

Summary
Raf Kinase Inhibitory Protein (RKIP or PEBP) is an inhibitor of the Raf/MEK/MAP kinase signaling cascade and a suppressor of cancer metastasis. We now show that RKIP associates with centrosomes and kinetochores and regulates the spindle checkpoint in mammalian cells. RKIP depletion causes decreases in the mitotic index, the number of metaphase cells and traversal times from nuclear envelope breakdown to anaphase, and an override of mitotic checkpoints induced by spindle poisons. Raf-1 depletion or MEK inhibition reverses the reduction in the mitotic index, whereas hyperactivation of Raf mimics the RKIP-depletion phenotype. Finally, RKIP depletion or Raf hyperactivation reduces kinetochore localization and kinase activity of Aurora B, a regulator of the spindle checkpoint. These results indicate that RKIP regulates Aurora B kinase and the spindle checkpoint via the Raf-1/MEK/ERK cascade, and demonstrate that small changes in the MAP kinase pathway can profoundly impact the fidelity of the cell cycle.
Introduction
Intracellular signal transduction pathways are critical to proper interpretation and integration of growth regulatory stimuli, and intricate mechanisms have evolved to ensure the fidelity of cell replication. Small changes that alter the magnitude of these signals can significantly impact cellular outcomes. Elucidating the nature of these signaling pathways and how they are modulated is central to understanding cell cycle control and the maintenance of genomic integrity.
One of the key players in the regulation of cell growth is the evolutionarily conserved MAP kinase (MAPK) pathway. The extracellular signal regulated kinases (ERKs) are a subfamily of MAPKs activated via a cascade involving Ras, Raf kinase, and MEK (Pearson et al., 2001). Activation of the ERK pathway is tightly controlled, and Raf-1 activation is a key regulatory step. Raf-1 activation involves multiple events including phosphorylation at activating sites S338 and Y341(Dhillon and Kolch, 2002). Raf-1 is also regulated by several proteins that modulate its activity, leading to different physiological outcomes. One regulator is Raf Kinase Inhibitory Protein (RKIP), also known as Phosphatidylethanolamine Binding Protein (PEBP) (Yeung et al., 1999).
RKIP is widely expressed and highly conserved, and many of its homologs regulate growth and differentiation signaling pathways (Trakul and Rosner, 2005). In mammalian cells, RKIP inhibits Raf-1 signaling to ERK 1,2, suppressing Raf-1-induced transformation (Yeung et al., 1999). RKIP can inhibit TNF-αinduced activation of IKKβin the NFκB cell survival pathway (Yeung et al., 2001). RKIP potentiates apoptosis induced by chemotherapeutic agents (Chatterjee et al., 2004; Jazirehi et al., 2004). Finally, RKIP suppresses metastasis in a human prostate cancer model, and this phenotype correlates with Raf-1 inhibition (Fu et al., 2003). Reduction in RKIP also correlates with metastatic progression in melanoma and breast cancer (Hagan et al., 2005; Schuierer et al., 2004).
RKIP blocks phosphorylation of regulatory sites on Raf-1 and inhibits Raf-1 activation (Trakul et al., 2005). Following cell stimulation, RKIP is phosphorylated on S153 by Protein Kinase C (PKC) causing dissociation of RKIP from Raf-1 (Corbit et al., 2003). Consistent with this mechanism, RKIP depletion from cells increases the amplitude and dose response of ERK activation and DNA (Trakul et al., 2005; Yeung et al., 1999). Upon release from Raf-1, phosphorylated RKIP inhibits GRK2, enhancing G protein-coupled receptor signaling (Lorenz et al., 2003). Thus, RKIP modulates the ERK signaling cascade both directly and via crosstalk, limiting the response of the cell to growth factor stimuli.
Although Raf-1 is activated during G1, some reports suggest that it also functions during the G2/M phase of the mammalian mitotic cell cycle. The activation of mitotic Raf-1 is Ras-independent but Pak-dependent (Ziogas et al., 1998). Pak phosphorylates S338 on Raf-1, a critical modification for Raf-1 activation in many systems (King et al., 1998). In Xenopus egg extracts, MAPK is not required for mitotic entry or exit, and MAPK activation promotes cell cycle arrest (Takenaka et al., 1997; Wang et al., 1997). By contrast, in mammalian cells, the Raf-1/ERK1,2 cascade may influence normal G2 progression and entry into mitosis (Hayne et al., 2000; Wright et al., 1999). Raf-1-activated ERK1c, an ERK variant, regulates mitotic Golgi fragmentation (Shaul and Seger, 2006). Finally, activated ERK1,2 is associated with kinetochores and spindle poles from prometaphase to anaphase and with the midbody at later stages of mitosis (Shapiro et al., 1998; Zecevic et al., 1998). Other kinases that are localized on centrosomes and/or kinetochores have been implicated in mitotic progression including Aurora A and B (Meraldi et al., 2004). Aurora B, an evolutionarily conserved kinase, is implicated in chromosomal alignment, cytokinesis, and spindle checkpoints. In complex with other “chromosomal passenger” proteins, Aurora B accumulates at inner centromeres during prometaphase and controls the interactions of microtubules with kinetochores. These observations raise the possibility that the Raf/MEK/ERK signaling cascade regulates mitosis via an interaction with mitotic kinases.
To address this question, we determined the cellular localization and effect of RKIP on mitotic progression in mammalian cells. Our results show that loss of RKIP leads to a decrease in mitotic index and metaphase cell number, and a defective spindle checkpoint through a mechanism involving enhanced Raf-1 activation and Aurora B kinase inhibition.
Results
pRKIP is Localized at Centrosomes and Kinetochores in Mitotic Cells
RKIP phosphorylation at S153 causes its dissociation from Raf-1 leading to Raf-1 activation. Since RKIP is a prostate cancer suppressor, we investigated whether RKIP is phosphorylated at S153 in proliferating prostate tumor cells. Immunocytochemistry using an anti-phosphoS153-RKIP (pRKIP) antibody shows selective staining of mitotic nonmetastatic (LNCaP) and metastatic (C4-2) prostate cells (Fig. 1A). We observed similar pRKIP immunostaining in areas of rapid cell proliferation within the developing brain and in the proliferative basal layer of skin as well as all mitotic cells tested (data not shown).
Figure 1.

pRKIP is Localized at Centrosomes and Kinetochores in Mitotic Cells (A) pRKIP in human prostate tumor cell lines. Arrows indicate centrosomes. (B) RKIP (Texas Red) and (C) pRKIP (FITC) in HeLa cells at different mitotic stages. (D) pRKIP (Texas Red) and Nek2 (FITC) in metaphase HeLa cell. (E) pRKIP (FITC) and 3F3/2 (rhodamine) in prometaphase Ptk-1 cell. Arrow: co-stained kinetochore. Scale bars, 10 μm.
