

Abstract
The ninth and tenth FIII domains (FIII9–10) of human fibronectin act in synergy to promote cell adhesion via the interaction with integrin receptors. Here we describe the functional and structural properties of a set of recombinant FIII9–10 mutants containing various alanine substitutions within the key synergistic site, DRVPHSRN in FIII9, either alone or in combination with another substitution (Leu1408 to Pro), on the opposite face of FIII9, that increases stability and the functional capacity of FIII9–10. We show that the introduction of mutations into the synergistic sequence of FIII9–10 has a negative effect on the adhesion of baby hamster kidney fibroblasts and results in reduced ability of these ligands to recognize integrin α5β1. Conformational stability of the FIII9 domain in the synergy site mutants is likewise reduced in comparison with native FIII9. The Leu1408 to Pro substitution in mutant FIII9–10 proteins carrying substitutions in the synergy site results in a substantial recovery of the adhesive activity of the mutants and affinity to α5β1 In keeping with the enhancement of functional activity, the Leu1408 to Pro substitution in the FIII9–10 synergy site mutants also causes a significant increase in conformational stability of FIII9. These observations imply a strong positive correlation between the biological activity and conformational stability of the assessed FIII9–10 mutants and suggest that a Leu1408 to Pro substitution restores the biological activity of the mutants via their ability to restore their conformational stability. We conclude that domain stability may be a major determinant of the synergistic potential of FIII9. Our data underscore the value of using more than one approach in such structure-function studies and the requirement for validating the global structural integrity of protein ligands in which sequences that disrupt function have been perturbed.
Fibronectin, a widely expressed extracellular matrix protein, contains multiple modular regions that interact with various members of the integrin family of heterodimeric cell-surface adhesion receptors. The central cell binding domain, which is known to recognize α5β1, αvβ3, αIIbβ3 and at least eight other integrins, is particularly well characterized (for a review, see Ref. 1). This region, spanning type III domains FIII8, FIII9, and FIII10, has become the focus of intensive research as a model for the universal and ubiquitous RGD-driven receptor-ligand interactions. The RGD tripeptide motif, residing on a protruding, flexible loop within the FIII10 module (2, 3), is essential for receptor binding and downstream cell adhesion events such as pp125FAK-, Rho-, and Rac-dependent signaling, cytoskeletal reorganization, and acquisition of spread morphology (4–6). Although it is presently not clear by which mechanism an RGD-containing ligand initiates a conformational change in the integrin headpiece and transmittal of the adhesive signal across the plasma membrane, the recent solution of the crystal structure of integrin αvβ3 in complex with an isolated RGD peptide by Xiong et al. (7) is an important first step toward elucidating the structural basis of the FIII10-integrin interaction.
The FIII9 domain adjacent to FIII10 is known to act in synergy with the latter in promoting integrin-mediated cell adhesion, drastically increasing the activity of a single FIII10 domain but not having any activity on its own (8). A number of sites within FIII9 have been proposed to be critical for its synergistic effect. The most well studied of these is the DRVPHSRN sequence or the so-called “synergy site,” the larger part of which is located on a surface loop of FIII9 lying approximately in plane with the RGD loop (3, 9) and thus being potentially accessible for contact with the integrin dimer. Previous studies have demonstrated that integrins binding to the FIII9–10 domain pair have a common requirement for the intact RGD motif, but they display distinct preferences for individual residues within the synergy site of FIII9 or may not need this site at all. For instance, while high affinity α5β1 binding is most sensitive to substitutions for Arg1379 (10), the most critical residues for αIIbβ3 appear to be Asp1373 and Arg1374 (11), whereas αvβ3 and α3β1 are indifferent to the presence of the FIII9 domain (11, 12). Other amino acids in the vicinity of the classical synergy site (notably Arg1369, Arg1371, Thr1385, and Asn1386) have also been shown to contribute to the synergistic activity of FIII9 (13), although no integrin specificity has been determined for these residues. Taken together, these findings support the notion that each distinct integrin-ligand interaction, despite exhibiting some overall topological similarities, has its idiosyncratic characteristics, which define the precise conditions for an efficient and specific binding to the synergistic FIII9–10 module pair.
Biophysical studies have revealed that in comparison with FIII10, the FIII9 domain is thermodynamically rather unstable (14) and displays much slower folding kinetics (15). Coupling to the neighboring FIII modules within fibronectin clearly plays an important role in stabilizing FIII9, since it exhibits around 3-fold higher conformational stability in module pairs FIII8–9 and FIII9–10 than on its own (16), and altering its distance from FIII10 by introducing interdomain linkers of varying length critically reduces its stability (14). The relationship between the thermodynamic properties of FIII9 and its synergistic integrin-dependent biological activity, however, remains little studied despite its likely important implications for understanding the mechanism of integrin-ligand complex formation. A previous investigation has shown that the decrease in FIII9 stability due to the insertion of the above-mentioned linkers between FIII9 and FIII10 coincides with the reduced adhesive activity of the domain pair and impaired induction of intracellular signaling (17). More recently, we were able to demonstrate a link between the enhancement of biological function of the FIII9–10 pair by the FIII8 module and its positive influence on the structural stability of FIII9 (16). In the latter study we also probed the role of the FIII9 synergy site in stabilizing the FIII8–9 pair within the FIII8–9–10 module tandem, documenting a negative impact of perturbing the synergy sequence in this three-domain context.
