

Abstract
The use of quantitative gene expression analysis for the diagnosis, prognosis, and monitoring of disease requires the ability to distinguish pathophysiological changes from natural variations. To characterize these variations in apparently healthy subjects, quantitative real-time PCR was used to measure various immune response genes in whole blood collected from blood bank donors. In a single-time-point study of 131 donors, of 48 target genes, 43 were consistently expressed and 34 followed approximately log-normal distribution. Most transcripts showed a limited dynamic range of expression across subjects. Specifically, 36 genes had standard deviations (SDs) of 0.44 to 0.79 cycle threshold (CT) units, corresponding to less than a 3-fold variation in expression. Separately, a longitudinal study of 8 healthy individuals demonstrated a total dynamic range (> 2 standard error units) of 2- to 4-fold in most genes. In contrast, a study of whole blood gene expression in 6 volunteers injected with LPS showed 15 genes changing in expression 10- to 90-fold within 2 to 5 h and returning to within normal range within 21 hours. This work demonstrates that (1) the dynamic range of expression of many immune response genes is limited among healthy subjects; (2) expression levels for most genes analyzed are approximately log-normally distributed; and (3) individuals exposed to an infusion of bacterial endotoxin (lipopolysaccharide), show gene expression profiles that can be readily distinguished from those of a healthy population. These results suggest that normal reference ranges can be established for gene expression assays, providing critical standards for the diagnosis and management of disease.
INTRODUCTION
Recent developments in gene and protein expression analysis technology have suggested that gene expression is a key indicator of an individual’s pathophysiologic status (1–4). Consequently, clinical application of gene expression technology will vastly improve on the current approaches for monitoring health and disease. Compelling associations between gene expression and disease have been demonstrated in many studies ranging from inflammatory disease to cancer. For instance, studies have pointed to abnormal gene expression in peripheral blood mononuclear cells in lupus patients compared with healthy controls (5,6). Other studies have found differences in gene expression patterns between cancerous liver or pancreatic tissue and nontumor liver and pancreatic tissues (7,8). Additionally, gene expression profiling of breast tumor biopsy tissue correlated with therapeutic response to treatment (9). Results from these studies demonstrate that measurements of gene expression can be used in the diagnosis and monitoring of disease. However, a key requirement for clinical application of gene expression technology is distinguishing between natural variations in gene expression among healthy subjects and changes associated with a disease condition. The establishment of a normal range of expression for a particular population is required as a “reference range” (10).
Immune function is controlled by a network of molecular and cellular pathways. It is well recognized that suppressed immune responses (for example, immunosuppressive therapies and AIDS) or excessive responses (for example, acute respiratory distress syndrome and autoimmunity) can contribute to disease. Thus, homeostatic control and tight regulation of responses are fundamental characteristics of the immune system. For example, in the absence of disease, body temperature remains relatively constant within an individual, suggesting that the body strives to hold its temperature close to a defended set point. During a response to infection, the inflammatory cytokines interleukin-1, interleukin-6, and tumor necrosis factor are released into the blood and bind with receptors in the hypothalamus, resulting in fever (11). However, immune cells also manufacture and release factors, such as interleukin-1 receptor antagonist and interleukin-10, that counteract the effects of pro-inflammatory cytokines and reduce body temperature (12,13). As a result, body temperature rises only moderately during many fever episodes, and returns to its previous set point upon clearance of the infection. This and other evidence (14) imply that inflammatory/immune genes may be tightly regulated. It is further hypothesized that immune system homeostasis would be reflected in a narrow range of expression levels or set points for key molecules in these pathways among healthy subjects.
In certain gene expression studies, reproducible patterns in subsets of genes have been noted in normal tissues (15–18). The majority of these studies have used microarrays to explore the patterns of expression in isolated blood cell fractions (15,18) or other target tissues, including retina (16) and skin (17). Some studies (16,19) have used replicate arrays to assess the relative contributions of technical and biological factors to the overall variation in measurement values. The results show interindividual variation for gene expression, as well as variation over time within an individual. In addition, gene expression can be sensitive to sources of technical variability, such as time after phlebotomy and method of RNA isolation (20–23). Even within a platform, such as microarray, considerable divergence is reported (24).