To investigate pRKIP localization in proliferating cells, we examined its expression at different stages of the cell cycle by immunostaining HeLa cells with anti-RKIP or anti-pRKIP antibodies. RKIP is constitutively expressed and widely distributed within the cell (Fig. 1B). In contrast, increased pRKIP staining is first observed in the nucleus of prophase cells and then throughout the cells after nuclear envelope breakdown (NEB) (Fig. 1C). Cellular immunostaining is maintained through anaphase, but by late telophase only the centrosomes remain detectably immunoreactive. During mitosis, anti-pRKIP immunoreactivity partially overlaps with that of the NIMA kinase Nek2, a marker for centrosomes (Fig. 1D). pRKIP is also localized at kinetochores, regions associated with the centromeres of chromosomes that regulate spindle attachment. In Ptk cells pRKIP co-localizes with the 3F3/2 epitope a marker for kinetochore proteins involved in the mitotic checkpoint (Campbell and Gorbsky, 1995) (Fig. 1E). Kinetochore localization of pRKIP in prometaphase and metaphase cells can also be seen in Fig. 5. The increase in pRKIP expression and a smaller enhancement in overall RKIP expression during mitosis is supported by immunoblotting of synchronized HeLa cell lysates. Upon cell progression from interphase to mitosis, there is an ∼ 2-fold increase in RKIP expression relative to tubulin. Analysis of the pRKIP:RKIP ratio confirms that pRKIP levels rise even higher (∼ 2 fold) relative to unphosphorylated RKIP (Fig. S1A and S1B), indicating that RKIP phosphorylation is specifically increased during mitosis.
Figure 5.
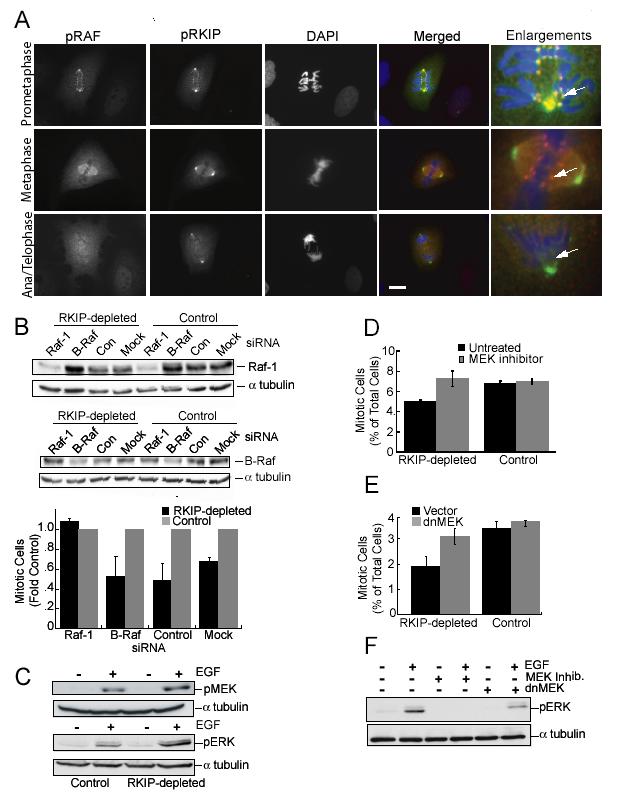
pRKIP Co-Localizes with Activated Raf-1, and Raf-1 Depletion or MEK Inhibition Reverses the Effect of RKIP Depletion on Mitotic Index(A) Localization of pS338 Raf-1 (rhodamine) and pRKIP (FITC) in prometaphase, metaphase, and anaphase/telophase Ptk-1 cells. Scale bar, 20 μm. (B) RKIP-depleted and control HeLa cells transfected with Raf-1, B-Raf, or control siRNA for 48 hrs, were immunoblotted for Raf-1 or B-Raf and mitotic cells counted. (C) Starved RKIP-depleted or control HeLa cells were untreated or treated with 100 ng/ml EGF for 5 min and immunoblotted for pERK and pMEK. (D) After synchronization RKIP-depleted or control HeLa cells were untreated or treated with 10 μM PD098059 for 4 hrs and mitotic cells counted. (E) RKIP-depleted and control HeLa cells were infected with dnMEK1 lentivirus or control lentivirus and mitotic cells counted. (F) RKIP-depleted HeLa cells were treated with 10 μM U0126, or infected with dnMEK1 lentivirus or control lentivirus, and stimulated with EGF as in (C). Lysates were immunoblotted for pERK. α-tubulin was the loading control (B, C, F). All error bars are +/-SE.
The timing of pRKIP appearance in mitosis was compared with that of cyclin B1 which is made in G2 and translocated to the nucleus during prophase. pRKIP is detected prior to cyclin B1 translocation and following cyclin B1 degradation during anaphase (Fig. S1C). Phosphorylation of Histone H3, which occurs in G2 phase and mitosis, precedes pRKIP association with the centrosomes, but loss of H3 phosphorylation during anaphase occurs before loss of pRKIP at centrosomes (data not shown). These results provide further evidence that pRKIP is elevated throughout mitosis.
RKIP Regulates Mitotic Progression
To assess the role of RKIP during mitosis, we depleted RKIP in several cell types by transient and stably expressed siRNAs. Transfected siRNA constructs suppress RKIP levels in a species-specific manner. Human 293T cells were cotransfected with HA-tagged rat RKIP (rRKIP) expression vector and either the PQY15 parent vector or shRNA vectors for human RKIP (hRKIP) or rRKIP and analyzed by immunoblotting with either anti-RKIP or anti-HA antibody. rRKIP shRNA suppresses exogenous HA-rRKIP but not endogenous hRKIP, whereas hRKIP shRNA suppresses endogenous hRKIP but not transfected HA-rRKIP (Fig. S2A). HeLa cells stably expressing rRKIP shRNA were used as controls in subsequent experiments.
To ensure that RKIP or pRKIP were detected by immunostaining, we analyzed RKIP-depleted H19-7 cells transfected with rRKIP or control siRNA. Although the results are an underestimate because nontransfected as well as transfected cells were counted, immunostaining of both RKIP and pRKIP in metaphase cells was decreased in RKIP-depleted H19-7 cells (Fig. S2B). Previous studies established that the anti-pRKIP antibody does not cross-react with unphosphorylated RKIP (Trakul et al., 2005). Thus, the smaller decrease in pRKIP relative to RKIP staining presumably reflects the fact that, since not all RKIP is depleted by siRNA, sufficient RKIP remains for phosphorylation by PKC. Reduction in total and centrosome-localized pRKIP was also observed in metaphase HeLa cells transfected with hRKIP siRNA (Fig. S2C). Thus, the reduced immunoreactivity in RKIP-depleted cells and distinct immunostaining patterns validate the specificity of the RKIP/pRKIP antibodies.
To determine whether the increase in pRKIP during mitosis reflects a regulatory role for RKIP in mitotic progression, we measured the effect of RKIP depletion on mitotic index. When rat H19-7 and Rat-1 cells were transfected with siRNA for rRKIP or control, RKIP siRNA reduced endogenous levels by 40-85% (Fig. 2A). Similarly, when HeLa cells were transiently transfected with hRKIP siRNA, the decrease in RKIP levels varied from 50-90%. Stably expressed hRKIP shRNA caused ∼ 50% decrease in overall RKIP expression in HeLa and H19-7 cells compared to controls (Fig. 2A) (Trakul et al., 2005). When the results of 11 experiments reflecting this variable range of RKIP depletion in the three different cell types were analyzed, a significant (30-80%) reduction in mitotic index was observed (Fig. 2A). Exogenous HA-rRKIP restored the mitotic index to wild type levels (Fig. 2B). Since RKIP depletion might affect the cell cycle at multiple stages, we analyzed the distribution of mitotic HeLa cells stably transfected with either empty vector, hRKIP shRNA (RKIP-depleted) or rRKIP shRNA (control). RKIP depletion caused a significant decrease only in metaphase cells (Fig. 2C). Transfection of rRKIP into the RKIP-depleted HeLa cells restored the normal distribution of metaphase cells; no consistent difference was observed between wild type and rRKIP-rescued cells (Fig. 2D; data not shown). These results demonstrate that RKIP regulates the number of mitotic cells in a proliferating cell population and, specifically, cell accumulation in metaphase.