Here, we have specifically addressed the question whether, and to what extent, do specific amino acid residues within the synergistic DRVPHSRN loop contribute to the global stability of FIII9 and how this relates to the functional performance of the FIII9–10 domain pair. We use a combination of biophysical, biochemical, and biological methods to show that the altered structural properties of various FIII9–10 synergy site mutants are strongly linked to their integrin α5β1-dependent function.
EXPERIMENTAL PROCEDURES
Construction of Mutant pGEX2T-FIII9 –10 Clones
Cloning of the wild type FIII9–10 domain pair into the pGEX2T vector (Amersham Biosciences) has been described elsewhere (8). Amino acid substitutions into the wild type FIII9–10 construct were introduced following the QuikChange™ protocol (Stratagene). The following mutations were made to the synergistic DRVPHSRN sequence of FIII9: Arg1379 to Ala (DRVPHSAN; mutant Ala1); Asp1373-Arg1374 to Ala-Ala and Ser1378-Arg1379-Asn1380 to Ala-Ala-Ala (AAVPHAAA; mutant Ala5), as highlighted in Fig. 1. Creation of mutant Ala3, containing the substitution of Ser1378-Arg1379-Asn1380 with Ala-Ala-Ala (DRVPHAAA), has been described previously (16). Constructs harboring mutations in the synergy site were further mutated in FIII9 by substituting Leu1408 with Pro (mutants Ala1-L1408P, Ala3-L1408P, and Ala5-L1408P). The latter substitution was also introduced into the wild type FIII9–10 pair, resulting in mutant L1408P (see Table I). The DNA sequence of all created mutants was confirmed using Sanger DNA sequencing methodology (Department of Biochemistry, University of Oxford).
Fig. 1. Ribbon diagram of the FIII9–10 domain pair.
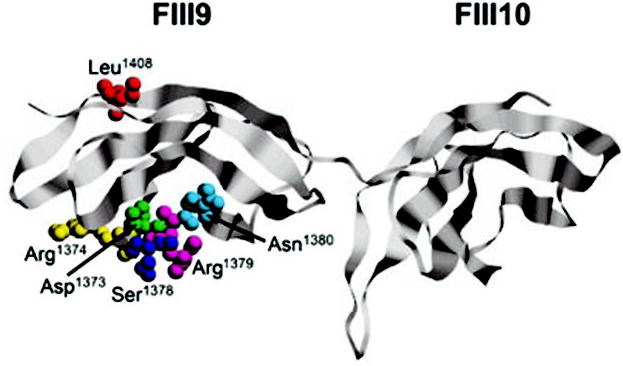
Atom co-ordinates were obtained from The Protein Data Bank (24) (PDB ID: 1FNF) and imaged using the program RasMol (www.umass.edu/microbio/rasmol). The substituted residues (colored) are shown in space-fill mode and labeled accordingly.
Table I.
Notations used for mutant FIII9 –10 proteins
| FIII9–10 mutant | Alanine substitutions in the 1373DRVPHSRN1380 sequence of FIII9 | Substitution of Leu1408 with Pro |
|---|---|---|
| Ala1 | DRVPHSAN | − |
| Ala3 | DRVPHAAA | − |
| Ala5 | AAVPHAAA | − |
| L1408P | DRVPHSRN | + |
| Ala1-L1408P | DRVPHSAN | + |
| Ala3-L1408P | DRVPHAAA | + |
| Ala5-L1408P | AAVPHAAA | + |
Expression and Purification of Recombinant FIII9 –10 Proteins
Wild type and mutant FIII9–10 constructs were expressed in Escherichia coli as glutathione S-transferase (GST)1 fusion proteins and purified as described previously (8). For equilibrium chemical denaturation studies, cleaved FIII proteins were obtained by thrombin digest of the respective GST fusion proteins as described elsewhere (16).
ELISA
Integrin α5β1 used in ELISA and surface plasmon resonance studies was purified from human placenta as described previously (16). ELISAs with plate-bound α5β1 and soluble FIII9 –10 ligands were carried out in the presence of divalent cations as detailed earlier (16). Assays were performed in duplicate, and background antibody binding in the absence of ligand was subtracted from the readings. Nonspecific binding of the GST fusion proteins to uncoated wells blocked with bovine serum albumin was measured separately for each ligand concentration point and subsequently subtracted from the corresponding values for total binding. Dose-response data from the assays were analyzed by non-linear regression using a sigmoidal curve fit (Prism, GraphPad Software). Assay results are expressed as the means of at least three independent experiments. Error bars represent S.E. (dose-response curves) or 95% confidence intervals (bar graphs).