In recent years, quantitative real-time (QRT) PCR has emerged as an effective and reproducible tool for transcript analysis (25). It measures relative abundances through PCR-based synthesis of target gene amplicons and activation of target-specific fluorescent probes. The amount of fluorescence generated during the exponential amplification phase provides robust comparative abundance measurements for different amplicons in the same or different wells (25). Whole blood contains representative populations of all the mature cells of the immune system as well as secretory proteins associated with cellular communications (26). The earliest observable changes of cellular immune activity are altered levels of gene expression within the various immune cell types (27). Therefore, QRT-PCR can be an effective technology for reproducibly quantifying gene expression in whole blood.
In studies reported here, we explored the variation among apparently healthy blood bank donors in the expression of a set of genes involved in immune responses. QRT-PCR was used to measure immune-related gene expression in whole blood samples, using procedures designed to sustain a high level of precision (repeatability and reproducibility). We tested the observed distribution of values to determine if it was consistent with sampling from a log-normal distribution, as has been asserted for many genes (28,29), and computed maximum likelihood estimates for the parameters of this distribution. We used statistical models to estimate the contributions of gender, age, and ethnicity to the overall differences in expression among subjects. By performing replicate measurements on longitudinal samples from a group of 8 donors, we computed relative proportions of variance arising from technical, temporal, and intersubject variability. Finally, to obtain limits for the dynamic range of expression achievable with a strong inflammatory stimulus, we performed time-course measurements for several immune response genes in a group of healthy volunteers challenged with an infusion of the bacterial endotoxin lipopolysaccharide (LPS).
MATERIALS AND METHODS
Donor Selection
Single-time-point blood samples from 131 blood donors satisfying American Red Cross blood bank standards (30) were obtained from 3 individual donor centers operated by Bonfils Blood Center, Denver, CO, USA. The samples were drawn on 3 different days over a 3-month period. Subject ages ranged from 22 to 69 years, with a median age of 44 years; age was not recorded for 61 subjects. Women (n = 64) and men (n = 67) were represented in about equal numbers. Ethnicity was reported as white/non-Hispanic for 109 subjects, Hispanic for 19, African-American for 2, and Asian/Pacific Islander for 1. No subjects in this study showed overt signs of disease that would make them ineligible to donate blood under American Red Cross standards. Because we cannot rule out undetected disease in the subjects, however, we refer to them as “apparently healthy” (18).
In addition, longitudinal samples were drawn from 8 volunteers (3 women, 5 men, age range 23 to 50 years) from the Denver area. Samples were collected from these donors approximately once per month for 6 to 8 months, yielding a total of 58 samples.
Samples from the blood donor subjects were collected under Western Institutional Review Board Study No. 20010324. The studies were also reviewed by the Lawrence Livermore National Laboratory Institutional Review Board. Written informed consent was obtained from all volunteers.
In a separate study, 6 healthy volunteers were injected intravenously over 1 min with a single dose (30 units/kg) of Gram-negative bacterial LPS, according to an approved protocol at Guys Hospital, London, UK. Blood samples were drawn and assayed before the LPS injection (0 h) and 2 and 5 h after LPS injection. Additional blood samples from 3 of 6 subjects (adult male volunteers who signed an informed consent form) were drawn and assayed 21 h after LPS injection. Medical history, physical examination, routine laboratory examination, and electrocardiogram were all normal. Subjects did not use any medication or have any significant illness within 8 weeks of the study.