Figure 2.

RKIP Depletion in Rat or Human Cells Reduces Mitotic Index and Metaphase Cells, and Regulates Mitotic Progression (A) RKIP was depleted by siRNA transient transfection of H19-7, HeLa or Rat-1 cells or stable shRNA (hRKIP) transfection of HeLa cells. Mitotic cell counts were normalized to cultures transfected with control siRNA (human lamin) or shRNA (rRKIP). Means +/- SE are from multiple Rat-1(2), H19-7(4), and HeLa(5) experiments. Upper panels are RKIP immunoblots from one experiment per cell type. (B) RKIP-depleted or control HeLa cells were co-transfected with GFP and HA-tagged rRKIP or control vector. Two days later GFP+ mitotic cells were counted. (C) RKIP-depleted, control, or empty vector HeLa cells were stained with Höechst. P = prophase, P-M = prometaphase, M = metaphase, and A/T = anaphase and telophase. *p=.01 by Student t-Test. (D) RKIP-depleted HeLa cells were transfected as in (B). Two days later GFP+ cells at different stages of mitosis were counted. (E) RKIP-depleted or control HeLa cells were synchronized. The interval from NEB to anaphase was determined by time-lapse photomicroscopy. (RKIP-depleted, n=11; Control, n=30) (F) RKIP-depleted or control HeLa cells were synchronized. Nine hours after release, cells at different mitotic stages were counted. All error bars are +/-SE.
A decrease in mitotic index could be due to apoptosis, cell stasis, or a faster rate of progression through mitosis. There was no difference in the growth of RKIP-depleted cells for 6 days compared to control (Fig. S2D), suggesting that a loss of cells by death or suppressed growth is not responsible for the decrease. Since the median time from NEB to anaphase measured in HeLa cells is approximately one hour, a relatively small difference in duration could make a big difference in mitotic cell number. To test this possibility, we synchronized RKIP-depleted HeLa cells and control cells using a double thymidine block, and then monitored the traversal time from NEB to anaphase. RKIP-depletion decreased the mean traversal time from 73 to 52 minutes (Fig. 2E). This difference is illustrated by a movie showing representative wild type and RKIP-depleted HeLa cells traversing mitosis at different rates (Fig. S3). Consistent with these data, analysis of the synchronized mitotic cell population at a single time point (9 hours) after release from thymidine block shows that more RKIP-depleted mitotic cells reach telophase and cytokinesis and fewer are in metaphase than control HeLa cells. (Fig. 2F). Taken together, these results indicate that RKIP regulates normal timing of mitosis from NEB to anaphase, and accumulation of cells in metaphase.
RKIP Depletion Overrides Mitotic Checkpoints Induced by Spindle Poisons
Cells accumulate in metaphase due to a mitotic checkpoint that restricts progression until chromosomes are properly attached to the spindle (Bharadwaj and Yu, 2004). This checkpoint, which regulates the ubiquitin/proteosome Anaphase Promoting Complex or cyclosome (APC/C), not only plays a role in normal anaphase initiation but is also triggered by spindle damage. Compounds that disrupt the mitotic spindle or its function reinforce the spindle checkpoint, and cells remain in metaphase. Cells with a defective checkpoint function override a mitotic poison-induced checkpoint more readily than wild type cells.
To determine if the reduction in metaphase cells upon RKIP depletion is related to spindle checkpoint relaxation, we treated HeLa cells with Taxol (paclitaxel), a microtubule stabilization drug that enables spindle attachment but prevents chromosome movement. HeLa cells treated with 1 nM Taxol accumulate in mitosis (Fig. 3A). By contrast, the increase in mitotic cells in response to Taxol in RKIP-depleted HeLa cells is substantially reduced. Mitotic checkpoint relaxation caused by RKIP depletion is not limited to highly tumorigenic, aneuploid cells such as HeLa. The mitotic index of nontransformed H19-7 cells stably expressing rRKIP shRNA is similarly decreased relative to control cells following 10 nM Taxol treatment (Fig. 3B).
Figure 3.

RKIP Depletion Decreases Cell Arrest in Taxol (A) RKIP-depleted or control HeLa cells were treated with Taxol for 5 hrs and mitotic cells counted. (B) RKIP-depleted or control H19-7 cells were Taxol-treated for 7 hrs and mitotic cells counted. Error bars are +/-SE. (C) RKIP-depleted or control HeLa cells were synchronized, treated with 1 nM Taxol at 2 hrs after release, and imaged beginning 5 hrs later. Mitotic cells reaching anaphase by indicated times are plotted. (D) Photomicrographs from (C). Arrowheads: RKIP-depleted and control cells that began mitosis at same time.
One possible explanation for this reduction in mitotic cells is that RKIP-depleted cells do not enter mitosis in the presence of Taxol. However, live cell time-lapse analysis indicates this is not the case When synchronized HeLa cells are treated with 1 nM Taxol, a larger fraction of RKIP-depleted cells progress to anaphase than control cells. For instance, by 11.5 hours after release from a double thymidine block, almost 40% of the mitotic RKIP-depleted cells, but only 23% of the control cells reached anaphase (Fig.3C). This difference is illustrated by time-lapse photographs showing two RKIP-depleted cells undergoing mitosis, cytokinesis, and cell division during the same time period that two control cells remain in metaphase (Fig. 3D). These results show that RKIP-depleted cells enter mitosis and override mitotic arrest at higher rates even when the checkpoint is enhanced by Taxol.
When microtubules are stabilized by high concentrations of Taxol, cells that escape mitotic arrest are characterized by decondensed chromatin in micronuclei. After treatment with 10 μM Taxol for 12 hours, the number of micronuclei in RKIP-depleted HeLa cells is >4 times the control cells (Fig. 4A). Cells exhibiting these micronuclei die within a couple days upon continued Taxol exposure (data not shown). These cells that escape arrest can also be visualized by chromosome preparations. Following mitotic shake-offs, chromosomal spreads of Taxol-arrested HeLa cells have well-defined chromosomal pairs, whereas cells that advanced through the checkpoint exhibit abnormal chromatin morphology (Fig. 4B). After 6 hours of 10 μM Taxol treatment, the number of mitotic cells that escape arrest is approximately twice as high in RKIP-depleted as control HeLa cells (Fig. 4B). Similar micronuclei and abnormal chromatin morphologies were observed in cells treated with inhibitors of Aurora B kinase after exposure to Taxol ((Hauf et al., 2003); data not shown). Chromosomal spreads of RKIP-depleted H19-7 cells exposed to Taxol also had fewer normal mitotic cells and more abnormal chromatin morphologies (data not shown). Thus, RKIP depletion increases chromosomal damage upon exposure to Taxol due to relaxation of the spindle checkpoint.
Figure 4.

