

Abstract
The ninth and tenth type III domains of fibronectin each contain specific cell binding sequences, RGD in FIII10 and PHSRN in FIII9, that act synergistically in mediating cell adhesion. We investigated the relationship between domain-domain orientation and synergistic adhesive activity of the FIII9 and FIII10 pair of domains. The interdomain interaction of the FIII9–10 pair was perturbed by introduction of short flexible linkers between the FIII9 and FIII10 domains. Incremental extensions of the interdomain link between FIII9 and FIII10 reduced the initial cell attachment, but had a much more pronounced effect on the downstream cell adhesion events of spreading and phosphorylation of focal adhesion kinase. The extent of disruption of cell adhesion depended upon the length of the interdomain linker. Nuclear magnetic resonance spectroscopy of the wild type and mutant FIII9–10 proteins demonstrated that the structure of the RGD-containing loop is unaffected by domain-domain interactions. We conclude that integrin-mediated cell adhesion to the central cell binding domain of fibronectin depends not only upon specific interaction sites, but also on the relative orientation of these sites. These data have implications for the molecular mechanisms by which integrin-ligand interactions are achieved.
The regulated adhesion of cells to the extracellular matrix (ECM)1 is essential for the development and function of normal tissues, and aberrant regulation of cell adhesion is often associated with disease. Fibronectins (FNs) are adhesive proteins that are abundant in the ECM of many cell types and have a critical role in many biological processes (1). Twenty isoforms of human FN, the expression of which is developmentally regulated, can be generated as a result of alternative splicing of the primary FN transcript (2–4). The FN molecules are dimers of disulfide-linked 235-kDa monomers. Each monomer is composed of type I, type II, and type III domains (FI, FII, and FIII), identified as repeating amino acid motifs in the primary structure (5) (Fig. 1). These motifs occur in many diverse cellular and extracellular proteins (6). Separable, functional regions of the FN molecule have been identified that contain binding activities for other components of the ECM, including collagen, fibrin, and heparin (1). Cells bind to FN via the central cell binding domain (CCBD) spanning the eighth, ninth, and tenth FIII domains (FIII8–10) (7) and via the CS1 and CS5 sites in the alternatively spliced IIICS region (8, 9) (see Fig. 1).
Fig. 1. Domain structure of fibronectin.

The diagram illustrates the organization of domains within a FN monomer. Binding regions for other ECM components are indicated below the molecule and the alternatively spliced EDIIIA, EDIIIB, and IIICS regions above. The central cell binding domain (CCBD). The three types of FN structural domains are represented by symbols: ▪, FN type I domain; ○, FN type II domain; open box with number inside, FN type III domain. The site of the disulfide bridges linking two monomers is indicated by SS.
The adhesion of cells to FN in the ECM is mediated by the integrin family of transmembrane receptors (10–12). The minimal cell recognition sequence RGD in FIII10 has been shown previously to interact with a number of integrins including α5β1 (13), α3β1 (14), αvβ1 (15, 16), αvβ3 (17), αvβ6 (18, 19), α8β1 (20, 21), and αIIbβ3 (20, 21). Integrin-mediated cell adhesion to FN results in phosphorylation of focal adhesion kinase (FAK, also known as pp125FAK), organization of the actin cytoskeleton, and cell spreading (22–26). Given the ubiquity of expression of FNs and their extensive role in cell adhesion, the molecular mechanisms by which FN supports cell binding and induces the integrin signaling pathway are of great interest.
An earlier nuclear magnetic resonance (NMR) study of FIII10 demonstrated that the RGD sequence resides in a mobile loop between the F and G β strands of the domain (27). Synthetic peptides that contain the RGD sequence exhibit some cell adhesive activity but do not mimic the full adhesive function of the FN molecule (7, 28, 29). Additional sites have been identified recently that are required for maximal adhesive activity. One site has been mapped to the loop between the C′ and E β strands in the ninth FIII domain and contains the peptide sequence PHSRN (30–34). The PHSRN site has been shown to act synergistically with RGD in cell adhesion mediated by α5β1, αvβ3, and αIIbβ (32, 34, 35).
We have shown previously that the structural integrity of the FIII9–10 pair of domains is required for the synergistic activity of the two domains in supporting cell spreading (36). Here we have investigated the relationship between domain-domain interactions and the biological activity of the FIII9–10 pair in specific cell adhesion events. Our strategy was to modify the strength of the domain-domain interaction and the relative mobility of the two domains by introducing flexible polyglycine interdomain linkers of different lengths. The influence of the linkers on the structure of the FIII9–10 pair was analyzed by NMR and the affect on biological activity of FIII9–10 was assessed by the ability of immobilized wild type and mutant GST-FIII fusion proteins to support events involved in cell adhesion. We discuss the correlation between our biological data and the underlying structural changes as examined by NMR and thermodynamic methods.
EXPERIMENTAL PROCEDURES
Biophysical Studies
NMR measurements were performed on fully 15N-labeled samples of FIII10 and FIII9–10 at 25 °C in 20 mm sodium acetate, 5% D2O, pH 4.8. Protein concentrations were 4 mM (FIII10) or 1.5 mm (FIII9–10). Three-dimensional 15N-correlated 1H,1H NOESY spectra (37) were recorded on a home-built spectrometer operating at a proton frequency of 600 MHz.