Sample Handling, Purification of RNA, and Preparation of cDNA
Blood was collected from study subjects by standard phlebotomy methods using a 21-gauge butterfly needle and PAXgene Blood RNA Tubes (no. 762115; Qiagen, Valencia, CA, USA) to stabilize messenger RNA (mRNA) against degradation and prevent induction of new mRNA expression (23). Samples were gently mixed by inversion and sat at room temperature for 2 to 24 h to ensure complete nucleic acid stabilization. Samples were then frozen at −70 °C and batch-shipped on dry ice in compliance with International Air Transport Association (IATA) shipping regulations.
Total RNA from PAXgene Blood RNA samples was extracted within 30 days of collection using the PAXgene Blood RNA Kit (no. 762134; Qiagen). RNA samples were treated with RNase-free DNase I (no. 79254; Qiagen) for digestion of contaminating genomic DNA, using manufacturer-recommended protocols during the purification process. Purified RNA samples were placed at −80 °C for long-term storage.
First-strand cDNA was synthesized with random hexamer primers using TaqMan Reverse Transcription reagents (N808-0234; Applied Biosystems, Foster City, CA, USA). Approximately 250 ng RNA was added to a prepared reverse-transcription reagent mixture consisting of PCR Buffer II, 1×; MgCl2, 5.5 mM; random hexamers, 2.5 μM; dNTP blend, 2 mM; RNase inhibitor, 40 units; and MultiScribe Reverse Transcriptase, 125 units. Samples were incubated at ambient temperature for 10 min with subsequent incubation at 37 °C for 60 min. After the 37 °C incubation, samples were incubated at 90 °C for 10 min and immediately chilled on ice. Newly synthesized cDNA samples were then placed at −80 °C for storage. Prior to QRT-PCR analysis, each cDNA sample was quality control tested for RNA quantity and quality of target genes using quantitative PCR analysis (QPCR; ABI Prism 7700 Sequence Detection System, Applied Biosystems, Foster City, CA, USA) of the 18S rRNA and β-actin.
QRT-PCR Analysis of Target Genes
Primer/probe reagents were custom-designed to achieve 3 performance criteria: (1) single-gene specificity of amplification as tested by gel electrophoresis, (2) dilutional linearity of amplification performance over 2 orders of magnitude, and (3) optimal amplification efficiency of 100 ± 6%, to yield a 2-fold change in transcript per CT unit (31). Primer/probe sets were designed to span 90 to 120 base pairs, optimized for robust amplification and specificity, minimization of secondary hybridization, and consistent performance. Quality control testing of reagents and manufactured plates ensured that amplification specificity and efficiency remained within established metrics during storage and new synthesis of nucleotides.
Amplification specificity was tested by QRT-PCR with a custom cDNA standard template of induced whole blood and cell lines. Specificity was determined by the size, number, and DNA sequence of the amplified product. The size and number of amplified products was determined by agarose gel electrophoresis. Amplified products were electrophoresed on a 4% agarose gel to visualize the number of DNA bands present. The molecular weight of each band was determined by comparison to known molecular weight markers (no. PR-G1741; Fisher Scientific, Hampton, NH, USA). The presence of a single DNA band of the correct size suggested specific amplification of the intended gene sequence. In certain cases, the amplified product DNA sequence was compared with the published sequence. Primer/probe amplification of genomic DNA was investigated using purified genomic DNA rather than cDNA as the template for QRT-PCR. The formation of primer dimers and spurious amplification was also investigated using DEPC water as a “no template” control for the QRT-PCR assay.
Amplification efficiency of a primer/probe set was determined by a dilutional linearity assay, using 5 serial dilutions of the standard cDNA template and running PCR reactions on each dilution in replicates of 4. Two or more versions of each target gene primer/probe set were designed and tested to select for both amplification efficiency and specificity. Similarly, each new primer/probe reagent lot was monitored to ensure matched amplification specificity and efficiency to previous primer/probe reagent lots.
Target gene transcripts were analyzed by QRT-PCR for each cDNA preparation using 2× TaqMan Universal PCR Master Mix (no. 4305719; Applied Biosystems) and Source MDx’s proprietary primer-probe sets. Reactions were run in sets of 4 replicates per gene (24 gene targets in a 96-well plate) on an ABI Prism 7700 Sequence Detection System. Each well also contained the specific primers and probe set to measure 18S rRNA as an internal control. The amount of cDNA template added to each reaction was held to a relatively narrow range, as determined by the cDNA quality control measurement of 18S RNA.