RKIP Depletion Enhances Taxol-Induced Chromatin Damage and Decreases Mitotic Arrest in Nocodazole(A) RKIP-depleted or control HeLa cells were untreated or treated for 12 hrs with 10 μM Taxol, stained with Höechst, and micronucleated cells counted. Insets: chromatin of Taxol-arrested cells (left), or a Taxol-escaped micronucleated cell (right). (B) RKIP-depleted or control HeLa cells were treated with 10 μM Taxol for 6 hrs, and chromosome spreads from Taxol-arrested or escaped cells were counted. (C) RKIP-depleted or control HeLa cells were treated with 0 or 100 nM nocodazole for 6 hrs and mitotic cells counted. Chromosome spreads are shown. (D) RKIP-depleted or control HeLa cells were synchronized and released for indicated times into medium +/-200 ng/ml nocodazole and mitotic cells counted. (E) HeLa RKIP-depleted or control cells were treated with 200 ng/ml nocodazole for 5 hrs. Arrested cells were shaken off and replated in nocodazole +/-5 μM Aurora B kinase inhibitor (ZM) for 4 hrs. PhosphoHistone H3+ (mitotic) cells were counted. All error bars are +/-SE.
Spindle disruption by agents such as nocodazole that prevent microtubule polymerization also activates a spindle checkpoint (Bharadwaj and Yu, 2004). To determine whether RKIP depletion causes cells to advance through a nocodazole-activated checkpoint, unsynchronized cells were treated with nocodazole for 6 hours. No difference between control and RKIP-depleted cells was observed after short treatment (Fig. 4C). However, differences in checkpoint efficacy were detected following longer nocodazole treatment. Thus, after synchronization, RKIP-depleted and control cells initially enter M phase at the same time and have similar mitotic indices independent of nocodazole (Fig. 4D, top). As M phase progresses in the absence of nocodazole, fewer RKIP-depleted cells are found in mitosis, consistent with the results in unsynchronized cells. With prolonged nocodazole treatment, RKIP-depleted cells do not accumulate in mitosis to the same extent as control cells (Fig. 4D, bottom). To investigate this difference, unsynchronized cells were pretreated with nocodazole for 5 hours, and the cells arrested in mitosis were harvested by shake-off and then replated in nocodazole for an additional 4 hours. Analysis of the replated mitotic cells indicates that fewer of the RKIP-depleted cells (17%) remain arrested in nocodazole relative to control cells (34%) (Fig. 4E). Aurora B inhibition by ZM 447439 similarly causes resistance to mitotic arrest by long but not short nocodazole treatment (Ditchfield et al., 2003). When RKIP-depleted and control cells were treated with an Aurora B kinase inhibitor (ZM) upon replating, most of the cells progressed through mitosis (Fig. 4E). These data indicate that RKIP depletion, like Aurora B inhibitors, relaxes the spindle checkpoint invoked by long-term nocodazole treatment.
The Raf/MEK/ERK Pathway Regulates the Spindle Checkpoint
One possible mechanism by which RKIP regulates mitosis is through Raf-1 modulation. In G1, phosphorylation of RKIP at S153 enables release of bound RKIP from Raf-1 and subsequent Raf-1 activation. Raf-1 is also activated during mitosis, and S338 phosphorylation (p338) is associated with this activation. To determine whether RKIP and Raf-1 might interact during mitosis, we immunostained PtK-1 cells with antibodies to pRKIP and pS338Raf-1. Fig. 5A (cf. arrows) shows extensive co-localization of pRKIP and pS338 Raf-1, particularly on the centrosomes and kinetochores, during prometaphase. The specificity of the anti-pS338Raf-1 antibody was verified by competition with pS338 peptide and confirmed using other anti-pS338 Raf antibodies (data not shown). By metaphase, there is still co-localization in areas of diffuse staining including the spindle region. The most intense p338Raf-1 staining is no longer at the centrosomes but instead is associated with the kinetochores, while pRKIP is at the centrosomes but not kinetochores. Thus, activated Raf-1 is proximal to its downstream target, since activated ERK has also been localized to kinetochores, peaking at prometaphase and gradually disappearing by mid-anaphase. These results are consistent with an interaction between the inhibitor RKIP and Raf-1 during early mitosis that is disrupted upon phosphorylation of RKIP, dissociation of pRKIP and subsequent activation of Raf-1.
If enhanced Raf activation causes the decreased mitotic index in RKIP-depleted cells, then decreased Raf activity should rescue the phenotype. Since RKIP inhibits Raf-1 but not B-Raf activation (Trakul et al., 2005), Raf-1 should be the preferential target of RKIP action. Consistent with this hypothesis, depletion of Raf-1 but not of B-Raf by siRNAs restored the mitotic index to control levels (Fig. 5B). These results support a role for Raf-1 in mediating the effects of RKIP depletion.
The main signaling cascade downstream of Raf-1 consists of MEK and ERK1,2. As we observed previously for other cell types, RKIP depletion in HeLa cells leads to enhanced EGF-induced MEK and ERK activation (Fig. 5C). To determine whether ERK might be involved in spindle checkpoint regulation by RKIP and Raf, we pretreated cells with MEK inhibitor. Although some reports suggest that MEK is required for progression from G2 to M (Hayne et al., 2000; Liu et al., 2004; Wright et al., 1999), we did not observe G2 arrest upon MEK inhibition in our system. When control or RKIP-depleted HeLa cells were synchronized and treated 2 hours later with 10 μM PD098059 for an additional 4 hours, the number of mitotic cells in the RKIP-depleted cultures increased, approaching the level in control cells (Fig. 5D). Inhibitor concentrations up to 50 μM and increased exposure times produced similar results, and addition of PD098059 to cells arrested with 10 μM Taxol eliminated the difference in mitotic index between control and RKIP-depleted cells (data not shown). In an alternate test of the role of MEK, we infected HeLa cells with lentivirus coding for a dominant-negative kinase-dead MEK1 (dnMEK). Expression of dnMEK increased the fraction of RKIP-depleted mitotic cells to that of control cells (Fig. 5E) and partially inhibited EGF-induced ERK1,2 activation relative to a MEK inhibitor (Fig 5F). These results indicate that MEK inhibition can rescue the decrease in mitotic cells caused by RKIP depletion.
The localization of activated Raf-1 and ERK at kinetochores and the rescue of the mitotic defect in RKIP-depleted cells by suppression of Raf-1 or MEK suggest that enhanced Raf-1/ERK activation is responsible for the mitotic phenotype. To determine whether increased Raf activation is sufficient, we assessed the effect of conditionally activated Raf kinase in HeLa cells. HeLa cells were stably transfected with an expression vector for ΔRaf-1:ER, a fusion protein comprising the estrogen-binding domain of the estrogen receptor and the Raf-1 kinase domain (Samuels et al., 1993). Stimulation of Raf kinase in ΔRaf-1:ER-transfected cells with tamoxifen for one hour induced activation of ERK1,2 (data not shown). When control cells and ΔRa f-1:ER cells were treated with tamoxifen for 24 hours, the mitotic index decreased approximately 40% in the latter (Fig. 6A), comparable to that obtained upon RKIP depletion.
Figure 6.

