

Abstract
Integrins are an important family of signaling receptors that mediate diverse cellular processes. The binding of the abundant extracellular matrix ligand fibronectin to integrins α5β1 and αvβ3 is known to depend upon the Arg-Gly-Asp (RGD) motif on the tenth fibronectin FIII domain. The adjacent ninth FIII domain provides a synergistic effect on RGD-mediated integrin α5β1 binding and downstream function. The precise molecular basis of this synergy remains elusive. Here we have dissected further the function of FIII9 in integrin binding by analyzing the biological activity of the FIII9–10 interdomain interface variants and by determining their structural and dynamic properties in solution. We demonstrate that the contribution of FIII9 to both α5β1 and αvβ3 binding and downstream function critically depends upon the interdomain tilt between the FIII9 and FIII10 domains. Our data suggest that modulation of integrin binding by FIII9 may arise in part from its steric properties that determine accessibility of the RGD motif. These findings have wider implications for mechanisms of integrin-ligand binding in the physiological context.
The interaction of the abundant extracellular matrix molecule fibronectin with the integrin family of cell surface receptors mediates wide-ranging biological processes, including cell migration, invasion, and angiogenesis. Integrin-fibronectin interactions thus have a fundamental role in tissue development and homeostasis, and integrin receptor agonism and antagonism are implicated in the etiology of a number of diseases such as cancer and inflammatory disorders. However, the precise molecular bases of integrin-fibronectin interactions required for downstream biological function are not understood.
Integrins are heterodimeric cell surface receptors comprising one α and one β subunit. So far, 18 α and 8 β subunits have been identified, but the potential to form functional dimers is restricted. Some integrin subunits, notably αv and β1, are more promiscuous than others, such as α5, in the selection of subunits with which they can dimerize. Similarly, some integrins, such as αvβ3, can bind to more than a dozen different ligands including fibronectin, whereas the ligand repertoire of others, such as α5β1, is much more limited.
Fibronectin is a large dimeric glycoprotein (∼220 kDa) and is almost exclusively composed of repeating units falling into three structurally distinct categories, named type I (FI), type II (FII), and type III (FIII) domains. The central portion of the molecule contains a pair of FIII domains, designated FIII9 and FIII10, which constitute the best studied and most versatile integrin-binding region. The tripeptide adhesion motif Arg-Gly-Asp (RGD) resides on a surface loop in FIII10 and is the essential recognition site for at least 11 integrins. The adjacent FIII9 domain provides synergistic enhancement of binding to and signaling through several RGD-dependent integrins, including α5β1 (1-5) and αIIbβ3 (6, 7). In contrast, αvβ3 is generally believed not to require this synergistic effect (5, 7).
Numerous studies of the interactions of the FIII9–10 pair with integrins have demonstrated the functional importance of specific synergistic residues or sequences, notably the PHSRN motif, within FIII9 (2-4, 6-9). The structural basis for the synergy phenomenon, however, is elusive, and several different mechanisms have been proposed. The most obvious is a direct contribution from FIII9 to integrin binding (8, 10). Solution x-ray scattering and mutagenesis studies performed recently suggest the existence of direct interactions between FIII9 and the integrin α5 subunit (10). However, in a second study, electron micrographs did not reveal significant FIII9-integrin contact (11). Recent data from this laboratory suggest that the FIII9 domain can modulate binding via less specific, conformational effects affecting global domain stability (2, 3). In addition, partial structural uncoupling of FIII9 and FIII10 by the extension of the linker at the FIII9–10 interface leads to loss of synergistic function (12), indicating a requirement for adequate interdomain contact for receptor binding.
Although flexibility of the FIII9–10 linkage has been implicated in large scale conformational changes in native fibronectin (13), there is no direct evidence to support this, and the potential biological consequences of such structural changes are not known. Here we explore the possibility that modulation of the interdomain tilt between FIII9 and FIII10 may affect the domain pair's integrin binding and downstream biological activity. Our aim is to gain further structural insight into the mechanism of integrin recognition by fibronectin.
EXPERIMENTAL PROCEDURES
Construction and Expression of FIII Variants
Wild-type FIII9–10 and FIII10 clones were obtained by inserting the respective DNA fragments, amplified from the pGEX2T-FIII9–10 construct (14) and digested with NheI/HindIII, into the corresponding sites in the pRSET-A vector (Invitrogen). Creation of the FIII9–10 construct incorporating a Leu1408 to Pro mutation (designated here as FIII9′10) has been described previously (15). Further amino acid substitutions, Ala1340/Val1442 to Cys/Cys, were introduced into the latter construct according to the QuikChange™ protocol (Stratagene), resulting in mutant FIII9′10-CC. All FIII variants were expressed as His tag proteins in Escherichia coli strain BL21(DE3)pLysS (Promega) and purified using Ni2+-nitrilotriacetic acid Superflow resin (Qiagen). Isotopically labeled proteins were produced using modified M9 medium containing 5 g/liter 15NH4Cl and/or 2 g/liter uniformly labeled [13C]glucose (CK Gas Products). Purity of the proteins was assessed by SDS-PAGE and mass spectrometry.
The single disulfide bond present in the oxidized form of mutant FIII9′10-CC was reduced either by adding dithiothreitol at 100–400 times molar excess (for NMR and equilibrium unfolding experiments) or by alkylation with iodoacetamide (for cell adhesion and solid-phase assays). The oxidation state of mutant FIII9′10-CC was confirmed by mass spectrometry.
Enzyme-linked Immunosorbent Assays
Integrin α5β1 used in solid-phase binding assays was purified from human placenta as specified elsewhere (3). Purification of αvβ3 was performed in a similar manner using antibody clone HB11029 raised against the αvβ3 heterodimer. Binding of soluble FIII constructs to immobilized integrin was performed in the presence of divalent cations Mn2+, Mg2+, and Ca2+ as described earlier (3), except that horseradish peroxidase-conjugated anti-HisG antibody (Invitrogen) at 0.4 μg/ml was used for detection, followed by color development with the Sigma Fast™ tablet set. Data were analyzed by nonlinear regression as described (3). Assay results are expressed as the means ± S.E. (dose-response graphs) or the means + CI95% (bar graphs) of at least two independent determinations.