Construction of pGEXFIII9–10 Mutants
The constructs pGEXFIII10 and pGEXFIII9–10 have been described previously (36). The FIII9–10 linker mutant FIII9-PG-10, containing one additional proline and one glycine residue between threonine 1415 at the COOH terminus of FIII9, and valine 1416 at the NH2 terminus of FIII10 (according to the domain boundaries in Kornblihtt et al. (38)) was expressed from the construct pGEXFIII9-PG-10. For construction of pGEXFIII9-PG-10, FIII9, and FIII10 were amplified separately from FN cDNA (pFHIII (2)) by Pfu polymerase (Stratagene, La Jolla, CA) in the polymerase chain reaction. Oligonucleotide primers were designed that introduced a BglII site into the 5′ end of the amplified FIII9 cDNA, an XhoI site into the 3′ end of FIII10, and a SmaI site, CCCGGG, encoding the extra proline and glycine residues, into the FIII9–10 linker. The primers used were: FIII9 5′, TGAAGATCTGGTCTTGATTCCCCAACT, FIII9 3′, GGGTGTTGATTGTTGGCCAATCAATA; FIII10 5′, GGGGTTTCTGATGTTCCGAGGGA, FIII10 3′, TCACTCGAGTCATGTTCGGTAATTAATGGA. The polymerase chain reaction products were cleaved with either BglI (FIII9) or XhoI (FIII10), gel-purified using QIAEX II (Qiagen Ltd., Dorking, United Kingdom (UK)) according to the manufacturer’s instructions, and cloned into the BamHI/XhoI sites of pGEX4T (Pharmacia, Uppsala, Sweden). The sequence of the inserts in the recombinant clones was confirmed using the Sequenase 2.0 kit (Amersham International plc, Amersham, UK).
The mutant pGEXFIII9-P[G]5-10, encoding the fusion protein GFIII9-P[G]5-10, was generated by the introduction of the sequence GGAGGCGGAGGC encoding three glycine residues. Annealed oligonucleotides were inserted into the SmaI site of pGEXFIII9-PG-10. The sequence of the recombinant clones was confirmed as above. The position of insertion of the extended linkers in the mutant FIII9–10 proteins is shown in Fig. 2.
Fig. 2. Crystal structure of the FIII9–10 pair showing linker insertion point.

Ribbon structure of FIII9–10 pair from Leahy et al. (44). Linkers were inserted immediately before valine 1416 (arrow), the first valine of FIII10. The sequences GRGDS and PHSRN are displayed in ball-and-stick format.
Protein Expression
GST fusion proteins (GFIII9, GFIII10, GFIII9–10, GFIII9-PG-10, GFIII9-P[G]5-10) were expressed as described previously (36) with some modifications. Cultures of Escherichia coli transformed with pGEXFIII constructs were grown overnight in 8 × TY (6.4% tryptone T, 4.0% yeast extract, 0.5% NaCl), diluted 1 in 10 into fresh 8 × TY, and incubated with shaking for 1 h at 37 °C. Induction of protein expression was achieved by addition of 0.1 m isopropyl-β-d-thiogalactopyranoside and further incubation for 3 h at 37 °C. Cells were pelleted by centrifugation, resuspended in 0.02 volumes ice-cold PBS, and lysed by freeze-thawing and sonication. The cell debris was pelleted by centrifugation and the supernatant filtered through a 0.22-μm pore filter and nutated with 50% glutathione-Sepharose beads (Pharmacia Biotech Inc.) for 10 min at 20 °C. The fusion proteins were eluted from the glutathione-Sepharose beads with 10 mm glutathione, 50 mm Tris-Cl, pH 8, and dialyzed against PBS at 4 °C. Cleaved FN fragments FIII9, FIII10, FIII9–10, FIII9-PG-10, and FIII9-P[G]5-10 were produced by the addition of thrombin (2.5 units of thrombin/mg of protein) to GST-FIII fusion proteins adsorbed to the glutathione-Sepharose matrix. Purity of the proteins was assessed by SDS-PAGE and mass spectroscopy.
Cell Attachment and Spreading Assays
Baby hamster kidney (BHK) cells and human endometrial stromal fibroblasts (hESF) were used in cell attachment and spreading assays. Pure cultures of hESF were prepared by collagenase digestion of endometrial tissue followed by centrifugation through Percoll as described elsewhere (39). Cell adhesion and spreading assays were carried out as described previously (36). Essentially, the surface of duplicate wells of 96-well flat-bottomed, tissue culture-grade plates (Becton Dickinson, Oxford, UK) was coated with doubling dilutions of 100 μg ml−1 GST-FIII fusion proteins in PBS for 16 h at 4 °C and washed with PBS. Uncoated plastic was blocked by incubation in 1% bovine serum albumin in PBS for 1 h at 37 °C. Either BHK or hESF cells (104 in 100 μl in GMEM or Dulbecco’s modified Eagle’s medium, respectively) were inoculated into each well and incubated in 5% CO2 for 1 h at 37 °C. Cells were washed gently in PBS, fixed in 4% formaldehyde, 4% glutaraldehyde in PBS, and scored for spreading as described previously (36). Total cell attachment was assessed by staining with 0.1% crystal violet as described elsewhere (40). Bound dye was measured at A570 nm with a Titertek Multiscan plate reader.
For the inhibition assays, the surface of duplicate wells of a 96-well plate was coated with 5 μg ml−1 FN and washed and blocked as described above. Test protein diluted in 25 μl of PBS was incubated with 104 BHK cells in 25 μl of GMEM for 2 min. Cells and protein were then inoculated into the precoated wells equilibrated with 50 μl of GMEM. Cells were incubated, fixed, and analyzed as described above.