Surface Plasmon Resonance Studies
Real-time biomolecular interaction analysis was performed using a BIACORE 2000 instrument (Biacore, Uppsala, Sweden). All experiments were performed at room temperature using the standard HEPES-buffered saline as running buffer. Briefly, 9000–16,000 resonance units anti-GST antibody (Biacore) was immobilized on CM5 sensor chips (Biacore) according to the manufacturer’s instructions (BIAapplications Handbook, Biacore). A volume of 30 μl of different GST-tagged FIII9–10 proteins diluted in HEPES-buffered saline were then injected at 5 μl/min to reach a constant level of binding. For kinetic measurements, integrin α5β1 was injected for 10 min at three different concentrations (80, 160, and 320 nm) at a constant flow rate of 5 μl/min. Both the association and dissociation reactions were performed in Tris-buffered saline (25 mm Tris, 150 mm NaCl, pH 7.4) containing MnCl2, MgCl2, and CaCl2 at 2 mm each. The same injections of integrin were performed on a GST protein used as a reference. The sensor surfaces were regenerated with a short pulse of 10 mm glycine, pH 2.2. Data were analyzed by using the global fitting algorithm of the BIAevaluation 3.0 software package (Biacore), using a simple 1:1 kinetic model. At least two independent experiments were performed for each α5β1 -FIII9 –10 interaction. Error bars represent S.D.
Cell Adhesion Assays
Cell attachment and spreading assays were performed with baby hamster kidney (BHK) fibroblasts as detailed elsewhere (8). Cell attachment was quantified by staining adherent cells with 0.1% crystal violet as described previously (8). The data obtained are expressed as the means ± S.E. of at least three independent experiments.
Equilibrium Chemical Denaturation Studies
Equilibrium unfolding experiments were carried out on recombinant GST-cleaved FIII9–10 proteins, incubated in 0–4 m guanidinium hydrochloride (GdnHCl) in phosphate-buffered saline, as described previously (16). Measurements were performed at 25 °C on a Shimadzu RF5001PC spectrofluorimeter, using an excitation wavelength of 278 nm. Fluorescence emitted was recorded at wavelengths ranging from 360 to 425 nm. Data from the experiments were fitted for a two-state unfolding mechanism as described earlier (20). Error bars in the bar graphs represent 95% confidence intervals.
RESULTS
A Leu1408 to Pro Substitution Increases the Affinity of FIII9 –10 Mutants to α5β1
To evaluate the effect of perturbing the synergy site of FIII9 on integrin α5β1 recognition, solid-phase receptor binding assays were performed with wild type FIII9–10 ligand and mutants Ala1, Ala3, and Ala5 (Fig. 2), incorporating various substitutions within the DRVPHSRN sequence (see Table I). The apparent Kd of the interaction (ligand concentration at the midpoint of the dose-response curves shown in Fig. 2a) was observed to increase gradually as more substitutions were introduced, rising from ~2 nm for the wild type to ~5 nm for mutants Ala1 and Ala3 and ~15 nm for mutant Ala5 (Fig. 2b, closed bars on the left-hand panel). This dependence of high affinity α5β1 binding on the intact synergy site was in agreement with previous studies (16, 18) and further demonstrated the differential and co-operative effect of individual synergistic residues in securing efficient receptor recognition.
Fig. 2. Solid-phase binding of recombinant wild type and mutant FIII9–10 proteins to integrin α5β1 as measured by ELISA and SPR.
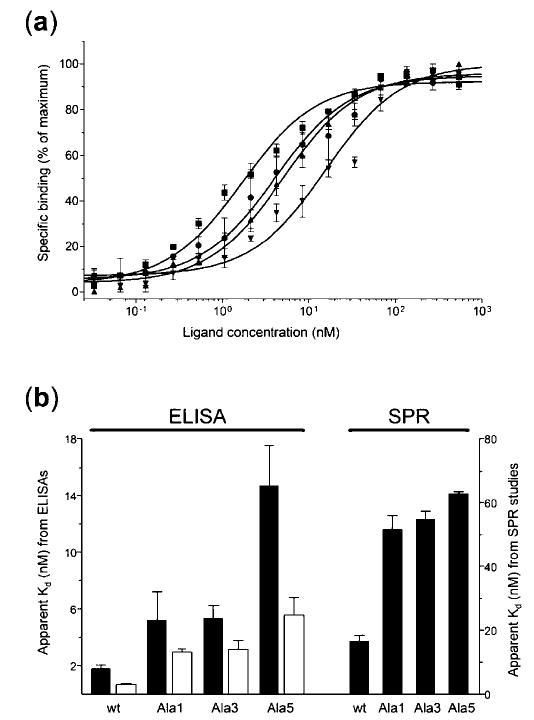
a, dose-response curves for wild type FIII9–10 (▪) and mutants Ala1 (•), Ala3 (▴), and Ala5 (▾). Results are normalized and expressed as percentages of maximum binding activity. b, comparison of the apparent Kd values for wild type FIII9 –10 (wt) and mutants Ala1 to Ala5 either without (closed bars) or with (open bars) a Leu1408 to Pro substitution, as derived from ELISAs (left-hand panel) and SPR studies (right-hand panel). Note that lower apparent Kd values reflect higher receptor affinity and vice versa.