Cell Adhesion Assays
Cell attachment and spreading assays with plate-bound recombinant proteins were performed using baby hamster kidney (BHK)1 fibroblasts as detailed elsewhere (14). The data presented are expressed as the means ± S.E. of at least three independent duplicate or triplicate determinations.
Equilibrium Unfolding Studies
Equilibrium chemical denaturation assays were carried out on FIII proteins incubated in the presence of 0 to ∼8 m guanidinium hydrochloride (GdnHCl) in 10 mm HEPES, 100 mm NaCl, pH 7.4, as described earlier (3). The data obtained were fitted for a two-state unfolding mechanism (16). The error bars represent CI95%.
NMR Sample Preparation
All NMR samples were prepared from freeze-dried 15N-labeled protein in a 50 mm sodium acetate buffer in 95/5% H2O/D2O. The pH was adjusted to 4.8 by the addition of HCl. 4% polyacrylamide gels with 3% cross-linking were prepared (17, 18) and radially compressed by an axial ratio of 1.45 using a custom-made device (19).
NMR Experiments
All NMR experiments were performed at 25, 30, or 35 °C on spectrometers operating at 1H frequencies of 600 and 750 MHz with triple-resonance triaxial gradient probes. Experiments used gradient-enhanced coherence selection (20) and water flip-back (21) wherever possible. For backbone assignment of FIII9′10, three-dimensional 15N-edited NOESY-HSQC (22) and TOCSY-HSQC (23) spectra with mixing times of 125 and 29.0 ms, respectively, and a TOCSY field strength of 11,900 Hz were recorded on a 1 mm sample at 600 MHz, as well as the three-dimensional triple-resonance experiments HNCA (24), HNCOCA (25), and CBCACONH (26) at 750 MHz. Hα, Hβ, and NH resonances were assigned using a combination of cross-peaks in the three-dimensional 15N-edited TOCSY and NOESY-HSQC spectra between Hα, Hβ to NH, and NH to NH resonances as well as through bond correlations between the Cα(i – 1) and Cα(i) observed in the HNCA and HNCACO. Characteristic Cα/Cβ shifts in the CBCACONH aided the identification of amino acids (27). Initial assignments of NH and 15N resonances of the FIII9′10-CC mutant were derived from FIII9′10, and these were confirmed through Hα,Hβ to NH, and NH to NH cross-peaks in the three-dimensional 15N-edited NOESY-HSQC.
Heteronuclear [1H]15N NOE spectra were acquired on 1 mm samples at 30 °C and 600 MHz with acquisition times of 40.4 ms for 15N and 82 ms for 1H. 1H saturation in the NOE experiment was effected by means of a train of 225° flip-angle pulses at 10-ms intervals for 4.5 s. 15N-T1 and T2 relaxation time constants of 0.25 mm uniformly 15N-labeled samples were measured at 25 °C as described (28). For each sample a series of eight 1H-15N autocorrelation spectra with acquisition times of 91.1 ms for 15N and 82 ms for 1H was recorded. Transverse relaxation was measured using a spin-echo sequence with a CPMG delay of 285 μs. Relaxation delays varied between 40 ms and 1.2 s and between 4.5 and 136 ms for the T1 and T2 series, respectively.
1H-15N residual dipolar couplings (RDCs) of 0.35 mm 15N-labeled samples in stretched polyacrylamide gels (19) were measured at 35 °C using the in-phase, anti-phase scheme (29) with acquisition times of 50.7 ms (15N) and 51 ms (1H). The reference 1H-15N scalar couplings in isotropic solution were recorded at 35 °C. The 1H-15N RDCs were obtained from the difference of the chemical shift of 15N resonances in the aligned and isotropic state. Errors were estimated to be 1.0 Hz for each of four measurements, resulting in a combined error of 2.0 Hz.
NMR Data Analysis
All NMR data were processed with Felix 2.3 (Biosym Technologies, Inc., San Diego, CA) and analyzed with NMR-view5 (30). Alignment tensors of the individual domains FIII9 and FIII10 for each of the three variants were obtained from the 1H-15N RDC data from residues with a [1H]15N NOE > 0.65 using the program MODULE (31) and known x-ray coordinates (32). Monte Carlo simulations were used for error estimation. MODULE was also used to perform rigid body reorientations of the domains around residue Ser1317 in the interdomain linker to obtain a best fit with the RDC data. Subsequently, the interdomain tilt and twist angles were calculated with mod22 (33) using the conserved Trp1347 and Trp1437 as a reference. Relaxation analysis and derivation of diffusion tensors was performed according to standard techniques (34) using software written in house or the program TENSOR (35).
RESULTS
Experimental Strategy
In order to probe the role of inter-domain orientation and flexibility in fibronectin function, we changed native domain-domain coupling by modifying the interface between FIII9 and FIII10, creating an extra interdomain covalent bond to restrict domain-domain mobility. The double cysteine mutant FIII9′10-CC harbors altered residues that are positioned on loops extending into the interdomain space from either side (Fig. 1). The mutant was engineered on a previously described modified FIII9–10 construct designated FIII9′10, which incorporates a Leu1408 → Pro substitution in FIII9 (Fig. 1) that confers better conformational stability and functional activity (15). The variant proteins were assessed for stability, analyzed by NMR, and tested in functional assays of integrin binding and biological activity.
Fig. 1.

Ribbon diagram of the FIII9–10 domain pair. Atomic coordinates were obtained from the Protein Data Bank (43) (Protein Data Bank code 1FNF) and imaged using RasTop (available on the World Wide Web at www.openrasmol.org). Residues substituted in this study, shown in their putative mutant conformation as rendered by Swiss PDB Viewer (available on the World Wide Web at spdbv.niehs.nih.gov), are displayed in ball-and-stick format and labeled accordingly using one-letter code. The dotted line designates disulfide bond present in oxidizing conditions. The synergistic PHSRN sequence and the RGD motif are indicated in yellow and blue, respectively.