Inhibition of Cell Adhesion by Integrin Antibodies
The surface of duplicate wells of a 96-well plate were coated with 10 μg ml−1 FN and blocked with bovine serum albumin as described above. GMEM (30 μl) was added to each well and the plate incubated for 40 min at 37 °C. BHK cells (104 cells in 50 μl of GMEM) were incubated with 1 μg of anti-α5 (clone SAM-1), α2 (clone AK7; both from Serotec, Kidlington, UK), α3 (clone P1B5) or αv (clone VNR147); both from Life Technologies, Inc., Paisley, UK) antibodies in 20 μl of PBS for 2 min at 20 °C. Cells in antibody solution and GMEM were then added to the wells and the attachment assays performed as descibed above. The results were expressed as the mean of three independent experiment plus or minus the standard error of the mean (S.E.).
Immunoprecipitation and Western Blotting of FAK
BHK cells were allowed to spread for 1 h on tissue culture plastic-coated with 100 μg ml−1 GST-FIII fusion proteins as descibed above. The cells were washed three times with PBS, lysed with 1% SDS, 10 mm Tris, pH 7.4, 1 mm phenylmethylsulfonyl fluoride, 10 μg ml−1 leupeptin for 20 min at 0 °C, and passed several times through a 25-gauge needle. Cleared lysates containing equal amounts of total cell protein were incubated with anti-FAK antibody (Transduction Laboratories, Lexington, KY) in immunoprecipitation buffer (10 mm Tris, pH 7.4, 150 mm NaCl, 1% (w/v) Triton X-100, 10% (w/v) glycerol, 1 mm EDTA, 1 mm Na3VO4, 1 mm phenylmethylsulfonyl fluoride, 10 μg ml−1 leupeptin) for 1 h at 4 °C. Agarose-conjugated secondary antibody was added and incubated for 1 h at 4 °C. Precipitates were washed three times with immunoprecipitation buffer, resuspended in SDS-PAGE sample buffer, and boiled for 3 min. Duplicate samples of proteins were subjected to 10% SDS-PAGE (41) and Western blotted onto nitrocellulose membranes (42). Membranes were incubated in either anti-phosphotyrosine antibody (PY20; Transduction Laboratories) or anti-FAK antibody followed by horseradish peroxidase-conjugated secondary antibody (Sigma, Poole, UK). Proteins were visualized by enhanced chemiluminescence (ECL) using the ECL kit (Amersham International plc) according to the manufacturer’s instructions. The bands were quantified by scanning densitometry and Intelligent Quantifier software (BioImage, Ann Arbor, MI), and the level of FAK phosphorylation was expressed as relative to total FAK.
RESULTS
Extension of the FIII9–10 Linker Leads to Reduced Interdo-main Interaction
Detailed NMR and thermodynamic studies of the five cleaved constructs (FIII9, FIII10, FIII9–10, FIII9-PG-10, and FIII9-P[G]5-10) are presented in a separate publication (45). Briefly, introduction of additional linker residues leads to a gradual increase in the mobility of the domains relative to each other, as indicated by increasing T2 relaxation times of the backbone amide 15N-nuclei and to decreasing thermodynamic stabilization energies. Our results indicate that domain-domain interactions are significantly perturbed, but not completely abolished, by introduction of additional linker residues. However, perturbation of the domain-domain interactions does not affect the structure of the RGD site in solution. Fig. 3 shows that the RGDS peptide segment in FIII10 and FIII9–10 has near identical chemical shifts of backbone and side chain resonances and a completely conserved pattern of proton-proton NOEs. These data indicate that the presence of FIII9 does not introduce any additional structural or motional constraints into the highly flexible RGDS peptide.
Fig. 3. NMR spectroscopy of the RGD site of FIII10 and FIII9–10.

For each residue a pair of strips from 1H,1H NOESY planes of three-dimensional 15N-correlated NOESY spectra of uniformly 15N-labeled FIII10 and FIII9–10 is shown. Strips were taken at the 15N frequency of the 15N,1H direct correlation peak. Left- and right-hand strips of each pair correspond to the spectra of FIII10 and FIII9–10, respectively. The positions of NH and CαH cross-peaks are indicated by rectangles and circles, respectively.
Cell Attachment to FIII9–10 Is Partially Inhibited by Disruption of the Physical Interaction between FIII9 and FIII10
The receptors that mediate the attachment of BHK cells to the FIII9–10 region of FN were defined by the use of function blocking monoclonal antibodies (Fig. 4). Attachment of BHK cells to FN was inhibited in the presence of anti-α5 and αv antibodies by 80 and 25%, respectively, indicating that binding of these cells to FN is primarily a function of integrin α5β1, with some αvβ3 activity.
Fig. 4. Inhibition of cell attachment by function-blocking anti-integrin antibodies.

Adhesion of BHK cells to 100 μg ml−1 FN (shaded columns)- or GFIII9–10 (white columns)-coated plastic in the presence of 10 μg ml−1 anti-α2, anti-α3, anti-α5, or anti-αv antibody.
The FIII domain fusion proteins GFIII10, GFIII9–10, the two modified fusion proteins GFIII9-PG-10 and GFIII9-P[G]5-10, and FN were assayed for their ability to support BHK cell attachment, as assessed by measurement of crystal violet incorporation into adherent cells on plastic coated with the immobilized proteins (Fig. 5A). The fusion proteins exhibited similar binding capacities to plastic as assessed by enzyme-linked immunosorbent assay (data not shown). At high coating concentration (100 μg ml−1) FN and GFIII9–10 exhibited similar levels of cell attachment activity. Attachment activities of GFIII10, GFIII9-PG-10, and GFIII9-P[G]5-10 were 50, 65 and 60%, respectively, that of GFIII9–10. Thus attachment activity of both the mutant FIII9–10 proteins was reduced to a level similar to that of GFIII10. At lower coating concentrations (e.g. 6 μg ml−1, Fig. 5A) the differences in attachment activity of all GST-FIII fusion proteins were more pronounced. GFIII9–10 exhibited 65% activity relative to FN, GFIII9-PG-10, 38%, GFIII9-P[G]5-10, 28%, and GFIII10, 10% that of FN.