The results obtained by ELISA were confirmed using surface plasmon resonance (SPR), allowing the monitoring of real-time kinetic parameters of the ligand-integrin complex formation. The apparent Kd values for the α5β1 -FIII9 –10 interaction calculated by this method showed a similar increase upon introduction of mutations into the synergy loop of FIII9, being lowest for native FIII9–10 (~17 nm) and highest for mutant Ala5 (~63 nm) (Fig. 2b, closed bars on the right-hand panel). This difference stemmed from both the slower association (on-rate) and faster dissociation (off-rate) of the integrin from the mutated FIII9–10 mutants, as compared with the wild type (data not shown). The discrepancy between the relative Kd values from these experiments and the ELISA data (the affinity of the least active mutant versus wild type decreasing ~4-and ~8-fold, respectively) is likely due to the different assay formats used. SPR analysis was conducted with captured fibronectin ligands and soluble integrin, whereas immobilized integrin and soluble fibronectin fragments were used for ELISAs. Also, in SPR studies the integrin binds to the captured ligand in constant flow conditions, whereas the ELISAs are conducted in the absence of flow, which may have contributed to the variance of the relative affinities of individual FIII9–10 ligands. Nevertheless, both techniques show a clear dependence of binding affinity on the native structure of the synergy loop and further suggest that this synergy requirement is maintained regardless of the particular method of presentation of the FIII9–10 ligand to the integrin.
Earlier studies in our laboratory have shown that a single Leu1408 to Pro mutation within the FIII9–10 domain pair is able to confer a significant increase in structural stability to the native FIII9 domain maintaining an intact synergy loop (19). We therefore sought to assess the functional consequences of this substitution in the context of wild type FIII9–10 as well as synergy site mutants Ala1, Ala3, and Ala5 with relatively poor integrin binding properties. ELISAs performed with the various FIII9–10 mutants and immobilized α5β1 (Fig. 2b, open bars) revealed that mutant L1408P bound to the integrin with ~3-fold higher affinity than the wild type (apparent Kd ≈ 0.7 nm), and the same substitution in mutant Ala5-L1408P resulted in a similar ~3-fold enhancement of function over Ala5. A modest improvement in α5β1 binding capacity was also seen with mutants Ala1-L1408P and Ala3-L1408P in comparison with mutants Ala1 and Ala3, respectively.
Leu1408 to Pro Mutation Restores Cell Adhesion Activity of FIII9 –10 Synergy Site Mutants
Having determined the solid-phase integrin binding properties of the aforementioned FIII9–10 mutants, we proceeded to test the ability of these fibronectin fragments to support integrin-mediated adhesion of BHK fibroblasts, which express integrins α5β1 and αvβ3 (17), as a physiologically more relevant measure of biological activity (Fig. 3). Quantification of cell attachment to protein-coated surfaces (Fig. 3a) revealed that wild type FIII9–10 and mutant Ala1 were significantly better adhesive substrates for BHK cells than mutants Ala3 and Ala5, the latter retaining only ~50 and ~30% of the wild type activity, respectively, at a coating concentration of 1 μm. Cell spreading, a secondary event in the process of cell adhesion, was even more sensitive to perturbations in the synergy loop; the level of cell spreading at the same coating concentration dropped to ~70 and ~40% of wild type FIII9–10 activity for Ala1 and Ala3, respectively, while the response for Ala5 was barely ~10% of that of the wild type (Fig. 3b).
Fig. 3. Quantification of cell adhesion promoting ability of FIII9–10 synergy site mutants.
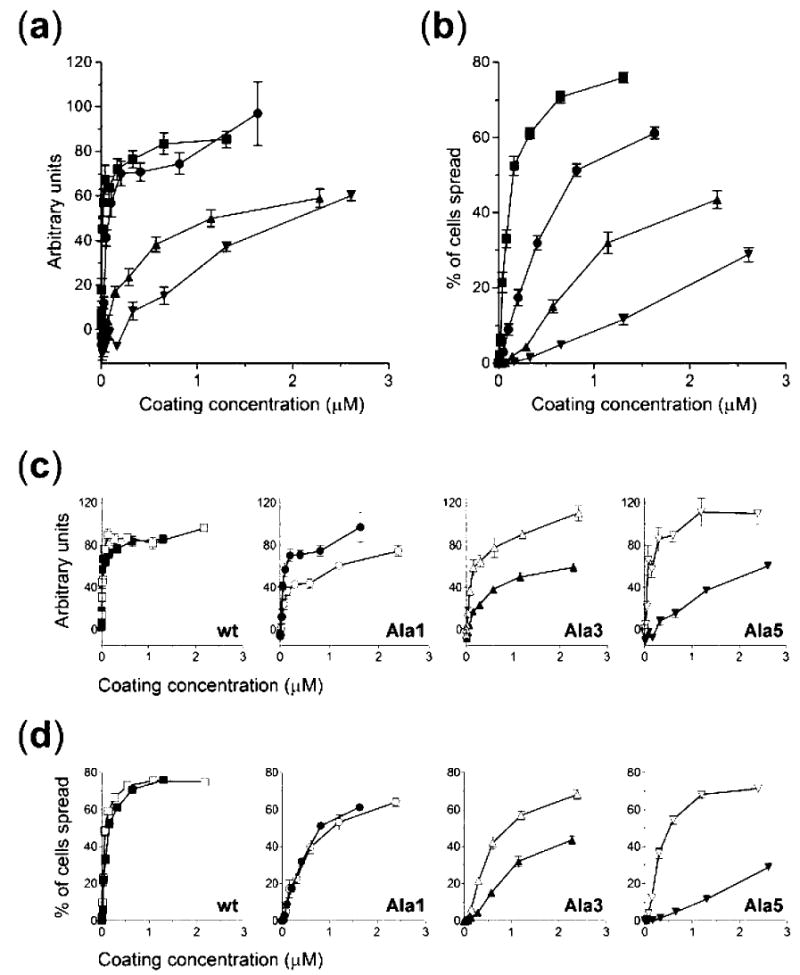
Attachment (a and c) and spreading (b and d) of BHK cells on surfaces coated with FIII9–10 (▪), Ala1 (•), Ala3 (▴), Ala5 (▾), L1408P (□), Ala1-L1408P (○), Ala3-L1408P (▵) or Ala5-L1408P (▿). Results are expressed in arbitrary units (a and c) or as percentages of cells spread (b and d). For clarity, separate graphs are shown in c and d for each mutant pair without (closed symbols) or with (open symbols) a Leu1408 to Pro substitution.