Introduction of an Interdomain Disulfide Bridge in FIII9–10 Increases FIII9 Stability
Wild type and mutant FIII9–10 proteins were first assessed for their global thermodynamic stability by performing chemical equilibrium unfolding experiments with GdnHCl as a denaturant (Fig. 2). FIII9′10-CC was tested both in its oxidized, disulfide-linked form (FIII9′10-CCox) and in the reduced form (FIII9′10-CCred). All tested proteins, except for FIII9′10-CCox, follow a two-state unfolding mechanism characteristic of native FIII9–10 (3, 36) and have very similar unfolding parameters for the FIII10 domain, whose conformational stability is known to be largely independent of the presence of FIII9 (3) (Fig. 2a, inset, and Table I). Analysis of the effect of the interface mutations on the unfolding of FIII9, however, reveals that mutant FIII9′10-CC shows an increased stability both in its reduced and oxidized forms (Fig. 2). We note that the moderate stability increase seen with FIII9′10-CCred may partly arise from an incomplete reduction of the interdomain disulfide bridge by dithiothreitol. The observed stability-promoting effect is, however, greatly enhanced in FIII9′10-CCox, such that unfolding of FIII9 can no longer be separated from that of FIII10, and the two domains follow a single transition curve (Fig. 2a, inset). The values for FIII9 stability in the variants, expressed as the GdnHCl concentration leading to 50% domain unfolding ([GdnHCl]), are 2.33, 3.12, and 4.43 m for FIII9′10, FIII9′10-CCred, and FIII9′10-CCox, respectively (Fig. 2b and Table I). These findings indicate that the unfolding process is sensitive to amino acid changes in the interface region between FIII9 and FIII10 and provide further support to the notion that the interdomain contact surface within FIII9–10 is an important determinant of the thermodynamic properties of FIII9 (36).
Fig. 2.

Thermodynamic stability of FIII9–10 proteins. a, normalized chemical denaturation curves for FIII9 unfolding, corresponding to the first transition region of the consecutive unfolding of FIII9 and FIII10 within wild type and mutant FIII9–10 variants, as depicted in the inset. Open squares, FIII9–10; filled squares, FIII9′10; blue circles, FIII9′10-CCred; red circles, FIII9′10-CCox. Denaturation data for FIII9′10-CCox are presented across the entire denaturation range encompassing both domains, since they unfold cooperatively. b, comparison of [GdnHCl]½ values, derived from regression analysis of the curves in a, for wild type (wt) FIII9–10, FIII9′10, FIII9′10-CCred, and FIII9′10-CCox (left to right).
Table I.
Equilibrium unfolding parameters for FIII9–10 variants
| FIII9–10 variant | Domain | [GdnHCl]½ | ΔG(H2O)a | m |
|---|---|---|---|---|
| M | kcal mol − 1 | kcal mol−1 m−1 | ||
| FIII9–10 | FIII9 | 1.28 | 3.38 | 4.02 |
| FIII10 | 5.01 | 8.28 | 1.77 | |
| FIII9′10 | FIII9 | 2.33 | 6.16 | 2.30 |
| FIII10 | 5.01 | 8.28 | 1.53 | |
| FIII9′10-CCred | FIII9 | 3.12 | 8.25 | 1.61 |
| FIII10 | 4.99 | 8.25 | 1.66 | |
| FIII9′10-CCox | FIII9–10 | 4.43 | 11.71b | 1.41 |
Due to potential errors associated with determining the individual slope (m) values for the linear transition region (16), the values of ΔG(H2O) were calculated from the average m values of 2.64 and 1.65 kcal mol−1 m−1 for FIII9 and FIII10, respectively.
Calculated from the average m value for FIII9.
Interdomain Disulfide Linkage Abolishes the Synergistic Cell Adhesion Activity of FIII9–10 and Reduces Its Affinity for Integrins α5β1 and αvβ3
The interface variants were next subjected to assays designed to quantify the impact of interdomain perturbations on function. Cell adhesion assays were performed with immobilized FIII9–10 proteins and BHK fibroblasts, which recognize fibronectin mainly via integrins α5β1 and αvβ3 (12). Integrin α5β1 accounts for most of the adhesion potential of BHK cells and is a major synergy-dependent receptor for fibronectin, whereas αvβ3 has been shown to bind the single FIII10 domain with high affinity and does not require FIII9 for full activity (5, 7). The functional activity of mutant FIII9′10-CCox was drastically reduced compared with FIII9′10 and was similar to that achieved with the single FIII10 domain (Fig. 3, a and b). This was the case both for cell attachment (Fig. 3a), a primary event of establishing integrin-dependent contact with the ligand, and the ensuing cell spreading (Fig. 3b), driven by extensive signaling and reorganization of the cytoskeletal architecture. In contrast, the adhesion-promoting ability of FIII9′10-CCred was indistinguishable from that of native FIII9–10 or FIII9′10 (Fig. 3, a and b).
Fig 3.
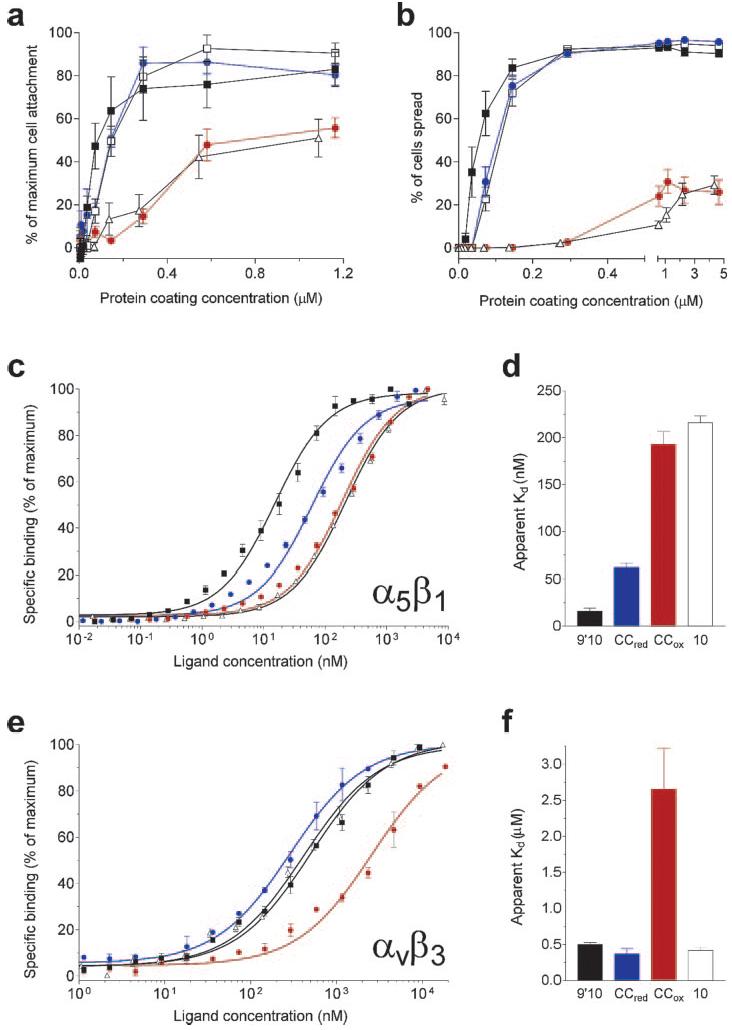
Effect of disulfide linkage on the biological activity of FIII9–10. a, attachment of BHK cells on surface-immobilized FIII9–10 variants and a single FIII10 domain. Results are expressed relative to the maximum level of attachment attained at higher concentrations of FIII9′10 (not shown). b, assessment of the ability of the FIII9–10 variants to support BHK cell spreading. c and e, normalized dose-response data for solid-phase binding of FIII proteins to surface-bound integrin α5β1 (c) or αvβ3 (e). Open squares, FIII9–10; filled squares, FIII9′10; blue circles, FIII9′10-CCred; red circles, FIII9′10-CCox; open triangles, FIII10. d and f, apparent Kd values derived from the curves in c and e, for binding of FIII9′10, FIII9′10-CCred, FIII9′10-CCox, and FIII10 (left to right) to integrin α5β1 (d) or αvβ3 (f).