Data Analysis
The difference between the fluorescence CT for the target gene and the endogenous control (18S rRNA) is presented as a ΔCT value (CT of target – CT of control]. For reference, a ΔCT of 2 is approximately equivalent to a 4-fold change in the amount of the transcript. For example, at baseline, TGFβ may have a ΔCT value of 16; after treatment, that ΔCT value may increase to 18. This change represents a 2 ΔCT difference or a decrease of 75% (1/4). The CT reporting system and estimation of relative gene expression are well described in the literature (32).
CT values above 37 were not used in the analysis, because they correspond to gene expression levels below the linear range of the assay. Values over this threshold were obtained for varying proportions of samples, depending on the gene and the study population examined. For the single-time-point samples, the mean and SD of the underlying ΔCT distribution were inferred by maximum likelihood estimation (MLE), under the assumption of a normal distribution, for genes having up to 50% of their CT values over the threshold. Distribution parameters and dynamic ranges were not computed for genes with more than 50% of CT values greater than 37.
Tests for Normality
Because ΔCT values are roughly proportional to the logarithm of the corresponding mRNA abundances, we used a combination of analytical methods to test ΔCT values for each gene for departures from normality.
The Anderson-Darling and Shapiro-Wilk tests were used to test the data against the null hypothesis that the observed values were sampled from a normal distribution, parameterized by the observed mean and standard error. These tests differ in their sensitivity to outliers and in the weight given to central versus outlying values. Smaller P values from these tests indicate rejection of the null hypothesis, i.e., deviation from normality.
We also generated plots of the quantiles of each gene’s ΔCT values against the corresponding quantiles of a standard normal distribution (Q-Q normal plots), together with histograms and normal density curves, to graphically characterize their deviations from normality.
Linear Mixed-Effect Model Analysis
Previous reports on longitudinal gene expression data sets (16,19) suggest that, for many genes, expression levels in repeated samples from the same subject are relatively stable compared with interindividual differences, even when the repeat samples are separated by time periods of several weeks. To quantify the relative magnitudes of intersubject versus temporal and technical variability in apparently healthy, untreated subjects, we fitted a linear mixed-effects (LME) model to the longitudinal study data. In this data set, each ΔCT measurement was associated with a gene g, subject i, sample index j, and replicate k. An LME model for these data is described by equation 1:
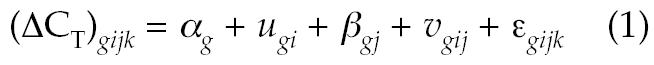 |
where αg is an intercept term dependent on the gene only, ugi is a random effect due to intersubject variability, βgj is a fixed effect due to systematic variations in processing affecting all samples drawn at the same time point, vgij is a random effect representing variability among samples from the same subject, and ɛgijk is an error term encompassing all residual sources of variability between replicates. The random effects ugi, vgij, and ɛgijk are assumed to be normally distributed with mean zero and variances σ2S, σ2T and σ2R, respectively. A restricted maximum likelihood (REML) algorithm (33) was used to fit the model parameters αg, βgj, σ2S, σ2T, and σ2R to the data.
