve as coactivators for many different DNA-binding transcriptional activator proteins, and limiting amounts of p300 and CBP have been proposed as a possible explanation for mutual antagonistic effects between different transcriptional activators that require these coactivators (48). Similarly, the antagonistic effect of the ligand-activated aryl hydrocarbon receptor on transcriptional activation by AR (49,50) and the mutually antagonistic effects of activated AR and TCF/LEF on each other (9,51,52) might be due to competition for limiting cofactors, such as CoCoA, which are involved in transcriptional activation of both members of these antagonistic pairs of transcription factors. Thus, the involvement of CoCoA, β-catenin, GRIP1, and CARM1 in multiple transcriptional regulatory pathways (e.g. the Wnt/TCF and AR pathways discussed here) also makes them candidates for competitive regulation among these different pathways.
Supplementary Fig. S2.
Domain requirements for CoCoA coactivator function with β-catenin or with GRIP1. CV-1 cells were transfected in 12-well plates with pGL3OT reporter plasmid (200 ng), pSG5.HA-LEF1 (1 ng), pSG5.HA-β-catenin (50 ng), and pSG5.HA-CoCoA full-length or fragments (200 ng) as indicated. Each graph shows the results from an independent repeat of the experiment shown in Fig. 6A.
Acknowledgments
We thank Mr. Daniel Gerke (University of Southern California) for expert technical assistance and Dr. Susan Groshen and the Biostatistics Core of the USC/Norris Cancer Center for assistance with statistical analyses. This work was supported by grant DK43093 to M.R.S. from the U.S. National Institutes of Health. J.H.K. was supported by a predoctoral fellowship from the University of Southern California/Norris Cancer Center Breast Cancer Research Training Program.
References
- 1.Morin PJ. Bioessays. 1999;21:1021–1030. doi: 10.1002/(SICI)1521-1878(199912)22:1<1021::AID-BIES6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 2.Giles RH, van Es JH, Clevers H. BiochimBiophysActa. 2003;1653:1–24. doi: 10.1016/s0304-419x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 3.Barker N, Clevers H. Bioessays. 2000;22:961–965. doi: 10.1002/1521-1878(200011)22:11<961::AID-BIES1>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 4.Billin AN, Thirlwell H, Ayer DE. MolCellBiol. 2000;20:6882–6890. doi: 10.1128/mcb.20.18.6882-6890.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu T, DeCostanzo AJ, Liu X, Wang H, Hallagan S, Moon RT, Malbon CC. Science. 2001;292:1718–1722. doi: 10.1126/science.1060100. [DOI] [PubMed] [Google Scholar]
- 6.Cavallo RA, Cox RT, Moline MM, Roose J, Polevoy GA, Clevers H, Peifer M, Bejsovec A. Nature. 1998;395:604–608. doi: 10.1038/26982. [DOI] [PubMed] [Google Scholar]
- 7.Brannon M, Brown JD, Bates R, Kimelman D, Moon RT. Development. 1999;126:3159–3170. doi: 10.1242/dev.126.14.3159. [DOI] [PubMed] [Google Scholar]
- 8.Truica CI, Byers S, Gelmann EP. Cancer Res. 2000;60:4709–4713. [PubMed] [Google Scholar]
- 9.Song LN, Herrell R, Byers S, Shah S, Wilson EM, Gelmann EP. MolCell Biol. 2003;23:1674–1687. doi: 10.1128/MCB.23.5.1674-1687.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jenster G. SeminOncol. 1999;26:407–421. [Google Scholar]
- 11.Heinlein CA, Chang C. EndocrRev. 2002;23:175–200. doi: 10.1210/edrv.23.2.0460. [DOI] [PubMed] [Google Scholar]
- 12.Glass CK, Rosenfeld MG. Genes Dev. 2000;14:121–141. [PubMed] [Google Scholar]
- 13.McKenna NJ, O'Malley BW. Cell. 2002;108:465–474. doi: 10.1016/s0092-8674(02)00641-4. [DOI] [PubMed] [Google Scholar]
- 14.Stallcup MR, Chen D, Koh SS, Ma H, Lee YH, Li H, Schurter BT, Aswad DW. Biochemical Society Transactions. 2000;28:415–418. [PubMed] [Google Scholar]
- 15.Xu J, Li Q. MolEndocrinol. 2003;17:1681–1692. [Google Scholar]
- 16.Stallcup MR, Kim JH, Teyssier C, Lee YH, Ma H, Chen D. JSteroid BiochemMolBiol. 2003;85:139–145. doi: 10.1016/s0960-0760(03)00222-x. [DOI] [PubMed] [Google Scholar]
- 17.Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, Stallcup MR. Science. 1999;284:2174–2177. doi: 10.1126/science.284.5423.2174. [DOI] [PubMed] [Google Scholar]
- 18.Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, Privalsky ML, Nakatani Y, Evans RM. Cell. 1997;90:569–580. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- 19.Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG. Nature. 1997;387:677–684. doi: 10.1038/42652. [DOI] [PubMed] [Google Scholar]
- 20.Yao TP, Ku G, Zhou N, Scully R, Livingston DM. ProcNatlAcadSciUSA. 1996;93:10626–10631. doi: 10.1073/pnas.93.20.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JH, Li H, Stallcup MR. MolCell. 2003;12:1537–1549. doi: 10.1016/s1097-2765(03)00450-7. [DOI] [PubMed] [Google Scholar]