We further sought to complement our cell-based studies with solid-phase receptor binding assays. Dose-response data from experiments on the FIII9′10-CC interface mutant and purified integrins α5β1 and αvβ3 (Fig. 3, c–f) show that the disulfide-linked FIII9′10-CCox loses the synergistic activity of its parent protein FIII9′10 for binding to α5β1. It exhibits effectively the same receptor affinity as the isolated FIII10 domain and has an apparent Kd value ∼15-fold higher than that of FIII9′10 (Fig. 3, c and d). In contrast, FIII9′10-CCred has a substantially higher affinity for α5β1, although it does not reach the level of binding achieved by FIII9′10 (Fig. 3, c and d). The FIII9′10-CCox variant also exhibits a markedly reduced affinity for integrin αvβ3, as compared with FIII9′10 (the apparent Kd is ∼5-fold higher), whereas both FIII9′10-CCred and the single FIII10 domain bind αvβ3 with an affinity similar to that of FIII9′10 (Fig. 3, e and f). These solid-phase observations are thus in general agreement with the cell adhesion data.
Our findings show that improved conformational stability of FIII9, as seen with mutant FIII9′10-CC, does not necessarily correlate with its synergistic biological activity. This prompted us to perform a detailed analysis of the structural basis of the loss of function of the disulfide-linked FIII9′10-CCox.
The Interdomain Disulfide Bridge Reduces the Average Tilt Angle between FIII9 and FIII10
The structural integrity of FIII9′10 and FIII9′10-CC was confirmed by solution state NMR. The 1H-15N backbone resonances of the FIII9′10-CC mutant were characteristic of folded molecules and showed only small chemical shift changes with respect to the FIII9′10 variant (data not shown). Local and global changes in structure and dynamics were further determined using chemical shift perturbation, RDCs, and heteronuclear relaxation measurements.
The chemical shift differences between FIII9′10 and FIII9′10-CC in the oxidized and reduced state are shown in Fig. 4. The only significant perturbations in both FIII9′10-CCox and FIII9′10-CCred with respect to FIII9′10 are restricted to the vicinity of the mutated residues (Fig. 4). This indicates that neither the introduction of this disulfide bond nor its reduction cause a significant structural change in the backbone structure of individual domains. This was expected because the disulfide bridge was designed to satisfy the distance and conformational constraints on the basis of crystal structure coordinates (32).
Fig 4.

Combined 15N and 1H chemical shift differences, Δδ, between FIII9′10 and FIII9′10-CCred (a) and FIII9′10 and FIII9′10-CCox (b). Δδ is given by Δδ = √((δ1H)2 + (δ15N/6)2). Values greater than 0.08, indicated by the horizontal dotted line, are considered to be significant. The filled horizontal bars denote the location of β-strands, and the vertical dotted lines mark the sites of the mutated residues Ala1340 and Val1442. The synergistic PHSRN sequence and the RGD motif are highlighted in yellow and blue, respectively.
Changes in the orientations of the NH bond vectors of FIII9′10 and the FIII9′10-CC variants were determined from the measurements of RDCs of NH bond vectors in two different alignment media. The results for the polyacrylamide medium are shown in Table II and illustrated in Fig. 5. The bicelle medium yields similar data (not shown). RDCs are very sensitive to angular reorientations of a particular bond or molecular fragment with respect to the molecular frame and are therefore well suited to investigate domain-domain orientations in solution. The RDCs of each domain in each variant were used to determine the alignment tensor of each domain individually. Subsequently, the domains of each construct were reoriented with respect to each other such that the individual alignment tensors coincided. There is good agreement between the RDC data and the values calculated from the x-ray structure (32), as shown by the quality factors, Q, in Table II, for the individual domains in all three FIII9–10 proteins. These values are typical for protein structures with a resolution of about 2 Å (37). The orientation of the main axis of the alignment tensors is in good agreement with that expected of an overall elongated shape for each domain in each protein. The rhombic terms (Ar) of the alignment tensors are small, making the estimation of the interdomain twist less certain than the tilt. There are small but measurable differences between the alignment tensors of individual domains within each protein (Table II). This indicates some degree of independent motion between the domains (see below).
Table II.