Fig. 5. Cell attachment to wild type and mutant linker FIII9–10 proteins.

BHK (A) and hESF (B) cell attachment at 100 μg ml−1 (shaded columns) and 6.25 μg ml−1 (white columns) coating concentration. Cell attachment to each of the test proteins was assessed by measurement of crystal violet incorporation at A570 nm and expressed relative to attachment on 100 μg ml−1 FN. The data shown represent the mean ± S.E. of at least four experiments.
The requirement for structural integrity of FIII9–10 for cell attachment via integrins in addition to α5β1 was also investigated in cell attachment assays using hESF cells, attachment of which is mediated by multiple integrins including α5β1.2 The data for these experiments are shown in Fig. 5B. At high coating concentrations (100 μg ml−1) the level of attachment to wild type GFIII9–10 was similar to that on FN. In addition, hESF cells attached efficiently to both GFIII9-PG-10, GFIII9-P[G]5-10, and GFIII10 coated at 100 μg ml−1, at a similar level to FN and GFIII9–10. At lower coating concentrations (e.g. 6 μg ml−1, Fig. 5B), while GFIII9–10 supported maximal attachment of stromal cells there was a stepwise decrease with the mutant FIII9–10 proteins with increase in the length of the interdomain linker. Immobilized GFIII10 supported approximately 10% the attachment on FN or GFIII9–10 at this concentration.
Cell Spreading Activity of FIII9–10 Is Progressively Reduced as the FIII9–10 Interdomain Linker Region Is Increased
The morphology of BHK cells plated onto FN, GFIII9–10, GFIII9-PG-10, GFIII9-P[G]5-10, GFIII10, or GST is shown in Fig. 6. Cells plated onto FN (Fig. 6A) exhibited a spread morphology. Cells plated onto GFIII9–10 were also spread, but not to the same extent as on FN (Fig. 6, compare B with A). The cells spread less on GFIII9-PG-10 (Fig. 6C), and very few cells exhibited a spread morphology on GFIII9-P[G]5-10 (Fig. 6D) or GFIII10 (Fig. 6E). Very few cells attached to GST (Fig. 6F) and those that did attach remained rounded.
Fig. 6. BHK cell adhesion to wild type and mutant linker proteins.
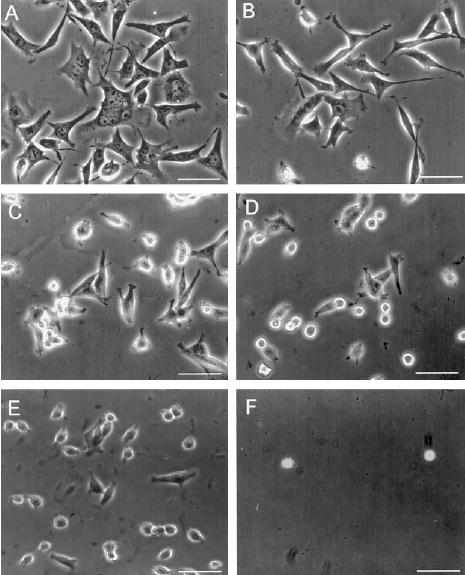
Phase contrast microscopy of BHK cells plated on FN (A) GFIII9–10 (B), GFIII9-PG-10 (C ), GFIII9-P[G]5-10 (D), GFIII10 (E), or GST (F) coated at 100 μg ml−1 protein. Bar = 25 μm.
Cells attached to plates coated with the GST-FIII fusion proteins and FN were scored for spreading, expressed as the percentage of attached cells that exhibited a spread morphology. Both BHK (Fig. 7A) and hESF (Fig. 7B) cells plated onto GFIII9–10 at a coating concentration of 100 μg ml−1 exhibited similar spreading to cells on FN. However, 50% spreading activity was obtained at a much lower coating concentration of FN (~0.2 μg ml−1) compared with 50% spreading activity on GFIII9–10 (12.5 μg ml−1). Both BHK and hESF cells spread poorly on GFIII10, which for both cell types supported less than 10% the spreading activity of GFIII9–10 at a coating concentration of 100 μg ml−1.
Fig. 7. Cell spreading on wild type and mutant linker proteins.

BHK cells (A, C) and hESF cells (B, D) were assayed for spreading on FN (•), GFIII9–10 (○), GFIII10 (×), GFIII9-PG-10 (▪), GFIII9-P[G]5-10 (□), or GST (▴) on doubling dilutions of each test protein (A and B). The percentage of cells spread on proteins coated at 100 μg ml−1 (shaded columns), and 6.25 μg ml−1 (white columns) is shown in C and D. The data shown represent the mean ± S.E. of at least four experiments.
Spreading of BHK cells on GFIII9-PG-10 coated at 100 μg ml−1 (Fig. 7C) was reduced to 70% compared with GFIII9–10, similar to the relative levels of cell attachment observed for these proteins. Spreading on the mutant GFIII-P[G]5-10, containing the longer linker, was substantially reduced at 100 μg ml−1, exhibiting levels of spreading activity less than 25% that of GFIII9–10. These differences were more pronounced at lower coating concentrations. At a coating concentration of 6.25 μg ml−1 (Fig. 7C) spreading activity of BHK cells on GFIII9–10 was approximately 30% that of FN, spreading on GFIII9-PG-10 was ~25% that of GFIII9–10, and there was no spreading on GFIII9-P[G]5-10.