The FIII9–10 mutants carrying an L1408P substitution were tested in these assays to determine whether this mutation was able to recover the adhesion promoting capability lost in the FIII9–10 synergy site mutants. The adhesive properties of mutant L1408P were enhanced in comparison with the native FIII9–10 at lower coating concentrations, confirming previous findings. Cell attachment on mutants Ala3-L1408P and Ala5-L1408P was restored to wild type levels, being ~2- and ~4-fold more efficient compared with Ala3 and Ala5, respectively, at 1μm coating concentration (Fig. 3c). Attachment to Ala1-L1408P, however, was reduced in comparison with Ala1. The spreading of BHK cells was likewise affected by the L1408P substitution (Fig. 3d). The activity of mutants Ala3-L1408P and Ala5-L1408P was significantly increased compared with Ala3 and Ala5 (~2- and ~8-fold, respectively, at 1 μm coating concentration). Cell spreading on mutant Ala1-L1408P remained at the same level as that supported by mutant Ala1.
Leu1408 to Pro Substitution Rescues Reduced FIII9 Stability in Synergy Site Mutants
Previous experiments with longer fibronectin fragments spanning domains FIII8 through FIII10 had indicated a role for the FIII9 synergistic sequence in contributing to the structural stability of the FIII8–9 pair (16). In this study, we explored further the correlation between the differential functional activity of FIII9–10 constructs harboring various mutations in the synergy site and their thermodynamic properties. Data from equilibrium unfolding experiments performed with FIII9–10 mutants using GdnHCl as a denaturant are presented in Fig. 4 and Table II. Global unfolding of the FIII9–10 domain pair is known to occur by a well defined two-step transition allowing for separate analysis of unfolding parameters for the individual FIII9 and FIII10 domains (14, 16). Since the stability of FIII10 does not depend significantly on the presence of FIII9 (16), only the first step of the denaturation curve, corresponding to the unfolding of FIII9 (14), was of interest here and is shown in isolation for clarity. The analysis used assumes a two-state domain unfolding mechanism as described by Pace and Scholtz (20). While FIII9 in mutant Ala1 displayed roughly the same unfolding properties as in wild type FIII9–10, the FIII9 module in mutant Ala3 was destabilised, an effect that was more pronounced for mutant Ala5 (Fig. 4a). The concentration of denaturant leading to 50% domain unfolding ([GdnHCl]½) dropped from 1.9 m for wild type FIII9 to 1.5 m for Ala3 and to 1.2 m for Ala5 (Fig. 4b, closed bars), and the respective negative shifts in the free energy of unfolding (ΔG(H2O)) relative to the wild type increased from −1.0 kcal mol−1 (Ala3) to −1.7 kcal mol−1 (Ala5) (Table II). These results thus demonstrate that the thermodynamic stability of FIII9 within the FIII9–10 pair is sensitive to specific amino acid substitutions within the synergy loop. When unfolding curves for FIII9 in mutants incorporating the Leu1408 to Pro substitution were assessed, a substantial shift to the right was seen for all Pro1408 mutants as compared with their Leu1408 counterparts (Fig. 4c). The corresponding increase in conformational stability of FIII9 as reflected by the respective [GdnHCl]½ and ΔG(H2O) values was ~1.5-fold for all Pro/Leu mutant pairs (Fig. 4b, open bars, and Table II).
Fig. 4. Comparison of thermodynamic stability parameters of related FIII9–10 species.