Raf-1 Activation Mimics RKIP-Depletion (A) Wild type or HeLa cells stably expressing ΔRaf-1:ER were treated for 24 hrs with tamoxifen and mitotic cells counted. (B) 1 μM tamoxifen was added to synchronized ΔRaf-1:ER HeLa cells at .5, 2.5, 4.5, 6.5, or 9 hrs after release. At 9 hrs mitotic cells were counted. (C) Mitotic stages of ΔRaf-1:ER HeLa cells treated as in (B). P = prophase, P-M = prometaphase, M = metaphase, A = anaphase, T = telophase, and CK = cytokinesis. P<.03 by Student t-Test for 6.5 hr tamoxifen treatment. (D) Synchronized wild type or ΔRaf-1:ER HeLa cells were incubated for 2.5 hrs, 1 μM tamoxifen added for 6.5 hrs and mitotic cells counted. (E) Wild type or ΔRaf-1:ER HeLa cells were treated with 0 or 1 μM tamoxifen for 19 hrs, 0 or 0.5 nM Taxol added for 5 hrs and mitotic cells counted. Error bars are +/-SE. (F) RKIP-depleted and control (left), or tamoxifen-treated ΔRaf-1:ER and wild type (right) HeLa cells were analyzed for Aurora B kinase activity. ZM: 1 μM Aurora kinase inhibitor. Error bars are +/-SD (left) or range (right). (G) Synchronized RKIP-depleted or control HeLa cells were released into 200 ng/ml nocodazole. Arrested (shake-off) or adherent cells were immunoblotted for Aurora B.
Both excessive and chronic Raf activation can trigger the G1 checkpoint in nontumorigenic cells. To verify that Raf kinase activation during or following DNA synthesis also decreased mitotic index, HeLa cells expressing ΔRaf-1:ER were released from a thymidine-induced G1/S block prior to addition of tamoxifen. The results show that Raf activated for at least 6.5 hours is sufficient to cause a maximal decrease in mitotic index (Fig. 6B).
Since RKIP depletion reduces the number of metaphase cells, we determined whether Raf kinase activation induces a similar phenotype. ΔRaf-1:ER and control cells were synchronized at G1/S and then treated with tamoxifen. Up to a 50% reduction in metaphase cells similar to that in RKIP-depleted cells was observed, reaching a maximum decrease at 6.5 hours of tamoxifen (Fig. 6C). Analysis at a single time point (9 hours) shows that more tamoxifen-treated mitotic ΔRaf-1:ER cells reached the late stages of mitosis than mitotic control cells (Fig. 6D), similar to RKIP-depleted cells (cf. Fig. 2F). These results indicate that Raf-1 activation or loss of RKIP reduces metaphase cell accumulation and faster progression through mitosis.
If enhanced activation of ERK upon RKIP depletion induces cells to pass through the mitotic checkpoint, then chronic activation of Raf kinase should also prevent arrest by spindle poisons such as Taxol. Consistent with this hypothesis, when cells expressing ΔRaf-1:ER were stimulated with tamoxifen prior to Taxol exposure, fewer cells accumulated in mitosis (Fig. 6E). In sum, these results show that hyperactivation of Raf-1 kinase either directly or via RKIP depletion overrides the spindle checkpoint.
RKIP Depletion Alters Localization and Kinase Activity of Aurora B at Kinetochores
Kinetochores form the interface between microtubules of the mitotic spindle and chromosomes and regulate chromosome movements during mitosis (Maiato et al., 2004). Among the proteins proximal to the centromeric heterochromatin that are involved in microtubule error correction are the passenger proteins that make up the active Aurora B kinase complex: Aurora B, Survivin, Borealin/Dasra B, and INCENP. Strikingly, a number of the mitotic phenotypes observed in RKIP-depleted cells resemble those of cells with suppressed Aurora B kinase activity (Ditchfield et al., 2003; Hauf et al., 2003; Honda et al., 2003). These common effects include a decrease in mitotic index, resistance to mitotic arrest by Taxol and chronic nocodazole treatment, and an increase in the number of “escaped” cells with abnormal chromatin morphologies in chromosome spreads from Taxol-treated cells. The major difference appears to be the robustness of the response since Aurora kinase inhibitors affect all cells acutely whereas the degree of RKIP depletion is variable. These results raise the possibility that RKIP depletion, via the Raf/MEK/ERK1,2 pathway, leads to inhibition of Aurora B kinase activity at the kinetochores.
To test this hypothesis, we assayed the kinase activity of Aurora B immunoprecipitated from nocodazole-arrested control or RKIP-depleted cells. The substrate was Histone H3 and, to quantitate nonspecific kinase activity, Aurora kinase inhibitor ZM 447439 (ZM) was added to duplicate reactions. Our results consistently showed at least a 2-fold decrease in Aurora B kinase activity in RKIP-depleted versus control cells (Fig. 6F). A decrease in Aurora B kinase activity was also observed in tamoxifen-activated ΔRaf-1:ER cells (Fig. 6F). These results demonstrate that either decreased RKIP or increased Raf activity can inhibit Aurora B kinase.
Loss of Aurora B kinase activity could result from a decrease in amount or activity of the enzyme. To distinguish between these possibilities, we analyzed lysates from nocodazole-arrested cells by immunoblotting for Aurora B. There was no difference in Aurora B levels between RKIP-depleted and control cells when either shake-off (mitotic) or adherent cells were analyzed (Fig. 6G). To investigate the activity of Aurora B at mitotic cell kinetochores, we immunostained RKIP-depleted and control cells with an antibody that recognizes an Aurora B autophosphorylation site required for activity (Yasui et al., 2004). CENP-A, a histone H3 isoform that is a centromere component, was quantified to normalize the results. Nocodazole-treated cells were analyzed for phosphoAurora B (pAurora B) and CENP-A expression by deconvolution microscopy. Relative staining intensities were quantitated, and results were plotted as a distribution to reflect the heterogeneous populations of RKIP-depleted as well as control cells. Comparison of the ratio of pAurora B: CENP-A staining at kinetochores for both types revealed a significant decrease in the median of the RKIP-depleted cell distribution compared to control cells (Fig. 7A). Examples of pAurora B and CENP-A costaining in control and RKIP-depleted cells illustrate this difference (Fig. 7A). RKIP-depleted prophase cells contained more activated ERK than did control cells (Fig. 7C) and pretreatment of the cells with the MEK inhibitor PD098059 eliminated the differences in the pAurora B:CENP-A distributions (Fig. 7B). These results provide further evidence that the MEK/ERK1,2 cascade regulates Aurora B kinase activation at kinetochores.
Figure 7.

RKIP Depletion Decreases Phosphorylated Aurora B Kinase and Phosphorylated CENP-A at Kinetochores(A) Synchronized RKIP-depleted or control HeLa cells were released into 200 ng/ml nocodazole for 9 hrs. After deconvolution microscopy, the ratio of pAurora B (Texas Red) to CENP-A (FITC) per cell was plotted. (B) Cells were treated as in (A) with 10 μM PD098059 for 7 hrs. (C) Synchronized RKIP-depleted or control HeLa cells were released for 9 hrs.. After deconvolution the ratio of pERK to CENP-A per prophase cell was plotted. (D) In nocodazole-treated (100ng/ml, 2hrs) RKIP-depleted or control HeLa cells pCENP-A and CREST were quantified at individual kinetochores. Data from 3 experiments with 19 Control and 21 RKIP-depleted cells (6-8 kinetochores per cell), *p<.02. (E) Cells treated as in (D) were immunostained for Aurora B and CREST. Data from 3 experiments with 19 Control and 16 RKIP-depleted cells (5-8 kinetochores per cell) were normalized to the control mean for each experiment, *p<.0003. Error bars are +/-SE (D,E). (F) In nocodazole-treated (200 ng/ml, 9hrs) RKIP-depleted or control HeLa cells, RKIP and pCENP-A were quantified by deconvolution microscopy. Y-axes: total pixel intensity x10-5. Lines and numbers are medians of cell populations (A,B, C, and F). For A to F, data were analyzed by the Wilcoxon Two-Sample test. (G) Low RKIP (arrow) and high RKIP (arrowhead) (FITC) cells in the RKIP-depleted population and their pCENP-A (Texas Red) immunoreactivities. RKIP-depleted and control cells below the RKIP median in (F) were ranked for RKIP and pCENP-A levels. Rankings were plotted for each cell. R is the coefficient of correlation. (H) Nocodazole-treated (100 ng/ml, 2hrs) HeLa RKIP-depleted and control cells were immunostained for pCENP-A and CENP-A. (I) Model for RKIP regulation of Aurora B kinase and the spindle checkpoint.