The number of hESF cells that spread on GFIII9–10 coated at 100 μg ml−1 was similar to BHK cells (Fig. 7, compare D with C). The number of hESF cells spread on GFIII9-PG-10 or GFIII9-P[G]5-10 was approximately 90 and 30%, respectively, of the number of cells spread on GFIII9–10. At a coating concentration of 6.25 μg ml−1, spreading of hESF cells on GFIII9–10 was 10% that of cells on FN. Cells plated onto GFIII9-PG-10 and GFIII9-P[G]5-10 exhibited 35 and 10%, respectively, levels of spreading on GFIII9–10. Thus spreading activity of hESF cells on FIII9-PG-10 was elevated compared with BHK cells. Spreading of hESF cells on GFIII10 at 100 μg ml−1 and at 6.25 μg ml−1 was 19 and 5%, respectively, relative to spreading on GFIII9–10, similar to levels found for BHK cells.
Activity of Wild Type and Mutant FIII9–10 Domains in Soluble Phase Assays
The capacity of purified wild type and mutant FIII domains to inhibit adhesion of BHK cells to fibronectin was also assessed in inhibition assays. The FIII domains were purified from GST adsorbed to glutathione-Sepharose beads by cleavage with thrombin and were tested for their capacity to inhibit attachment of BHK cells to immobilized FN coated on plastic at 5 μg ml−1. These data are shown in Fig. 8. Attachment of BHK cells to FN was completely abolished by the presence of 125 μm FIII9–10 in the culture. At a concentration of 125 μm, FIII9-PG-10 inhibited attachment of BHK cells to FN by 20%, and no significant inhibition was observed with FIII9-P[G]5-10 within the molar range used in these experiments. Relative to the controls, in which no protein was added to the cells prior to plating out, 50% inhibition of spreading of BHK cells on FN was achieved in the presence of 12.5 μm FIII9–10 and 125 μm FIII9-PG-10. No inhibition of cell spreading was observed with FIII9-P[G]5-10. At a concentration of 125 μm, FIII10 and GRGDS peptide both inhibited attachment of BHK cells to FN by 20 and 30%, respectively, and had a small affect on cell spreading. Thus insertion of two amino acids in the interdomain linker of FIII9–10 both immobilized and in solution had different affects on cell attachment and cell spreading.
Fig. 8. Inhibition of BHK cell adhesion by wild type and mutant FIII9–10 domains.

Attachment (A) and spreading (B) of BHK cells to FN (coated at 10 μg ml−1) was assessed in the presence of doubling dilutions of FIII10 (×), FIII9–10 (○), FIII9-PG-10 (▪), FIII9-P[G]5-10 (□), or GRGDS peptide (▵).
Phosphorylation of FAK Is Strictly Dependent upon Structural Integrity of FIII9–10
Given that perturbation of the FIII9–10 had a profound effect on cell spreading, and given that integrin-mediated cell spreading appears to be preceded by FAK phosphorylation (11), we examined the relationship between cell spreading and FAK phosphorylation in response to adhesion to the wild type and mutant FIII9–10 proteins. The relative amounts of phosphorylated FAK in BHK cells plated onto immobilized wild type or mutant GFIII9–10 domains were determined by immunoprecipitation of FAK and subsequent Western blotting with anti-phosphotyrosine antibodies as shown in Fig. 9A. The intensity of bands identified with anti-phosphotyrosine antibodies and representing phosphorylated FAK was expressed relative to total FAK as shown in Fig. 9B. Introduction of the short interdomain linker in GFIII9-PG-10 reduced levels of FAK phosphorylation by one-third. The longer linker in GFIII9-P[G]5-10 led to a further reduction in FAK phosphorylation, giving levels approximately 30% of that obtained with cells plated onto GFIII9–10 and similar to that of cells plated on GFIII10.
Fig. 9. Phosphorylation of FAK in BHK cells on wild type and mutant FIII9–10 fusion proteins.

Total FAK and phosphorylated FAK (coprecipitated with anti-FAK antibodies) from BHK cells plated on plastic coated with 100 μg ml−1 fusion proteins were detected by Western blotting (A). The percentage of phosphorylation of FAK on the mutant proteins relative to phosphorylation on GFIII9–10 (100%) was determined by densitometric analyses of the Western blots in A (B).
For comparison of cell attachment, cell spreading and FAK phosphorylation in response to GFIII10 and the mutant FIII9–10 proteins, the levels of activity obtained in each assay were expressed relative to those obtained for FIII9–10 (Table I). Phosphorylation of FAK and cell spreading activities were tightly correlated and affected to the same degree by insertion of additional linker sequence, whereas levels of cell attachment were consistently higher.
Table I. Comparison of FAK phosphorylation, cell spreading, and cell attachment on wild type and mutant FIII9–10 proteins.