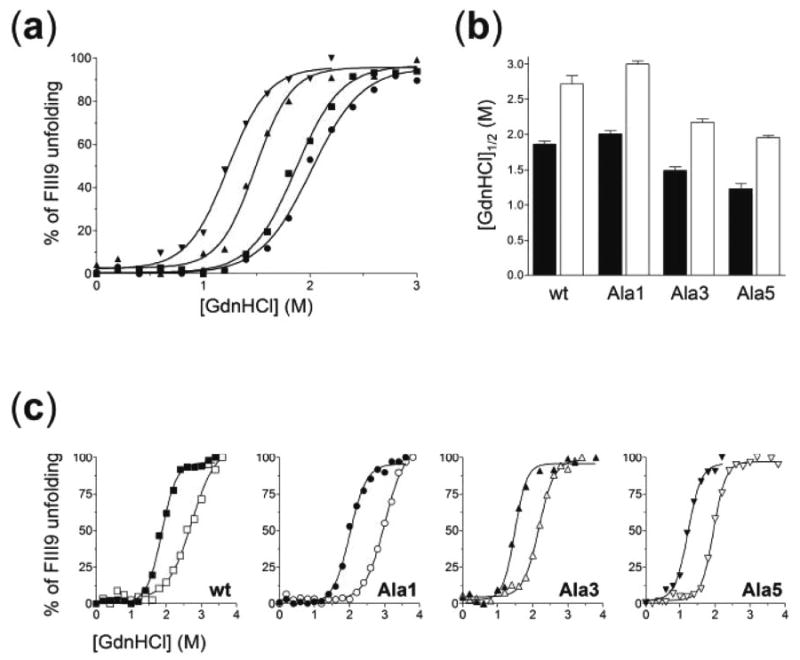
Equilibrium denaturation of FIII9 versus GdnHCl concentration in wild type and mutant FIII9–10 proteins (a and c). The symbols and graph display in c are the same as in Fig. 3. b, comparison of [GdnHCl]½ values for the unfolding of FIII9 in FIII9–10 proteins without (closed bars) or with (open bars) a Leu1408 to Pro substitution.
Table II.
Equilibrium unfolding parameters for the first denaturation step of the wild type and mutant FIII9 –10 domain pairs
| FIII9–10 construct | [GdnHCl]½ | ΔG((H2O))a | m |
|---|---|---|---|
| m | kcal mol−1 | kcal mol−1 m−1 | |
| Wild type | 1.87 | 4.86 | 2.71 |
| Ala1 | 2.01 | 5.23 | 2.44 |
| Ala3 | 1.49 | 3.87 | 2.96 |
| Ala5 | 1.23 | 3.20 | 2.81 |
| L1408P | 2.72 | 7.07 | 1.73 |
| Ala1-L1408P | 3.00 | 7.80 | 2.34 |
| Ala3-L1408P | 2.17 | 5.64 | 2.60 |
| Ala5-L1408P | 1.96 | 5.10 | 3.20 |
Due to potential errors associated with determining the individual slope (m) values for the linear transition region, the values of ΔG(H2O) were calculated from an average m value of 2.60 kcal mol−1 m−1 (20).
Finally, the observed differential thermodynamic stability of the FIII9–10 mutants was compared with their functional activity by performing correlation analysis. Mutant Ala1-L1408P was excluded from linear regression calculations due to its anomalously high stability/activity ratio. The results of the analysis, plotted in Fig. 5, reveal a strong correlation between the structural stability of the FIII9–10 proteins and mean biological activity (Pearson correlation coefficient r = 0.92; p = 0.004).
Fig. 5. Correlation between conformational stability and biological activity of FIII9–10 mutants.
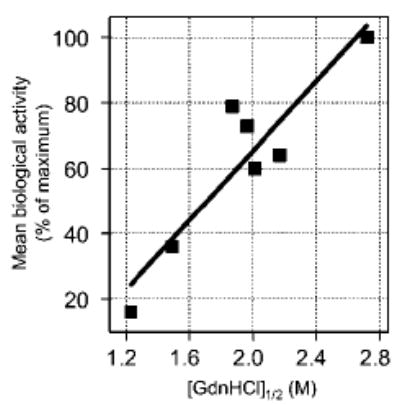
The values of [GdnHCl]½ for the unfolding of FIII9 in Ala5, Ala3, wild type FIII9 –10, Ala5-L1408P, Ala1, Ala3-L1408P, and L1408P (symbols from left to right), plotted against their respective mean biological activity values, are shown. The latter was defined for each protein as the arithmetic mean of normalized relative values for ELISA-derived apparent Kd, cell attachment at 1 μm coating concentration, and cell spreading at the same concentration and expressed as percentages of maximum biological activity attained. The line represents the results of linear regression analysis (r2 = 0.84), indicating a good correlation between the data sets.
DISCUSSION
The current study was aimed toward elucidating the structural basis for the synergistic biological activity of FIII9 in the FIII9 –10 module pair of fibronectin. By the use of a range of methods, we have established a distinct role for the DRVPH-SRN synergy sequence within the FIII9 domain in maintaining the overall conformational stability of FIII9. Our findings demonstrate that: (i) perturbing the native configuration of the FIII9 synergy site, required for efficient integrin α5β1 binding and cell adhesion, leads to impaired domain stability; (ii) the Leu1408 to Pro mutation can compensate for loss of biological activity of the synergy site mutants; and (iii) a strong correlation exists between the global structural integrity and synergistic function of the FIII9 –10 pair.