Alignment tensors and interdomain orientations of FIII9–10 variants
| FIII9–10 variant | Domain | Alignment tensors in PAGa |
Interdomain orientations in PAGa |
||||||
|---|---|---|---|---|---|---|---|---|---|
| A a b | A r b | α b | β b | γ b | Q c | Tiltd | Twistd | ||
| degrees | degrees | ||||||||
| FIII9′10 | FIII9 | 13.6 ± 0.3 | 3.1 ± 0.3 | −74 | 172 | 5 | 0.26 | 28 ± 1 | 349 ± 4 |
| FIII10 | 15.2 ± 0.3 | 2.9 ± 0.3 | 174 | 176 | −129 | 0.31 | |||
| FIII9′10-CCred | FIII9 | 12.6 ± 0.3 | 2.5 ± 0.2 | −64 | 175 | 16 | 0.24 | 21 ± 1 | 340 ± 5 |
| FIII10 | 11.1 ± 0.3 | 1.8 ± 0.2 | −91 | 173 | −24 | 0.35 | |||
| FIII9′10-CCox | FIII9 | 8.5 ± 0.4 | 1.6 ± 0.3 | 176 | 7 | 111 | 0.33 | 5 ± 2 | 355 ± 6 |
| FIII10 | 9.3 ± 0.4 | 3.6 ± 0.3 | −62 | 168 | 17 | 0.39 | |||
| FIII9 – 10 (x-ray) | 16 | 326 | |||||||
Fig 5.

RDCs and interdomain orientations of FIII9′10 variants. a, b, c, and d correspond to FIII9′10; e, f, g, and h correspond to FIII9′10-CCred; and i, j, k, and l correspond to FIII9′10-CCox. The left panels a, e, i, and b, f, and j show for each variant the same sections of the spectra used to measure the apparent 1JNH couplings in isotropic solution (a, e, and i) and in a strained polyacrylamide gel (b, f, and j). The values of the 1JNH couplings are given in Hz, and for clarity, the resonances are labeled with amino acid names only in a. The middle panels c, g, and k show the correlation between the observed and calculated RDCs, and the right panels d, h, and l show the resulting interdomain orientation of the corresponding domain pairs.
The analysis of the interdomain orientations (Table II) shows that the time-averaged interdomain tilt angle for FIII9′10 is 28 ± 1°, whereas FIII9′10-CCox shows a reduced tilt angle of 5 ± 1°. The tilt angle in FIII9′10-CCred is restored to 21 ± 1°. The values of the twist angles obtained for the three constructs range from 340 ± 5° to 355 ± 6° and are experimentally almost indistinguishable. These measurements thus indicate that the breaking and formation of the interdomain disulfide bond in FIII9′10-CC is associated with a significant change in the interdomain tilt angle but not interdomain twist. These observations correlate well with the data obtained from cell adhesion and integrin binding assays showing restoration of function for reduced FIII9′10-CC.
The Synergistic PHSRN Loop in FIII9 Is Relatively Rigid
Interdomain motion as well as modulation of the RGD loop mobility has been implicated in integrin binding and activation by the FIII9–10 fragment of fibronectin (32, 36). We therefore studied the dynamics of FIII9′10, FIII9′10-CCox, and FIII9′10-CCred by 15N relaxation. Local internal dynamics of the backbone NH bond vectors were derived from the [1H]15N NOE (Fig. 6). Values of the [1H]15N NOE of a backbone NH below its maximum value of 0.83 indicate the presence of subnanosecond motions. The data show that residues in β-sheets are comparatively rigid, whereas the N and C termini and a number of the loop regions display increased mobility. The RGD loop in FIII10 is clearly one of the most dynamic regions of the molecule, in agreement with previous studies on the single human FIII10 domain (38) and mouse FIII9–10 (39).
Fig. 6.
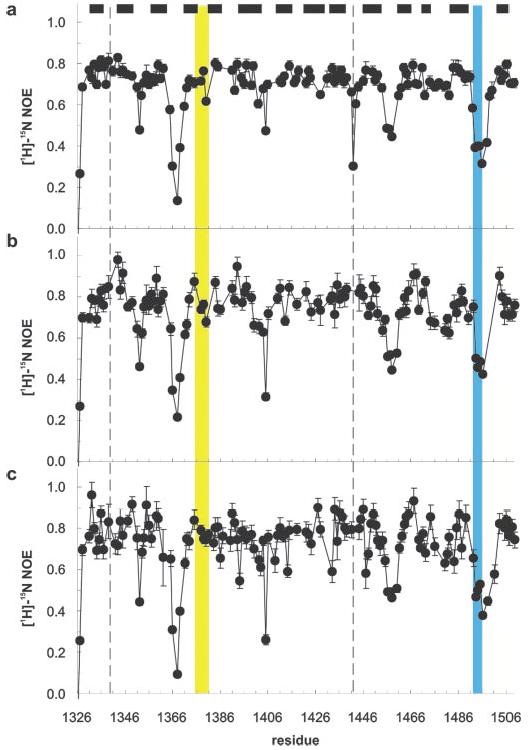
Backbone dynamics of FIII9′10 variants. Shown is a plot of [1H]15N NOE for FIII9′10 (a), FIII9′10-CCred (b), and FIII9′10-CCox (c). The filled bars denote the location of β-strands, and the dotted lines mark the sites of the mutated residues Ala1340 and Val1442. The synergistic PHSRN sequence and the RGD motif are highlighted in yellow and blue, respectively.
We found that the loop containing the PHSRN synergy site in FIII9′10 is rigid compared with the RGD loop and other extended loop regions. The local dynamics of the backbone NH vectors are very similar in FIII9′10 and the oxidized and reduced form of FIII9′10-CC (Fig. 6). In particular, this applies to the RGD loop and the synergy site of the variants. This indicates that the local dynamics of the individual domains on a subnanosecond time scale is virtually unaffected by the introduction of the disulfide bond.
The Interdomain Dynamics of the FIII9′10 Variants Are Similar
Comparison of experimentally determined rotational diffusion tensors and the calculated diffusion tensor of a rigid model provides information on interdomain dynamics. The diffusion tensors of FIII9′10 and FIII9′10-CCox were obtained from an analysis of the T1 and T2 relaxation times (Fig. 7). The errors in the FIII9′10-CCred data were larger, probably due to the presence of dithiothreitol, so that determination of a diffusion tensor was considered unreliable. In FIII9′10 the isotropic correlation times, determined using TENSOR (see “Experimental Procedures”), were 11.9 ns for FIII9 and 12.8 ns for FIII10, and in FIII9′10-CCox, they were 12.4 and 13.6 ns, respectively. For individual modules in both FIII9′10 and FIII9′10-CCox, a prolate diffusion tensor with an axial ratio (D∥/D⊥) of ∼1.8 ± 0.3 yielded an adequate description of the data, indicating that in solution both molecules are cigar-shaped. The small difference in correlation times of the individual domains in each protein and the fact that the axial ratio of the long axis to the short axis (D∥/D⊥) in both FIII9′10 and FIII9′10-CCox is smaller than that expected from a hydrated rigid structure suggest that both proteins have a similar degree of interdomain motion.