Levels of cell attachment, cell spreading, and FAK phosphorylation (from Figs. 5, 8, and 9) on the linker GFIII9–10 proteins are expressed relative to the levels obtained for GFIII9–10, the latter taken as 1 for each parameter measured.
| Protein | FAK phosphorylation | Cell spreading | Cell attachment |
|---|---|---|---|
| GFIII9–10 | 1.00 | 1.00 | 1.00 |
| GFIII9-PG-10 | 0.67 | 0.65 | 0.73 |
| GFIII9-P[G]5-10 | 0.29 | 0.29 | 0.65 |
| GFIII10 | 0.14 | 0.10 | 0.55 |
DISCUSSION
Previous studies have identified the FN peptide sequences RGD in FIII10 and PHSRN in FIII9 as integrin binding sites (32, 34) and have demonstrated the importance of structural integrity and contiguity of the FIII9–10 pair for synergistic cell adhesive activity of the two domains (36). In this study we have further dissected the intra- and intermolecular interactions involved in the cell adhesion activity of the FIII9–10 pair. We show that 1) precise spatial positioning of the two binding sites in FIII9 and FIII10 is critical for synergistic activity, and 2) downstream cell adhesion events of FAK phosphorylation and cell spreading are more sensitive than cell attachment to disruption of the spatial relationship of FIII9 and FIII10.
A number of experimental approaches have been used to dissect the cell adhesive activity of FN. The synergistic site containing PHSRN was recently identified in two important experiments (32, 34). In the case of α5β1-mediated cell adhesion, Aota et al. (32) used hybrid constructs consisting of the eigth and tenth FIII domains. In this experiment putative synergy regions from FIII9 were swapped into the corresponding position in FIII8 in the FIII8–10 hybrid. The synergistic site was mapped in this way to the loop between the C′ and E β strands in FIII9. In a second study by Bowditch et al. (34) the sequence DRVPHSRNSIT was identified as the candidate synergistic site for αIIbβ3-mediated cell adhesion, based upon the ability of the peptide to inhibit binding of a 20-kDa fragment of FN containing FIII9 and FIII10 to the platelet-specific integrin αIIbβ3.
Akiyama et al. (35) recently reported that cell adhesive capacity of FN fragments derived from the CCBD is enhanced when the fragments are presented in solution by nonfunction blocking antibodies adsorbed onto plastic. In consideration of the possible steric and conformational effects of the presenting antibody molecule (the mass of which is up to one order of magnitude larger than the FN fragments we tested) on the adhesive properties of the FN fragments, we chose an alternative method in which the GST-FIII fusion proteins were adsorbed directly onto the plastic. A further consideration was that in situ, FN exists as one of many closely associated components of the ECM, and dimeric FN is presumably immobilized within the matrix, by virtue of its interaction with other ECM components, rather than being mobile above the surface. Ugarova et al. (43). reported that in solution only one of the two FIII10 cell binding sites in dimeric FN is accessible to a monoclonal antibody specific for that site, whereas adsorption of dimeric FN to polystyrene results in a conformational change in the molecule resulting in accessibility of both sites to the antibody, thus having possible implications for our interpretation of solid and soluble phase assays.
The structure of the proteins tested was also taken into account. Recombinant FIII9–10 fragments, in which the domain boundaries were strictly maintained as defined previously (27, 38), were used in this study. The strategy we adopted to investigate the molecular mechanisms involved in the adhesion of cells to FIII9–10 was thus chosen so that the nature of the synergistic interaction between FIII9 and FIII10 could be examined under conditions that mimic as closely as possible, within the limitations of the assays, the molecular configuration of the sites as they exist within the native FN molecule in the ECM.
We dissected the molecular mechanisms involved in the adhesive activity of FIII9 and FIII10 by assaying cell attachment as an indicator of primary, integrin-ligand interaction, and FAK phosphorylation and cell spreading as indicators of downstream cell adhesion events, in response to wild type and mutant linker FIII9–10 proteins. The introduction of just two amino acid residues in the interdomain linker between FIII9 and FIII10 in the mutant protein GFIII9-PG-10 resulted in some decrease in cell attachment but substantial decreases in phosphorylation of FAK and cell spreading. The inclusion of four more amino acids in the interdomain linker had little additional effect on cell attachment. Levels of FAK phosphorylation and cell spreading were, however, reduced even further. The data presented here show that in contrast to cell attachment, signaling via FAK phosphorylation and cell spreading are highly dependent upon the physical interaction between FIII9 and FIII10. Furthermore, the close correlation between FAK phosphorylation and cell spreading activities on the wild type and mutant proteins suggests that these events are tightly coupled. While FIII10 is sufficient for integrin binding, our data further demonstrate that the precise structural relationship between FIII9 and FIII10 must be maintained for facilitation of downstream cell adhesion events. The foregoing arguments suggest that there is a temporally resolvable sequence of FN-mediated adhesion events that can be tested experimentally.
There are a number of possible physical effects of the interdomain linkage that might account for reduced biological activity of the mutant linker FIII9–10 proteins. One reason could be that the mutant FIII protein structure may be incorrectly folded. Thermodynamic data (described in further detail in Spitzfaden et al. (45)) show that FIII10 has a significant stabilizing effect on FIII9. This effect is greatest in FIII9–10 and is progressively reduced as the interdomain linker is extended, but the change in ΔGeq from FIII9–10 to FIII9-P[G]5-10 is relatively small compared with the change from FIII9-P[G]5-10 to free FIII9. Additionally, comparison of 1H,15N NMR spectra shows that the polypeptide backbones of FIII9 and FIII10 in the wild type and mutant domain pairs are almost identical (45). These data indicate that the mutant FIII9–10 proteins have a three-dimensional, folded structure comparable with the wild type FIII9–10.
Another possible explanation for reduced biological activity of the mutants could be possible steric interference between the RGD-containing loop in FIII10 and another part of the molecule. Whereas the overall mobility of FIII10 increases in the mutant proteins compared with the wild type (T2 data (45)), there are no significant changes in the chemical shifts and proton-proton NOE effects of backbone amides and side chain α-carbons in the RGDS peptide (Fig. 3). Therefore, the RGD loop appears to form no new atomic contacts as FIII9 and FIII10 are separated, neither are existing contacts broken, which rules out the possibility of steric interference.