We have used alanine scanning mutagenesis within the synergy site to establish the structural importance of residues previously reported to play a key role in integrin-dependent adhesive processes (10, 11, 13). In our experiments, the reduction in biological activity for a single Arg1379 mutant (Ala1) is very similar to that observed recently by Redick et al. (13). Further loss of function upon mutating both the N- and C-terminal portions of the DRVPHSRN sequence (in Ala5) is also in good general agreement with the latter report. Further substitutions of amino acids outside the DRVPHSRN site have been shown to have an effect in combination with Arg1379 in FIII7–10 fragments (13), although it is not clear to what extent these substitutions affect local or global foldedness. While confirming earlier findings, we show here that the perturbations introduced in the FIII9 –10 mutants not only affect the functionality of the synergy site but also have a strong negative effect on the thermodynamic stability of the FIII9 domain. Equilibrium denaturation experiments reveal that even though the synergy site resides on a solvent-exposed surface loop protruding from the main domain core, it contributes significantly to the overall foldedness of FIII9, as synergy site mutants carrying multiple substitutions unfold at much lower denaturant concentrations than the wild type. The fact that this effect is not observable for the single Arg1379 mutant points to the involvement of more extensive interactions between the core and the synergy loop in conferring stability to FIII9.
The structure-function correlation seen with the synergy site mutants is further reinforced by the finding that a single independent substituted residue (Pro for Leu1408) on the other side of the FIII9 module (shown in Fig. 1), conceivably unable to make a direct contact with either the synergy loop itself or the integrin receptor, essentially reverses the effect of inactivating the synergy site both in terms of biological function and structural characteristics of the FIII9 –10 pair. Such indirect influence on integrin α5β1 -dependent activities is therefore most likely exerted via global stabilization of the FIII9 module.
In a previous report, we studied the influence of the FIII8 module within the FIII8 –9 –10 domain tandem upon the stability of FIII9, showing a causal link between FIII8-induced enhancement of synergistic biological activity and restoration of structural integrity of an FIII9 –10 pair containing a dysfunctional synergy site (16). On the basis of these findings, we suggested that the requirement for the DRVPHSRN sequence may only be critical for integrin binding in the case of an isolated FIII9 –10 pair. However, here we show that a Leu1408 to Pro substitution within FIII9 –10 achieves a similar effect without the need for an additional N-terminal domain. Taken together, these two studies imply that any sufficiently strong influence producing an increase in the global stability of FIII9, be it a single critical amino acid substitution or a whole additional domain, may overcome the requirement for an intact synergy site in securing efficient integrin recognition. This kind of conformation-dependent receptor competency appears to play an important role in the regulation of adhesion-related cellular responses. The latter notion may be relevant in the light of the evidence that some integrins appear to modulate synergy site binding as a function of their activation state. For instance, α5β1 may lose its dependence on the synergy sequence in the presence of stimulatory antibodies or Mn2+ ions (21), while α4β1 and α4β7 only recognize FIII9 when constitutively activated (22). Thus the requirement for the synergy site may to a large extent depend on the specific overall conformation of both the fibronectin ligand and the integrin receptor.
Earlier studies utilizing synergy site-derived peptides in isolation (11) or in the context of a foreign FIII scaffold (10) have shown that mutations in the DRVPHSRN sequence can affect integrin binding and cell adhesion even in the absence of a native FIII9 fold. This suggests that while maintaining conformational stability of FIII9 may be an important function of the synergy loop, it is likely that this is not the only mechanism whereby this region fulfils its synergistic potential and that, as proposed by a number of previous reports, direct and specific molecular interactions between residues in the synergy site and the ligand binding pocket of the receptor are also crucial for biological activity. As suggested previously (16), full uncoupling of these two mechanisms of action may not be possible, and both need to be taken into account when modeling the establishment and maintenance of fibronectin-integrin interactions.
The specific stability parameters of the FIII9 –10 domain pair appear to depend upon the particular conditions in which domain unfolding is evoked. For instance, while chemical denaturation unfolding experiments employed in this and other studies show that the FIII9 module is thermodynamically much less stable than FIII10, steered molecular dynamics simulations using forced mechanical stretching of the module pair predict that FIII9 may be mechanically more stable than FIII10 (23). The apparent contradiction between the two data sets may be explained by consideration of the fact that forced unfolding is a kinetic process that queries the height of energetic barriers separating different folded states rather than the equilibrium energies measured by chemical unfolding. Given that the forced unfolding method mimicks the physiologically highly relevant stretching of the fibronectin molecule under tension and that the FIII9 domain responds differently in different unfolding scenarios, it would be interesting to investigate how the synergy site mutants employed in the current study would affect the mechanical (as opposed to thermodynamic) stability of FIII9 and which specific intradomain interactions are most vulnerable to disruption by stretching.
In conclusion, this report provides strong evidence in support of the hypothesis that domain stability is an important determinant of integrin α5β1 -dependent synergistic biological potential of the FIII9 module. Our observation that relatively minor amino acid substitutions produce unexpectedly large effects in the thermodynamic stability of FIII9 thus highlights the requirement for careful structural evaluation of modular proteins participating in similar receptor-ligand interactions.