Fig. 7.
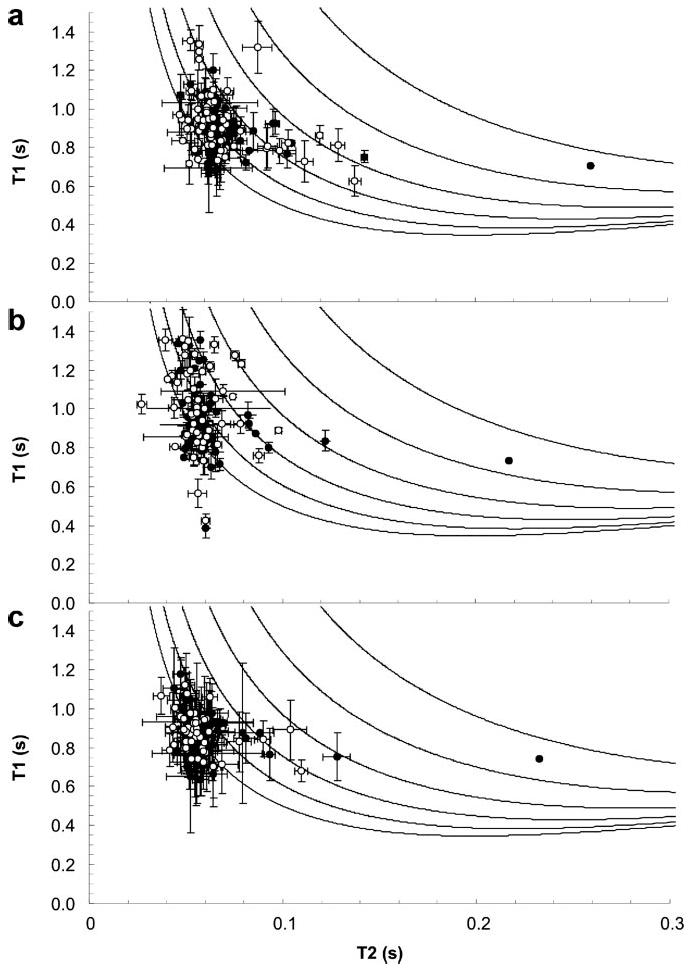
15N-T1 and 15N-T2 relaxation time constants for FIII9′10 (a), FIII9′10-CCred (b), and FIII9′10-CCox (c). The closed circles indicate residues from the FIII9 domain, and open circles show residues from the FIII10 domain. The lines are theoretical T1 and T2 values calculated using the Lipari-Szabo model (44) with order parameters, S2, of 1, 0.9, 0.8, 0.7, 0.6, and 0.5.
We conclude from these structural analyses that the primary effect of the introduction of the disulfide bond between residues Cys1340 and Cys1442 at the interdomain interface of FIII9′10 is a reduction in the interdomain tilt angle. Structural and dynamic changes are localized to the vicinity of the mutated residues. The average interdomain orientation in FIII9′10 in solution shows a greater tilt angle than the x-ray structure, with the RGD site more prominently exposed by the tilt. In contrast, the tilt angle in FIII9′10-CCox is significantly reduced. All FIII9′10 variant proteins appear to have a similar degree of interdomain flexibility.
DISCUSSION
The structure of integrin αvβ3 complexed with an Arg-Gly-Asp peptide (40) provided groundbreaking insight into how integrins interact with specific active motifs within their ligands, but the precise molecular mechanism by which large extracellular matrix proteins bind RGD-dependent integrins remains unclear. A major issue is the nature of the synergistic effect of FIII9 on RGD-mediated binding of fibronectin to integrin α5β1. Here we provide evidence for a further structural determinant for fibronectin-integrin interactions by demonstrating that the FIII9-FIII10 interdomain tilt has a profound effect upon integrin binding and function.
There are currently a number of different and conflicting models to explain how the synergy between FIII9 and FIII10 is achieved. Several lines of evidence suggest that a synergy site in FIII9, containing the residues PHSRN, exerts an effect by making direct contact with integrins such as α5β1 (4, 8, 9, 12). These include observations that the distance between the RGD loop and the PHSRN loop and the degree of interdomain coupling are important for optimal integrin activity (12, 36). Point mutations in the synergy site have been shown to be important for both integrin binding and downstream function (2-4, 6, 7, 9). However, some of the effects of these mutants can be reversed by the addition of contiguous FIII domains or by stability-conferring mutations in FIII9 (2, 3). This suggests that FIII9 provides long range stabilization of critical integrin-binding motifs in the FIII9–10 pair and that the presence of correctly folded FIII9 is required to ensure proper orientation of binding sites in FIII10. This hypothesis is consistent with electron micrographs of the FIII7–10 string of domains in complex with a truncated α5β1 that suggest that FIII9 is not in direct contact with the integrin (11). In consideration of these observations and of differences in the integrin-binding kinetics (on-rates) between native FIII7–10 and its synergy site mutants, Takagi et al. proposed that FIII9 exerts its effects indirectly via a modulation of the RGD loop conformation or by electrostatic steering (11). This interpretation, however, is not in agreement with a recent x-ray scattering and mutagenesis study of a truncated α5β1 construct in complex with FIII6–10 that indicated a region of direct contact between the synergy site and the β-propeller domain of α5 (10).