- 22.Li H, Kim JH, Koh SS, Stallcup MR. JBiolChem. 2004;279:4212–4220. doi: 10.1074/jbc.M311374200. [DOI] [PubMed] [Google Scholar]
- 23.Koh SS, Li H, Lee YH, Widelitz RB, Chuong CM, Stallcup MR. JBiolChem. 2002;277:26031–26035. doi: 10.1074/jbc.M110865200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hecht A, Vleminckx K, Stemmler MP, van Roy F, Kemler R. EMBO J. 2000;19:1839–1850. doi: 10.1093/emboj/19.8.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takemaru KI, Moon RT. JCell Biol. 2000;149:249–254. doi: 10.1083/jcb.149.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun Y, Kolligs FT, Hottiger MO, Mosavin R, Fearon ER, Nabel GJ. ProcNatlAcadSciUSA. 2000;97:12613–12618. doi: 10.1073/pnas.220158597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben Ze'ev A. ProcNatlAcadSciUSA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tetsu O, McCormick F. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 29.Verma UN, Surabhi RM, Schmaltieg A, Becerra C, Gaynor RB. ClinCancer Res. 2003;9:1291–1300. [PubMed] [Google Scholar]
- 30.Miller JR. Genome Biol. 2002;3:REVIEWS3001.1–15. doi: 10.1186/gb-2001-3-1-reviews3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R. Development. 1995;121:3529–3537. doi: 10.1242/dev.121.11.3529. [DOI] [PubMed] [Google Scholar]
- 32.Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM. EMBO J. 1999;18:5931–5942. doi: 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Romagnolo B, Berrebi D, Saadi-Keddoucci S, Porteu A, Pichard AL, Peuchmaur M, Vandewalle A, Kahn A, Perret C. Cancer Res. 1999;59:3875–3879. [PubMed] [Google Scholar]
- 34.Barker N, Hurlstone A, Musisi H, Miles A, Bienz M, Clevers H. EMBO J. 2001;20:4935–4943. doi: 10.1093/emboj/20.17.4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen SL, Dowhan DH, Hosking BM, Muscat GE. Genes Dev. 2000;14:1209–1228. [PMC free article] [PubMed] [Google Scholar]
- 36.Ma H, Hong H, Huang SM, Irvine RA, Webb P, Kushner PJ, Coetzee GA, Stallcup MR. MolCellBiol. 1999;19:6164–6173. doi: 10.1128/mcb.19.9.6164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rogatsky I, Luecke HF, Leitman DC, Yamamoto KR. ProcNatlAcadSciUSA. 2002;99:16701–16706. doi: 10.1073/pnas.262671599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee SK, Kim HJ, Na SY, Kim TS, Choi HS, Im SY, Lee JW. JBiolChem. 1998;273:16651–16654. doi: 10.1074/jbc.273.27.16651. [DOI] [PubMed] [Google Scholar]
- 39.Sheppard KA, Rose DW, Haque ZK, Kurokawa R, McInerney E, Westin S, Thanos D, Rosenfeld MG, Glass CK, Collins T. MolCell Biol. 1999;19:6367–6378. doi: 10.1128/mcb.19.9.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soutoglou E, Papafotiou G, Katrakili N, Talianidis I. JBiolChem. 2000;275:12515–12520. doi: 10.1074/jbc.275.17.12515. [DOI] [PubMed] [Google Scholar]
- 41.Belandia B, Parker MG. JBiolChem. 2000;275:30801–30805. doi: 10.1074/jbc.C000484200. [DOI] [PubMed] [Google Scholar]
- 42.Beischlag TV, Wang S, Rose DW, Torchia J, Reisz-Porszasz S, Muhammad K, Nelson WE, Probst MR, Rosenfeld MG, Hankinson O. MolCell Biol. 2002;22:4319–4333. doi: 10.1128/MCB.22.12.4319-4333.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vo N, Goodman RH. JBiolChem. 2001;276:13505–13508. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- 44.An W, Kim J, Roeder RG. Cell. 2004;117:735–748. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 45.Chen SL, Loffler KA, Chen D, Stallcup MR, Muscat GE. JBiolChem. 2002;277:4324–4333. doi: 10.1074/jbc.M109835200. [DOI] [PubMed] [Google Scholar]
- 46.Kim JH, Stallcup MR. JBiolChem. 2004;279:49842–49848. doi: 10.1074/jbc.M408535200. [DOI] [PubMed] [Google Scholar]
- 47.Meyer ME, Gronemeyer H, Turcotte B, Bocquel MT, Tasset D, Chambon P. Cell. 1989;57:433–442. doi: 10.1016/0092-8674(89)90918-5. [DOI] [PubMed] [Google Scholar]
- 48.Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose DW, Glass CK, Rosenfeld MG. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- 49.Kizu R, Okamura K, Toriba A, Kakishima H, Mizokami A, Burnstein KL, Hayakawa K. ArchToxicol. 2003;77:335–343. doi: 10.1007/s00204-003-0454-y. [DOI] [PubMed] [Google Scholar]
- 50.Morrow D, Qin C, Smith R, III, Safe S. JSteroid BiochemMolBiol. 2004;88:27–36. doi: 10.1016/j.jsbmb.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 51.Mulholland DJ, Read JT, Rennie PS, Cox ME, Nelson CC. Oncogene. 2003;22:5602–5613. doi: 10.1038/sj.onc.1206802. [DOI] [PubMed] [Google Scholar]
- 52.Chesire DR, Isaacs WB. Oncogene. 2002;21:8453–8469. doi: 10.1038/sj.onc.1206049. [DOI] [PubMed] [Google Scholar]