Acknowledgments
We are grateful to Richard Grant for constructing the mutants Ala1 and Ala5 and to Anton van der Merwe for assistance with Biacore studies.
Footnotes
This work was supported by the Wellcome Trust and the Medical Research Council.
The abbreviations used are: GST, glutathione S-transferase; ELISA, enzyme-linked immunosorbent assay; BHK, baby hamster kidney; GdnHCl, guanidinium hydrochloride; SPR, surface plasmon resonance.
References
- 1.Johansson S, Svineng G, Wennerberg K, Armulik A, Lohikangas L. Front Biosci. 1997;2:126 –146. doi: 10.2741/a178. [DOI] [PubMed] [Google Scholar]
- 2.Main AL, Harvey TS, Baron M, Boyd J, Campbell ID. Cell. 1992;71:671–678. doi: 10.1016/0092-8674(92)90600-h. [DOI] [PubMed] [Google Scholar]
- 3.Leahy DJ, Aukhil I, Erickson HP. Cell. 1996;84:155–164. doi: 10.1016/s0092-8674(00)81002-8. [DOI] [PubMed] [Google Scholar]
- 4.Guan JL, Trevithick JE, Hynes RO. Cell Regul. 1991;2:951–964. doi: 10.1091/mbc.2.11.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burridge K, Turner CE, Romer LH. J Cell Biol. 1992;119:893–903. doi: 10.1083/jcb.119.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hotchin NA, Kidd AG, Altroff H, Mardon HJ. J Cell Sci. 1999;112:2937–2946. doi: 10.1242/jcs.112.17.2937. [DOI] [PubMed] [Google Scholar]
- 7.Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL, Arnaout MA. Science. 2002;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 8.Mardon HJ, Grant K. FEBS Lett. 1994;340:197–201. doi: 10.1016/0014-5793(94)80137-1. [DOI] [PubMed] [Google Scholar]
- 9.Copié V, Tomita Y, Akiyama SK, Aota S, Yamada KM, Venable RM, Pastor RW, Krueger S, Torchia DA. J Mol Biol. 1998;277:663–682. doi: 10.1006/jmbi.1998.1616. [DOI] [PubMed] [Google Scholar]
- 10.Aota S, Nomizu M, Yamada KM. J Biol Chem. 1994;269:24756–24761. [PubMed] [Google Scholar]
- 11.Bowditch RD, Hariharan M, Tominna EF, Smith JW, Yamada KM, Getzoff ED, Ginsberg MH. J Biol Chem. 1994;269:10856–10863. [PubMed] [Google Scholar]
- 12.Akiyama SK, Aota S, Yamada KM. Cell Adhes Commun. 1995;3:13–25. doi: 10.3109/15419069509081275. [DOI] [PubMed] [Google Scholar]
- 13.Redick SD, Settles DL, Briscoe G, Erickson HP. J Cell Biol. 2000;149:521–527. doi: 10.1083/jcb.149.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spitzfaden C, Grant RP, Mardon HJ, Campbell ID. J Mol Biol. 1997;265:565–579. doi: 10.1006/jmbi.1996.0736. [DOI] [PubMed] [Google Scholar]
- 15.Plaxco KW, Spitzfaden C, Campbell ID, Dobson CM. J Mol Biol. 1997;270:763–770. doi: 10.1006/jmbi.1997.1148. [DOI] [PubMed] [Google Scholar]
- 16.Altroff H, van der Walle CF, Asselin J, Fairless R, Campbell ID, Mardon HJ. J Biol Chem. 2001;276:38885–38892. doi: 10.1074/jbc.M105868200. [DOI] [PubMed] [Google Scholar]
- 17.Grant RP, Spitzfaden C, Altroff H, Campbell ID, Mardon HJ. J Biol Chem. 1997;272:6159 –6166. doi: 10.1074/jbc.272.10.6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mould AP, Askari JA, Aota S, Yamada KM, Irie A, Takada Y, Mardon HJ, Humphries MJ. J Biol Chem. 1997;272:17283–17292. doi: 10.1074/jbc.272.28.17283. [DOI] [PubMed] [Google Scholar]
- 19.van der Walle, C. F., Altroff, H. & Mardon, H. J. (2002) Protein Eng. 15, in press [DOI] [PMC free article] [PubMed]
- 20.Pace, C. N. & Scholtz, J. M. (1997) in Protein Structure, A Practical Approach (Creighton, T. E., ed) pp. 299 –321, IRL Press, Oxford
- 21.Danen EH, Aota S, van Kraats AA, Yamada KM, Ruiter DJ, van Muijen GN. J Biol Chem. 1995;270:21612–21618. doi: 10.1074/jbc.270.37.21612. [DOI] [PubMed] [Google Scholar]
- 22.Yin Z, Giacomello E, Gabriele E, Zardi L, Aota S, Yamada KM, Skerlavaji B, Doliana R, Colombatti A, Perris R. Blood. 1999;93:1221–1230. [PubMed] [Google Scholar]
- 23.Craig D, Krammer A, Schulten K, Vogel V. Proc Natl Acad Sci U S A. 2001;98:5590 –5595. doi: 10.1073/pnas.101582198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]