Similar results were obtained in nocodazole-arrested HeLa cells immunostained for phosphorylated CENP-A (pCENP-A), a substrate of Aurora B required for proper kinetochore assembly (Zeitlin et al., 2001). Analysis of pCENP-A: CENP-A staining indicated that pCENP-A is reduced in RKIP-depleted cells relative to controls. (See Fig. 7H for a cell with minimal pCENP-A expression at kinetochores.) To quantitate this difference, we co-stained cells for CREST, a centromere marker, as an internal control. pCENP-A:CREST staining intensity at individual kinetochores decreased approximately 25% in RKIP-depleted cells relative to control cells (Fig. 7D). This decrease in CENP-A phosphorylation corresponds to a decrease in Aurora B localization at kinetochores when individual kinetochores costained with antibodies to Aurora B and CREST (Fig. 7E) were analyzed. Finally, we investigated the activity of Aurora B kinase at kinetochores in metaphase cells. Immunostaining of control and RKIP-depleted HeLa cells for both Aurora B kinase and pCENP-A and analysis by deconvolution microscopy revealed that the RKIP-depleted cells are a heterogeneous population with at least three phenotypes (Fig. S4). Approximately 40% of the cells resembled wild type controls, about 25% of the cells lacked most of the CENP-A phosphorylation, and about one-third of the cells had reduced pCENP-A staining. Thus, RKIP-depleted cells exhibit less Aurora B kinase activity at the kinetochores than control cells.
If RKIP depletion reduces phosphorylation of pCENP-A, then cells with less RKIP should have less pCENP-A. To determine whether there is a correlation between RKIP expression levels and CENP-A phosphorylation, nocodazole-arrested cells were analyzed by co-immunostaining with an anti-RKIP monoclonal antibody and an anti-pCENP-A polyclonal antibody. As observed for pAurora B and for pCENP-A at single kinetochores, the median pCENP-A staining was significantly decreased in RKIP-depleted relative to control cells (Fig. 7F). Analysis of the cells below the RKIP median by linear regression showed a good correlation between the levels of RKIP expression and CENP-A phosphorylation in individual RKIP-depleted cells (Fig. 7G). In contrast, no correlation could be detected for the control cells. These results provide further evidence that RKIP depletion is indeed responsible for the phenotypes observed.
Discussion
The results presented here describe a role for Raf Kinase Inhibitory Protein in the spindle checkpoint. The potential role of RKIP in mitosis was suggested by the association of pS153RKIP with mitotic centrosomes and kinetochores in a variety of cell types and tissues. RKIP depletion leads to a decrease in mitotic index, an acceleration in timing of the metaphase/anaphase transition and a defect in the spindle checkpoint in HeLa and H19-7 cells. Consistent with localization of pS338 Raf-1 at kinetochores during mitosis, Raf activation can mimic and MEK inhibition can rescue the effects of RKIP depletion. Finally, RKIP depletion causes decreased localization of phosphorylated Aurora B at kinetochores and loss of kinase activity. These results demonstrate that loss of RKIP, through hyperactivation of the Raf/MEK/ERK1,2 signaling cascade, regulates the mitotic checkpoint via inhibition of Aurora B kinase (Fig. 7I). Several studies have shown that excessive activation of Raf and MAP kinase in G1 leads to upregulation of cyclin-dependent kinase inhibitors, culminating in cell arrest or senescence (Woods et al., 1997; Zhu et al., 1998). As a negative regulator that controls the amplitude and dose response of Raf-1 kinase activity rather than the absolute on or off state, RKIP moderates the extent of ERK activation. Thus, these results indicate that Raf and RKIP play a key role in the spindle checkpoint by controlling the range of ERK1,2 signaling.
The RKIP depletion phenotype including override of the spindle checkpoint corresponds to the phenotype of cells following Aurora B inhibition. The spindle checkpoint delays chromosome segregation in response to problems with spindle attachment or tension at the kinetochores (Bharadwaj and Yu, 2004). Activation of this mitotic checkpoint is a dynamic, multistep process involving a number of proteins including Mad2, BubR1, and Bub3 that inhibit the ubiquitin ligase activity of the APC/C complex by preventing Cdc20 association. The spindle checkpoint is not only triggered by spindle damage but also plays a role in the initiation of anaphase in every cell. The passenger protein complex consisting of Aurora B, INCENP, Survivin and Borealin/Dasra B is required for maintaining the integrity of mitotic regulation, including phosphorylation of histone H3 and its variant CENP-A, spindle assembly, chromatin-induced stabilization of microtubules, and mitotic arrest in response to microtubule poisons such as Taxol (Vagnarelli and Earnshaw, 2004). The passenger proteins stabilize Aurora B localization to the inner centromere and are required for kinetochore-associated Aurora B kinase activity. Therefore, a decrease in Aurora B localization and kinase activity at the kinetochore should alter the integrity of the spindle assembly checkpoint.
The specific mechanism by which RKIP regulates Aurora B kinase activity remains to be determined. Aurora B kinase activity could be inhibited directly by ERK1,2 phosphorylation of Aurora B or indirectly by mechanisms such as phosphorylation of other proteins required for recruitment of the kinase to the kinetochores. However, no phosphorylation of Aurora B kinase by ERK was observed, indicating the mechanism is not direct (data not shown). Aurora A phosphorylates CENP-A during prophase, and a requirement for pCENP-A in Aurora B recruitment to the kinetochores has been reported (Kunitoku et al., 2003). Thus, a mechanism involving inhibition of Aurora A might account for the observed results; however, recent studies show localization in the absence of CENP-A (Klein et al., 2006) indicating that the role of Aurora A/CENP-A in Aurora B activation is not yet resolved.
The results we have obtained provide a potential explanation for growth and tumor-regulating functions of RKIP that have recently been described. Treatment of cells with chemotherapeutic agents such as Taxol can enhance RKIP expression in the arrested cells and potentiate apoptosis (Chatterjee et al., 2004). Our results suggest that the increase in RKIP may not be due to induction but rather to the normal increase that occurs during mitosis. If RKIP promotes arrest or apoptosis due to the mitotic checkpoint, then higher levels of RKIP should increase cell death. Conversely, depletion of RKIP should lead to slippage of cells through the checkpoint resulting in fewer arrested or apoptotic cells and an increase in aneuploidy depending upon the specific cell type. In fact, expression of oncogenic Ras, an upstream activator of Raf-1, has been shown to promote chromosome instability via ERK (Saavedra et al., 1999). Consistent with this possibility, RKIP was recently shown to function as a metastasis suppressor in prostate cancer (Fu et al., 2003). In xenografts, metastatic PC3 cells that overexpressed RKIP showed a marked decrease in the number of mice that developed metastases (70%), and the expression of RKIP inversely correlated with Raf-1 and ERK activity. A decrease in RKIP expression also correlates with melanoma and breast cancer tumor progression (Hagan et al., 2005; Schuierer et al., 2004).