In addition, it is useful to quantify the contributions to intersubject variability arising from subject characteristics such as sex, age, and ethnicity. All 3 of these parameters were recorded for 68 subjects in the single-time-point study. Expression data for these subjects was fitted to the LME model described by equation 2:
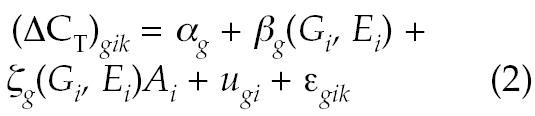 |
where αg is an intercept term dependent on the gene only, Gi, Ai, and Ei are the sex, age, and ethnicity of subject i, βg(G,E) is a gene-specific offset for the given sex and ethnicity, ζg(G,E) is the slope of a linear age effect depending on both sex and ethnicity, ugi is a random effect due to intersubject variability not explained by age, sex, or ethnicity, and ɛgik is an error term encompassing all residual sources of variability between replicate PCR reactions for a given sample. After fitting this model, the percentage contribution of sex, age, and ethnicity effects to the intersubject variance for gene g was estimated by equation 3:
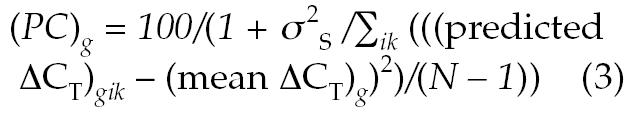 |
where N is the total number of measurements for gene g, σ2S is the variance parameter estimated for the distribution of the random subject effects, predicted ΔCT is the value predicted from the fixed effects portion of equation 2, and mean ΔCT is computed over all measurements for gene g.
All data analyses were performed using the R open source programming environment for statistical computation (34). LME models were programmed using the R package “nlme” (33).
RESULTS
Most Genes Exhibit Limited Dynamic Range of Expression Across Subjects in Single-Time-Point Measurements
A series of studies were undertaken to examine the expression of immune-related gene transcripts in whole blood of apparently healthy subjects. In the largest single-time-point study, blood was collected from 131 blood donors following the American Red Cross donor standards and analyzed for the expression of 48 inflammation- and immune-related gene transcripts. These transcripts encode cell surface molecules, such as CD4, CD14, CD19, and ICAM-1; signaling molecules, such as PTGS2 (COX2), PLA2G7, and NF-κB; cytokines, such as IL-1B and TGFβ; proteinases, such as ELA2; and proteinase inhibitors (see Table 1). The overall range of CT values for the 48 genes studied is plotted in Figure 1. The bars in the plot encompass the central 90% of the observed values (i.e., they extend from the 5th to the 95th percentiles), whereas the whiskers on either end of the bar extend to the extreme values. For genes with expression levels sampled from a log-normal distribution, the ends of the bars would correspond to 1.64 SD on either side of the mean CT.
Table 1.
Genes with detectable expression in healthy blood donor samples, together with statistical summaries of ΔCT distribution, expression fold changes corresponding to 2 standard deviations of ΔCT distribution, and P values for normality tests.
| HUGO Designation | Gene Name and Aliases | n | Mean | Median | SD | Fold Change ± 2 SD | Shapiro– Wilk | Anderson– Darling |