The most likely explanation for the strong influence of the linkers upon biological activity is that synergy of the integrin-binding sites within the FIII9–10 pair is absolutely dependent upon specific interdomain interactions and therefore maintenance of the relative orientation of the RGD and PHSRN peptides. The data we present here are consistent with previous reports (34, 44) that the RGD and PHSRN sites in FIII9 and FIII10 are not in physical contact. The importance of the FIII9–10 interdomain interface is surprising considering that in the crystal structure the interdomain area is only 333 Å2 (44) and relatively small in comparison with interface areas between other FN FIII domains. Furthermore, solution NMR measurements revealed that specific contacts between the two modules are scarce. Our data nevertheless show that synergistic activity not only requires a specific distance between RGD and PHSRN, but also precise spatial positioning of the two sites.
In the wild type FIII9–10 domain pair, the orientation of the integrin binding sites with respect to each other is dependent upon the twist and tilt angles between FIII9 and FIII10. These angles are maintained by specific interdomain interactions at the interface between the two domains and by the torsional restraints of the interdomain amino acid residues. Partial disruption of this interface results in random flexing and rotating of the two domains between active and inactive conformations, and this effect is amplified in the longer linker mutant. It is therefore possible that the incremental nature of the changes observed in cell adhesion on immobilized FIII9–10 wild type and mutant proteins is a result of stabilization of a subset of active conformations. However, our data showing lower activity of the mutant in inhibition assays is in agreement with Aota et al. (32) who found that substitution mutants of PHSRN also showed lower activity in inhibition assays than in direct adhesion assays. Our biophysical data clearly show that there is a significant breakdown of the specific interdomain contacts after the addition of two extra linker residues, resulting in increased mobility of the two domains relative to each other. The differences in biological activity of the mutant proteins in the direct adhesion and inhibition assays can be explained if, in the direct adhesion assays, the two integrin-binding peptides are fixed in the optimal relative orientation for binding the integrin, whereas in solution the mutants oscillate between optimal and suboptimal orientation. Thus it would be expected that immobilized fibronectin competes more effectively for the integrin receptor than does soluble protein. This is not surprising in view of the proposed effect on FN of adsorption to surfaces (43).
In conclusion, our findings demonstrate a requirement for the precise positioning of the RGD loop and the synergy site in FIII9–10 for cell adhesion and strongly suggest that the different synergistic regions of the CCBD have separable, and possibly sequential, functions in cell adhesion. These data have implications for our understanding of the inter- and intramolecular mechanisms involved in integrin-mediated adhesion and signaling.
Footnotes
This work was supported by a Medical Research Council grant (to H. J. M.).
The abbreviations used are: ECM, extracellular matrix; FN, fibronectin; CCBD, central cell binding domain; BHK, baby hamster kidney; hESF, human endometrial stromal fibroblast; PBS, Dulbecco’s phosphate-buffered saline; GMEM, Glasgow minimum essential medium; PCR, polymerase chain reaction; RGD, Arg-Gly-Asp; GRGDS, Gly-Arg-Gly-Asp-Ser; PHSRN, Pro-His-Ser-Arg-Asn; NOE(SY), nuclear Overhauser effect (spectroscopy).
H. J. Mardon, unpublished data.
References
- 1.Hynes, R. O. (1990) Fibronectins, Springer-Verlag, New York
- 2.Kornblihtt AR, Vibe-Pedersen K, Baralle FE. EMBO J. 1984;3:221–226. doi: 10.1002/j.1460-2075.1984.tb01787.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hynes RO, Schwarzbauer JE, Tamkun JW. CIBA Found Symp. 1984;108:75–92. doi: 10.1002/9780470720899.ch6. [DOI] [PubMed] [Google Scholar]
- 4.Hynes RO. Annu Rev Cell Biol. 1985;1:67–90. doi: 10.1146/annurev.cb.01.110185.000435. [DOI] [PubMed] [Google Scholar]
- 5.Petersen TE, Thogersen HC, Skorstengaard K, Vibe-Pedersen K, Sottrup-Jensen L, Magnusson S. Proc Natl Acad Sci U S A. 1983;80:137–141. doi: 10.1073/pnas.80.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell ID, Spitzfaden C. Structure. 1994;2:333–337. doi: 10.1016/s0969-2126(00)00034-4. [DOI] [PubMed] [Google Scholar]
- 7.Obara M, Kang MS, Yamada KM. Cell. 1988;53:649–657. doi: 10.1016/0092-8674(88)90580-6. [DOI] [PubMed] [Google Scholar]