We have demonstrated previously that biological function and integrin binding activity of FIII9–10 correlates directly with thermodynamic stability of FIII9 (2, 3). However, in this study, we have shown that introduction of a disulfide bond across the interdomain interface between the FIII9 and FIII10 domains increases the stability of the FIII9–10 pair and at the same time attenuates not only the synergistic effect of FIII9 on α5β1 binding but also the ability of the FIII9–10 pair to recognize αvβ3. Integrin binding activity is regained by reduction of the disulfide bond. This strengthens the notion that the effect of manipulating the interdomain interface is specific and distinct from stability effects. Furthermore, we are able to define precisely the effect of the introduction of the disulfide bond on the structure of the FIII9–10 pair. The characterization of the solution structure and dynamics of FIII9′10 and its double cysteine variants reveals that the introduction of the interdomain disulfide bridge reduces the average interdomain tilt angle from ∼28° in the parent protein FIII9′10 to ∼5° in FIII9′10-CCox, in effect straightening the protein structure. These changes correlate with the reduction in biological activity and integrin α5β1 and αvβ3 binding observed with this variant. Given the weight of evidence to suggest that FIII9 is not required for activation of αvβ3, these data are unexpected, since they demonstrate that FIII9 prevents binding to both α5β1 and αvβ3 when the interdomain tilt is reduced. This raises the possibility that FIII9 may exert steric hindrance that abrogates binding of αvβ3 as well as α5β1 when FIII9–10 adopts a straightened conformation. We also note that changes in the local structure and dynamics of FIII9 and FIII10 are restricted to the vicinity of the mutations, thus reinforcing the importance of the altered interdomain tilt, rather than local structural effects, for the modulation of integrin binding.
Conformational stability of FIII9 is known to be important for integrin-dependent function of FIII9–10 (2, 3). The data we present here suggest either that the large increase in stability of FIII9 that we observe in the disulfide-linked variant is detrimental to integrin binding and activation or that any positive effect on integrin binding gained by the stability increase is more than offset by the negative effect of the altered orientation of FIII9. One explanation of our data is that the introduction of the interdomain interface disulfide bond creates a fixed orientation for the FIII9 synergy site with respect to the RGD loop in FIII10 that is not readily accommodated by the surface of the α5 subunit. The observed low flexibility of the loop in FIII9 carrying the PHSRN synergy site would further complicate any conformational adaptation. This would be consistent with a direct interaction of the synergy site residues with the β-propeller domain of α5 (10). However an alternative explanation is that the RGD loop is more accessible in some domain orientations than others, although it is observed to remain mobile in all of the variants analyzed. According to this scenario, the reduction in the tilt between FIII9 and FIII10 introduces a steric effect by FIII9 that precludes sufficient interaction of the RGD loop with its binding pocket at the integrin αvβ3 surface. Binding of the disulfide-linked FIII9′10-CCox to integrin α5β1 is reduced to a level similar to that achieved by FIII10, suggesting that the less tilted FIII9 domain does not block binding of the RGD motif to α5β1 but restricts any synergistic steering effect on integrin recognition. This may in part explain how integrin specificity can arise in response to changes in interdomain orientation. One possibility is that the RGD binding pocket on α5β1 is less deep and more accessible to the ligand than the corresponding interface on αvβ3, thus making α5β1 more tolerant of steric interference by the reoriented FIII9. This scenario does not exclude the use of motifs in FIII9 for direct integrin binding but can readily explain the reduced binding of FIII9′10-CCox to a synergy-independent integrin such as αvβ3.
Whatever the mechanism, we predict that such changes in the interdomain tilt between FIII9 and FIII10 can be achieved in situ because of the interdomain flexibility, which is inferred to be unusually high for this domain pair (32, 41). Previous structural data suggest that the integrin headpiece comprising an α subunit β-propeller and a β subunit I-like domain does not undergo large conformational changes upon binding to fibronectin-derived ligands (10, 11, 40). Flexibility of the FIII9–10 pair, as revealed here and inferred from previous studies on human (11, 36) and mouse (39) FIII9–10, may thus allow conformational changes that are required to accommodate binding to the integrin headpiece.
In summary, we have demonstrated that a change in fibronectin domain tilt can act as a switch between high and low integrin binding activity. Such changes in the interdomain tilt angle may be important physiologically in accommodating structural differences between the low activity, compact form of soluble plasma fibronectin and the high activity, fibrillar form of matrix fibronectin (13, 42). In addition, changes in the FIII9–10 interdomain tilt may occur in situ in response to binding of other extracellular matrix components at proximal and/or distal sites on the fibronectin molecule. We therefore propose that modulation of interdomain tilt provides an additional mechanism for fine-tuning fibronectin-integrin interactions.
Footnotes
This work was supported by the Medical Research Council and the Wellcome Trust. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: BHK, baby hamster kidney; GdnHCl, guanidinium hydrochloride; HSQC, heteronuclear single quantum correlation; NOE, nuclear Overhauser effect; NOESY, NOE spectroscopy; RDC, residual dipolar coupling; TOCSY, total correlation spectroscopy.