It has been suggested that partial suppression of the spindle checkpoint rather than its total elimination is more likely to lead to cancer since complete inactivation could result in cell death (Bharadwaj and Yu, 2004). RKIP depletion leads to such a partial suppression of the spindle checkpoint. Interestingly, RKIP itself does not induce cell death unless overexpressed or mutated to prevent dissociation from Raf-1 (data not shown). Conversely, loss of endogenous RKIP or enhanced Raf kinase activation leads to a spindle checkpoint defect that enables cells to escape Taxol-induced arrest more easily. Cells proceed through division or die depending on the dose, suggesting that RKIP levels in cancer cells can influence the Taxol regimen needed for toxicity. These data indicate that Raf-1 kinase activity must be tightly regulated during mitosis, and RKIP plays a key role in modulating this activity. Cells lacking RKIP should display an increase in chromosomal abnormalities and genetic changes when under oncogenic or toxic stress, providing one mechanism for enhancing their metastatic potential.
Experimental Procedures
Cell Lines and Synchronization
Cells were grown at 37°C (HeLa, Rat-1, Ptk-1, LN CaP, C4-2B, HEK293), or 33°C (H19-7) in 5% CO2 in high glucose DMEM with 10% FBS, 50 u/ml penicillin, and 5μg/ml streptomycin. Cells stably expressing shRNA RKIP vectors were described (Trakul et al., 2005). 293T cells were cotransfected with pCMV-VSV-G, pCMV and dnMEK1 or control vector to produce lentivirus. HeLa cells were synchronized at G1/S by two 17-hr treatments with 2.5 mM thymidine separated by a 7-hr recovery period. See Supplemental Data for details.
Mitotic Indices and Staging
For mitotic cell counting and staging, cells were fixed as above, DNA was stained with Höechst 33342 or 4’, 6-diamidino-2-phenylindole (DAPI), and 300-500 cells were counted in triplicate. For green fluorescent protein (GFP) or FITC-luciferase transfected cells, triplicate counts of ≥100 green cells were done. Höechst staining or chromosome spreads identified cells that escaped a 10 μM Taxol-induced mitotic checkpoint. Arrested and “escaped” cells were counted in triplicate (≥100 nuclei each). Control cells were treated DMSO.
In vitro Kinase Assays and Aurora Kinase Inhibition
For Aurora B kinase assays, synchronized cells were released into 200 ng/ml nocodazole for 9 hrs, harvested by shake-off, lysed, and immunoprecipitated. The kinase assay contained 20μl of immunoprecipitate with 5 μg of histone H3 (Roche), 10 μM ATP, 2.5 μCi [γ-32P]ATP and 1 μM ZM447439 or DMSO (30 min, 37°C). See Supplemental Data for details.
Image Acquisition and Quantitation
Time-lapse photomicroscopy employed a 20X objective on a Zeiss Axiovert 100TV in an environmental box (37°C, 5% CO2) at the University of Chicago BSD Digital Light Microscope Facility.
For deconvolution and quantification, z-stacks were acquired and deconvolved in Openlab (Improvision). Measurements were performed on composites of deconvolved images.
For quantification at individual kinetochores digital images from single optical sections were captured using a Nikon E800 epifluorescence microscope and processed with IPLab Scientific Imaging Software (Scanalytics Inc.).See Supplemental Data for details.
Acknowledgements
We thank Drs. Hyunsook Lee, Anning Lin, Erich A. Nigg, Jonathan Pines, and Stephen Taylor for helpful discussions, Keiko Yoshida, Shirley Bond, and Suzana Gomes for technical assistance, William Earnshaw, Stephen Taylor, Sheila Stewart, Mark Abe, Yuru Liu, and AstraZeneca (ZM 447439) for generous gifts of reagents. This work was supported by NIH grants NS 033858 (MRR), CA105299 (PS), a Cornelius Crane Trust for Eczema Research gift (MRR), and a Boehringer Ingelheim PhD scholarship (URK).
Footnotes
- EGF
- epidermal growth factor
- ERK
- extracellular signal regulated kinase
- MAPK
- MAP kinase
- MEK
- MAP/ERK kinase
- PEBP
- phosphatidylethanolamine binding protein
- PKC
- protein kinase C
- RKIP
- Raf Kinase Inhibitory Protein
References
- Bharadwaj R, Yu H. The spindle checkpoint, aneuploidy, and cancer. Oncogene. 2004;23:2016–2027. doi: 10.1038/sj.onc.1207374. [DOI] [PubMed] [Google Scholar]
- Campbell MS, Gorbsky GJ. Microinjection of mitotic cells with the 3F3/2 anti-phosphoepitope antibody delays the onset of anaphase. J Cell Biol. 1995;129:1195–1204. doi: 10.1083/jcb.129.5.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee D, Bai Y, Wang Z, Beach S, Mott S, Roy R, Braastad C, Sun Y, Mukhopadhyay A, Aggarwal BB, et al. RKIP sensitizes prostate and breast cancer cells to drug-induced apoptosis. J Biol Chem. 2004;279:17515–17523. doi: 10.1074/jbc.M313816200. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Trakul N, Eves EM, Diaz B, Marshall M, Rosner MR. Activation of Raf-1 Signaling by Protein Kinase C through a Mechanism Involving Raf Kinase Inhibitory Protein. J Biol Chem. 2003;278:13061–13068. doi: 10.1074/jbc.M210015200. [DOI] [PubMed] [Google Scholar]
- Dhillon AS, Kolch W. Untying the regulation of the Raf-1 kinase. Arch Biochem Biophys. 2002;404:3–9. doi: 10.1016/s0003-9861(02)00244-8. [DOI] [PubMed] [Google Scholar]
- Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, Mortlock A, Keen N, Taylor SS. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–280. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z, Smith PC, Zhang L, Rubin MA, Dunn RL, Yao Z, Keller ET. Effects of raf kinase inhibitor protein expression on suppression of prostate cancer metastasis. J Natl Cancer Inst. 2003;95:878–889. doi: 10.1093/jnci/95.12.878. [DOI] [PubMed] [Google Scholar]
- Hagan S, Al-Mulla F, Mallon E, Oien K, Ferrier R, Gusterson B, Garcia JJ, Kolch W. Reduction of Raf-1 kinase inhibitor protein expression correlates with breast cancer metastasis. Clin Cancer Res. 2005;11:7392–7397. doi: 10.1158/1078-0432.CCR-05-0283. [DOI] [PubMed] [Google Scholar]
- Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL, Peters JM. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161:281–294. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayne C, Tzivion G, Luo Z. Raf-1/MEK/MAPK pathway is necessary for the G2/M transition induced by nocodazole. J Biol Chem. 2000;275:31876–31882. doi: 10.1074/jbc.M002766200. [DOI] [PubMed] [Google Scholar]
- Honda R, Korner R, Nigg EA. Exploring the functional interactions between Aurora B, INCENP, and survivin in mitosis. Mol Biol Cell. 2003;14:3325–3341. doi: 10.1091/mbc.E02-11-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazirehi AR, Vega MI, Chatterjee D, Goodglick L, Bonavida B. Inhibition of the Raf-MEK1/2-ERK1/2 signaling pathway, Bcl-xL down-regulation, and chemosensitization of non-Hodgkin’s lymphoma B cells by Rituximab. Cancer Res. 2004;64:7117–7126. doi: 10.1158/0008-5472.CAN-03-3500. [DOI] [PubMed] [Google Scholar]