- 8.Humphries MJ, Akiyama SK, Komoriya A, Olden K, Yamada KM. J Cell Biol. 1986;103:2637–2647. doi: 10.1083/jcb.103.6.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mould AP, Komoriya A, Yamada KM, Humphries MJ. J Biol Chem. 1991;266:3579–3585. [PubMed] [Google Scholar]
- 10.Hynes RO. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 11.Loftus JC, Smith JW, Ginsberg MH. J Biol Chem. 1994;269:25235–25238. [PubMed] [Google Scholar]
- 12.Ruoslahti E, Pierschbacher MD. Science. 1987;238:491–497. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]
- 13.Pytela R, Pierschbacher MD, Ruoslahti E. Cell. 1985;40:191–198. doi: 10.1016/0092-8674(85)90322-8. [DOI] [PubMed] [Google Scholar]
- 14.Takada Y, Huang C, Hemler ME. Nature. 1987;326:607–609. doi: 10.1038/326607a0. [DOI] [PubMed] [Google Scholar]
- 15.Vogel BE, Tarone G, Giancotti FG, Gailit J, Ruoslahti E. J Biol Chem. 1990;265:5934–5937. [PubMed] [Google Scholar]
- 16.Dedhar S, Gray V. J Cell Biol. 1990;110:2185–2193. doi: 10.1083/jcb.110.6.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith JW, Vestal DJ, Irwin SV, Burke TA, Cheresh DA. J Biol Chem. 1990;265:11008–11013. [PubMed] [Google Scholar]
- 18.Busk M, Pytela R, Sheppard D. J Biol Chem. 1992;267:5790–5796. [PubMed] [Google Scholar]
- 19.Weinacker A, Chen A, Agrez M, Cone RI, Nishimura S, Wayner E, Pytela R, Sheppard D. J Biol Chem. 1994;269:6940–6948. [PubMed] [Google Scholar]
- 20.Gardner JM, Hynes RO. Cell. 1985;42:439–448. doi: 10.1016/0092-8674(85)90101-1. [DOI] [PubMed] [Google Scholar]
- 21.Plow EF, McEver RP, Coller BS, Woods VL, Jr, Marguerie GA, Ginsberg MH. Blood. 1985;66:724–727. [PubMed] [Google Scholar]
- 22.Burridge K, Petch LA, Romer LH. Curr Biol. 1992;2:537–539. doi: 10.1016/0960-9822(92)90020-b. [DOI] [PubMed] [Google Scholar]
- 23.Burridge K, Turner CE, Romer LH. J Cell Biol. 1992;119:893–903. doi: 10.1083/jcb.119.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guan JL, Trevithick JE, Hynes RO. Cell Regul. 1991;2:951–964. doi: 10.1091/mbc.2.11.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kornberg L, Earp HS, Parsons JT, Schaller M, Juliano RL. J Biol Chem. 1992;267:23439–23442. [PubMed] [Google Scholar]
- 26.Schaller MD, Parsons JT. Curr Opin Cell Biol. 1994;6:705–710. doi: 10.1016/0955-0674(94)90097-3. [DOI] [PubMed] [Google Scholar]
- 27.Main AL, Harvey TS, Baron M, Boyd J, Campbell ID. Cell. 1992;71:671–678. doi: 10.1016/0092-8674(92)90600-h. [DOI] [PubMed] [Google Scholar]
- 28.Pierschbacher MD, Ruoslahti E. Nature. 1984;309:30–33. doi: 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- 29.Dufour S, Duband JL, Humphries MJ, Obara M, Yamada KM, Thiery JP. EMBO J. 1988;7:2661–2671. doi: 10.1002/j.1460-2075.1988.tb03119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagai T, Yamakawa N, Aota S, Yamada SS, Akiyama SK, Olden K, Yamada KM. J Cell Biol. 1991;114:1295–1305. doi: 10.1083/jcb.114.6.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aota S, Nagai T, Yamada KM. J Biol Chem. 1991;266:15938–15943. [PubMed] [Google Scholar]
- 32.Aota S, Nomizu M, Yamada KM. J Biol Chem. 1994;269:24756–24761. [PubMed] [Google Scholar]
- 33.Bowditch RD, Halloran CE, Aota S, Obara M, Plow EF, Yamada KM, Ginsberg MH. J Biol Chem. 1991;266:23323–23328. [PubMed] [Google Scholar]
- 34.Bowditch RD, Hariharan M, Tominna EF, Smith JW, Yamada KM, Getzoff ED, Ginsberg MH. J Biol Chem. 1994;269:10856–10863. [PubMed] [Google Scholar]
- 35.Akiyama SK, Aota S, Yamada KM. Cell Adhes Commun. 1995;3:13–25. doi: 10.3109/15419069509081275. [DOI] [PubMed] [Google Scholar]
- 36.Mardon HJ, Grant KE. FEBS Lett. 1994;340:197–201. doi: 10.1016/0014-5793(94)80137-1. [DOI] [PubMed] [Google Scholar]
- 37.Fesik SW, Zuiderweg ERP. J Magn Reson. 1988;78:588–593. [Google Scholar]
- 38.Kornblihtt AR, Umezawa K, Vibe-Pedersen K, Baralle FE. EMBO J. 1985;4:1755–1759. doi: 10.1002/j.1460-2075.1985.tb03847.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fernandez-Shaw S, Shorter SC, Naish CE, Barlow DH, Starkey PM. Hum Reprod. 1992;7:156–161. doi: 10.1093/oxfordjournals.humrep.a137609. [DOI] [PubMed] [Google Scholar]
- 40.Kueng W, Silber E, Eppenberger U. Anal Biochem. 1989;182:16–19. doi: 10.1016/0003-2697(89)90710-0. [DOI] [PubMed] [Google Scholar]
- 41.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 42.Towbin H, Staehelin T, Gordon J. Proc Natl Acad Sci U S A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ugarova TP, Zamarron C, Veklich Y, Bowditch RD, Ginsberg MH, Weisel JW, Plow EF. Biochemistry. 1995;34:4457–4466. doi: 10.1021/bi00013a039. [DOI] [PubMed] [Google Scholar]
- 44.Leahy DJ, Ikramuddin A, Erickson HP. Cell. 1996;84:155–164. doi: 10.1016/s0092-8674(00)81002-8. [DOI] [PubMed] [Google Scholar]
- 45.Spitzfaden C, Grant RP, Mardon HJ, Campbell ID. J Mol Biol. 1997;265:565–579. doi: 10.1006/jmbi.1996.0736. [DOI] [PubMed] [Google Scholar]