REFERENCES
- 1.Obara M, Yoshizato K. Exp. Cell Res. 1995;216:273–276. doi: 10.1006/excr.1995.1033. [DOI] [PubMed] [Google Scholar]
- 2.Altroff H, Choulier L, Mardon HJ. J. Biol. Chem. 2003;278:491–497. doi: 10.1074/jbc.M209992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altroff H, van der Walle CF, Asselin J, Fairless R, Campbell ID, Mardon HJ. J. Biol. Chem. 2001;276:38885–38892. doi: 10.1074/jbc.M105868200. [DOI] [PubMed] [Google Scholar]
- 4.Redick SD, Settles DL, Briscoe G, Erickson HP. J. Cell Biol. 2000;149:521–527. doi: 10.1083/jcb.149.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akiyama SK, Aota S, Yamada KM. Cell Adhes. Commun. 1995;3:13–25. doi: 10.3109/15419069509081275. [DOI] [PubMed] [Google Scholar]
- 6.Kauf AC, Hough SM, Bowditch RD. Biochemistry. 2001;40:9159–9166. doi: 10.1021/bi010503x. [DOI] [PubMed] [Google Scholar]
- 7.Bowditch RD, Hariharan M, Tominna EF, Smith JW, Yamada KM, Getzoff ED, Ginsberg MH. J. Biol. Chem. 1994;269:10856–10863. [PubMed] [Google Scholar]
- 8.Mould AP, Askari JA, Aota S, Yamada KM, Irie A, Takada Y, Mardon HJ, Humphries MJ. J. Biol. Chem. 1997;272:17283–17292. doi: 10.1074/jbc.272.28.17283. [DOI] [PubMed] [Google Scholar]
- 9.Aota S, Nomizu M, Yamada KM. J. Biol. Chem. 1994;269:24756–24761. [PubMed] [Google Scholar]
- 10.Mould AP, Symonds EJ, Buckley PA, Grossmann JG, McEwan PA, Barton SJ, Askari JA, Craig SE, Bella J, Humphries MJ. J. Biol. Chem. 2003;278:39993–39999. doi: 10.1074/jbc.M304627200. [DOI] [PubMed] [Google Scholar]
- 11.Takagi J, Strokovich K, Springer TA, Walz T. EMBO J. 2003;22:4607–4615. doi: 10.1093/emboj/cdg445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grant RP, Spitzfaden C, Altroff H, Campbell ID, Mardon HJ. J. Biol. Chem. 1997;272:6159–6166. doi: 10.1074/jbc.272.10.6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson KJ, Sage H, Briscoe G, Erickson HP. J. Biol. Chem. 1999;274:15473–15479. doi: 10.1074/jbc.274.22.15473. [DOI] [PubMed] [Google Scholar]
- 14.Mardon HJ, Grant KE. FEBS Lett. 1994;340:197–201. doi: 10.1016/0014-5793(94)80137-1. [DOI] [PubMed] [Google Scholar]
- 15.van der Walle CF, Altroff H, Mardon HJ. Protein Eng. 2002;15:1021–1024. doi: 10.1093/protein/15.12.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pace CN, Scholtz JM. In: Protein Structure: A Practical Approach. Creighton TE, editor. IRL Press; Oxford, UK: 1997. pp. 299–321. [Google Scholar]
- 17.Sass HJ, Musco G, Stahl SJ, Wingfield PT, Grzesiek S. J. Biomol. NMR. 2000;18:303–309. doi: 10.1023/a:1026703605147. [DOI] [PubMed] [Google Scholar]
- 18.Ishii Y, Markus MA, Tycko R. J. Biomol. NMR. 2001;21:141–151. doi: 10.1023/a:1012417721455. [DOI] [PubMed] [Google Scholar]
- 19.Chou JJ, Gaemers S, Howder B, Louis JM, Bax A. J. Biomol. NMR. 2001;21:377–382. doi: 10.1023/a:1013336502594. [DOI] [PubMed] [Google Scholar]
- 20.Kay LE, Keifer P, Saarinen T. J. Am. Chem. Soc. 1992;114:10663–10665. [Google Scholar]
- 21.Grzesiek S, Bax A. J. Am. Chem. Soc. 1993;115:12593–12594. [Google Scholar]
- 22.Kay LE, Marion D, Bax A. J. Magn. Reson. 1989;84:72–84. [Google Scholar]
- 23.Driscoll PC, Clore GM, Marion D, Wingfield PT, Gronenborn AM. Biochemistry. 1990;29:3542–3556. doi: 10.1021/bi00466a018. [DOI] [PubMed] [Google Scholar]
- 24.Kay LE, Ikura M, Tschudin R, Bax A. J. Magn. Reson. 1990;89:496–514. doi: 10.1016/j.jmr.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Grzesiek S, Bax A. J. Magn. Reson. 1992;96:432–440. [Google Scholar]
- 26.Grzesiek S, Bax A. J. Am. Chem. Soc. 1992;114:6291–6293. [Google Scholar]
- 27.Grzesiek S, Bax A. J. Biomol. NMR. 1993;3:185–204. doi: 10.1007/BF00178261. [DOI] [PubMed] [Google Scholar]
- 28.Farrow NA, Zhang O, Forman-Kay JD, Kay LE. J. Biomol. NMR. 1994;4:727–734. doi: 10.1007/BF00404280. [DOI] [PubMed] [Google Scholar]
- 29.Ottiger M, Delaglio F, Bax A. J. Magn. Reson. 1998;131:373–378. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- 30.Johnson BA, Blevins RA. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 31.Dosset P, Hus JC, Marion D, Blackledge M. J. Biomol. NMR. 2001;20:223–231. doi: 10.1023/a:1011206132740. [DOI] [PubMed] [Google Scholar]
- 32.Leahy DJ, Aukhil I, Erickson HP. Cell. 1996;84:155–164. doi: 10.1016/s0092-8674(00)81002-8. [DOI] [PubMed] [Google Scholar]
- 33.Bork P, Downing AK, Kieffer B, Campbell ID. Q. Rev. Biophys. 1996;29:119–167. doi: 10.1017/s0033583500005783. [DOI] [PubMed] [Google Scholar]
- 34.Werner JM, Campbell ID, Downing AK. Methods Mol. Biol. 2001;173:285–300. doi: 10.1385/1-59259-184-1:285. [DOI] [PubMed] [Google Scholar]
- 35.Dosset P, Hus JC, Blackledge M, Marion D. J. Biomol. NMR. 2000;16:23–28. doi: 10.1023/a:1008305808620. [DOI] [PubMed] [Google Scholar]
- 36.Spitzfaden C, Grant RP, Mardon HJ, Campbell ID. J. Mol. Biol. 1997;265:565–579. doi: 10.1006/jmbi.1996.0736. [DOI] [PubMed] [Google Scholar]
- 37.Bax A, Kontaxis G, Tjandra N. Methods Enzymol. 2001;339:127–174. doi: 10.1016/s0076-6879(01)39313-8. [DOI] [PubMed] [Google Scholar]
- 38.Main AL, Harvey TS, Baron M, Boyd J, Campbell ID. Cell. 1992;71:671–678. doi: 10.1016/0092-8674(92)90600-h. [DOI] [PubMed] [Google Scholar]
- 39.Copie V, Tomita Y, Akiyama SK, Aota S, Yamada KM, Venable RM, Pastor RW, Krueger S, Torchia DA. J. Mol. Biol. 1998;277:663–682. doi: 10.1006/jmbi.1998.1616. [DOI] [PubMed] [Google Scholar]
- 40.Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL, Arnaout MA. Science. 2002;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 41.Sharma A, Askari JA, Humphries MJ, Jones EY, Stuart DI. EMBO J. 1999;18:1468–1479. doi: 10.1093/emboj/18.6.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ugarova TP, Zamarron C, Veklich Y, Bowditch RD, Ginsberg MH, Weisel JW, Plow EF. Biochemistry. 1995;34:4457–4466. doi: 10.1021/bi00013a039. [DOI] [PubMed] [Google Scholar]
- 43.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lipari G, Szabo A. J. Am. Chem. Soc. 1982;104:4546–455. [Google Scholar]