- King AJ, Sun H, Diaz B, Barnard D, Miao W, Bagrodia S, Marshall MS. The protein kinase Pak3 positively regulates Raf-1 activity through phosphorylation of serine 338. Nature. 1998;396:180–183. doi: 10.1038/24184. [DOI] [PubMed] [Google Scholar]
- Klein UR, Nigg EA, Gruneberg U. Centromere Targeting of the Chromosomal Passenger Complex Requires a Ternary Subcomplex of Borealin, Survivin, and the N-Terminal Domain of INCENP. Mol Biol Cell. 2006 doi: 10.1091/mbc.E05-12-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunitoku N, Sasayama T, Marumoto T, Zhang D, Honda S, Kobayashi O, Hatakeyama K, Ushio Y, Saya H, Hirota T. CENP-A phosphorylation by Aurora-A in prophase is required for enrichment of Aurora-B at inner centromeres and for kinetochore function. Dev Cell. 2003;5:853–864. doi: 10.1016/s1534-5807(03)00364-2. [DOI] [PubMed] [Google Scholar]
- Liu X, Yan S, Zhou T, Terada Y, Erikson RL. The MAP kinase pathway is required for entry into mitosis and cell survival. Oncogene. 2004;23:763–776. doi: 10.1038/sj.onc.1207188. [DOI] [PubMed] [Google Scholar]
- Lorenz K, Lohse MJ, Quitterer U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature. 2003;426:574–579. doi: 10.1038/nature02158. [DOI] [PubMed] [Google Scholar]
- Maiato H, DeLuca J, Salmon ED, Earnshaw WC. The dynamic kinetochore-microtubule interface. J Cell Sci. 2004;117:5461–5477. doi: 10.1242/jcs.01536. [DOI] [PubMed] [Google Scholar]
- Meraldi P, Honda R, Nigg EA. Aurora kinases link chromosome segregation and cell division to cancer susceptibility. Curr Opin Genet Dev. 2004;14:29–36. doi: 10.1016/j.gde.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu B, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (map) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- Saavedra HI, Fukasawa K, Conn CW, Stambrook PJ. MAPK mediates RAS-induced chromosome instability. J Biol Chem. 1999;274:38083–38090. doi: 10.1074/jbc.274.53.38083. [DOI] [PubMed] [Google Scholar]
- Samuels ML, Weber MJ, Bishop JM, McMahon M. Conditional transformation of cells and rapid activation of the mitogen-activated protein kinase cascade by an estradiol-dependent human Raf-1 protein kinase. Mol Cell Biol. 1993;13:6241–6252. doi: 10.1128/mcb.13.10.6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuierer MM, Bataille F, Hagan S, Kolch W, Bosserhoff AK. Reduction in Raf kinase inhibitor protein expression is associated with increased Ras-extracellular signal-regulated kinase signaling in melanoma cell lines. Cancer Res. 2004;64:5186–5192. doi: 10.1158/0008-5472.CAN-03-3861. [DOI] [PubMed] [Google Scholar]
- Shapiro PS, Vaisberg E, Hunt AJ, Tolwinski NS, Whalen AM, McIntosh JR, Ahn NG. Activation of the MKK/ERK pathway during somatic cell mitosis: direct interactions of active ERK with kinetochores and regulation of the mitotic 3F3/2 phosphoantigen. J Cell Biol. 1998;142:1533–1545. doi: 10.1083/jcb.142.6.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaul YD, Seger R. ERK1c regulates Golgi fragmentation during mitosis. J Cell Biol. 2006;172:885–897. doi: 10.1083/jcb.200509063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka K, Gotoh Y, Nishida E. MAP kinase is required for the spindle assembly checkpoint but is dispensable for the normal M phase entry and exit in Xenopus egg cell cycle extracts. J Cell Biol. 1997;136:1091–1097. doi: 10.1083/jcb.136.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trakul N, Menard RE, Schade GR, Qian Z, Rosner MR. Raf kinase inhibitory protein regulates Raf-1 but not B-Raf kinase activation. J Biol Chem. 2005;280:24931–24940. doi: 10.1074/jbc.M413929200. [DOI] [PubMed] [Google Scholar]
- Trakul N, Rosner MR. Modulation of the MAP kinase signaling cascade by Raf kinase inhibitory protein. Cell Res. 2005;15:19–23. doi: 10.1038/sj.cr.7290258. [DOI] [PubMed] [Google Scholar]
- Vagnarelli P, Earnshaw WC. Chromosomal passengers: the four-dimensional regulation of mitotic events. Chromosoma. 2004;113:211–222. doi: 10.1007/s00412-004-0307-3. [DOI] [PubMed] [Google Scholar]
- Wang XM, Zhai Y, Ferrell JE., Jr. A role for mitogen-activated protein kinase in the spindle assembly checkpoint in XTC cells. J Cell Biol. 1997;137:433–443. doi: 10.1083/jcb.137.2.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods D, Parry D, Cherwinski H, Bosch E, Lees E, McMahon M. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol. 1997;17:5598–5611. doi: 10.1128/mcb.17.9.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JH, Munar E, Jameson DR, Andreassen PR, Margolis RL, Seger R, Krebs EG. Mitogen-activated protein kinase kinase activity is required for the G(2)/M transition of the cell cycle in mammalian fibroblasts. Proc Natl Acad Sci USA. 1999;96:11335–11340. doi: 10.1073/pnas.96.20.11335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui Y, Urano T, Kawajiri A, Nagata K, Tatsuka M, Saya H, Furukawa K, Takahashi T, Izawa I, Inagaki M. Autophosphorylation of a newly identified site of Aurora-B is indispensable for cytokinesis. J Biol Chem. 2004;279:12997–13003. doi: 10.1074/jbc.M311128200. [DOI] [PubMed] [Google Scholar]
- Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–177. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- Yeung KC, Rose DW, Dhillon AS, Yaros D, Gustafsson M, Chatterjee D, McFerran B, Wyche J, Kolch W, Sedivy JM. Raf kinase inhibitor protein interacts with NF-kappaB-inducing kinase and TAK1 and inhibits NF-kappaB activation. Mol Cell Biol. 2001;21:7207–7217. doi: 10.1128/MCB.21.21.7207-7217.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecevic M, Catling AD, Eblen ST, Renzi L, Hittle JC, Yen TJ, Gorbsky GJ, Weber MJ. Active MAP kinase in mitosis: localization at kinetochores and association with the motor protein CENP-E. J Cell Biol. 1998;142:1547–1558. doi: 10.1083/jcb.142.6.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlin SG, Shelby RD, Sullivan KF. CENP-A is phosphorylated by Aurora B kinase and plays an unexpected role in completion of cytokinesis. J Cell Biol. 2001;155:1147–1157. doi: 10.1083/jcb.200108125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Woods D, McMahon M, Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998;12:2997–3007. doi: 10.1101/gad.12.19.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziogas A, Lorenz IC, Moelling K, Radziwill G. Mitotic Raf-1 is stimulated independently of Ras and is active in the cytoplasm. J Biol Chem. 1998;273:24108–24114. doi: 10.1074/jbc.273.37.24108. [DOI] [PubMed] [Google Scholar]