|---|---|---|---|---|---|---|---|---|
| ADAM17 | A Disintegrin and Metalloproteinase Domain 17 | 129 | 18.56 | 18.55 | 0.63 | 2.39 | 0.7512 | 0.5395 |
| APAF1 | Apoptotic Protease Activating Factor 1 | 131 | 16.46 | 16.48 | 0.54 | 2.13 | 2.1E-05 | 0.0150 |
| C1QA | Complement Component 1, Q Subcomponent, Alpha Polypeptide | 128 | 20.25 | 20.21 | 0.92 | 3.57 | 0.0939 | 0.0879 |
| CD14 | CD14 Antigen | 129 | 13.92 | 14.01 | 0.63 | 2.41 | 1.1E-05 | 8.0E-07 |
| CD19 | CD19 Antigen | 131 | 18.19 | 18.09 | 0.78 | 2.94 | 1.4E-05 | 1.1E-07 |
| CD4 | CD4 Antigen | 131 | 14.80 | 14.84 | 0.49 | 1.98 | 0.0064 | 3.8E-04 |
| CD86 | CD86 Anitigen; B7–2 Protein | 128 | 17.64 | 17.68 | 0.51 | 2.04 | 3.1E-05 | 6.6E-04 |
| CD8A | CD8 Antigen, Alpha Polypeptide, p32 | 130 | 15.74 | 15.72 | 0.67 | 2.54 | 0.0653 | 0.8402 |
| CXCL1 | Chemokine (C–X–C Motif) Ligand 1 (GRO–1) | 131 | 20.01 | 20.00 | 0.67 | 2.53 | 0.1150 | 0.1522 |
| CYBB | Cytochrome B–245 Beta Polypeptide | 130 | 13.98 | 14.02 | 0.57 | 2.21 | 0.0058 | 0.0542 |
| DPP4 | Dipeptidylpeptidase IV (CD26) | 131 | 18.33 | 18.35 | 0.61 | 2.34 | 0.1253 | 0.0602 |
| EGR1 | Early Growth Response 1 | 130 | 20.42 | 20.49 | 0.65 | 2.47 | 0.0074 | 0.0013 |
| ELA2 | Elastase 2, Neutrophil | 126 | 19.90 | 19.78 | 1.29 | 5.95 | 2.1E-04 | 1.4E-04 |
| GCLC | Glutamate–Cysteine Ligase, Catalytic Subunit | 128 | 18.86 | 18.90 | 0.64 | 2.41 | 5.6E-07 | 2.9E-05 |
| HMGB1 | High–Mobility Group Box 1 | 130 | 16.28 | 16.25 | 0.69 | 2.59 | 0.0055 | 0.0524 |
| HMOX1 | Heme Oxygenase (Decycling) 1 | 131 | 16.45 | 16.50 | 0.67 | 2.53 | 0.0028 | 0.0045 |
| HSPA1A | Heat Shock Protein 1A, 70 kDa | 129 | 13.83 | 13.88 | 0.80 | 3.01 | 3.7E-08 | 1.2E-06 |
| ICAM1 | Intercellular Adhesion Molecule 1 | 131 | 17.68 | 17.71 | 0.55 | 2.15 | 0.0969 | 0.0514 |
| IFI16 | Interferon γ-Inducible Protein 16 | 130 | 16.75 | 16.72 | 0.84 | 3.20 | 0.0441 | 0.1004 |
| IL10 | Interleukin 10 | 75 | 22.87 | 22.94 | 0.75 | 2.81 | 9.0E-04 | 0.0070 |
| IL15 | Interleukin 15 | 129 | 21.45 | 21.45 | 0.70 | 2.65 | 0.0051 | 0.0275 |
| IL18 | Interleukin 18 (Interferon γ-Inducing Factor) | 130 | 20.05 | 20.05 | 0.54 | 2.11 | 0.0517 | 0.0625 |
| IL18BP | IL–18 Binding Protein | 131 | 16.74 | 16.72 | 0.44 | 1.84 | 0.2787 | 0.6132 |
| IL1B | Interleukin 1β | 130 | 16.67 | 16.67 | 0.79 | 2.99 | 0.0011 | 0.0200 |
| IL1R1 | Interleukin 1 Receptor, Type I | 125 | 21.08 | 21.06 | 0.98 | 3.90 | 0.5969 | 0.9508 |
| IL1RN | Interleukin 1 Receptor Antagonist | 129 | 16.88 | 16.91 | 0.67 | 2.54 | 0.1494 | 0.1755 |
| IL8 | Interleukin 8 | 97 | 21.01 | 20.86 | 1.46 | 7.53 | 0.0321 | 0.1185 |
| LTA | Lymphotoxin α | 114 | 20.05 | 19.99 | 0.65 | 2.45 | 3.4E-04 | 0.0014 |
| MMP9 | Matrix Metalloproteinase 9 | 129 | 15.91 | 16.01 | 1.16 | 4.97 | 3.8E-05 | 1.9E-06 |
| MNDA | Myeloid Cell Nuclear Differentiation Antigen | 130 | 12.54 | 12.51 | 0.64 | 2.44 | 0.1193 | 0.1563 |
| MPO | Myeloperoxidase | 131 | 21.20 | 21.22 | 0.77 | 2.92 | 0.7944 | 0.7479 |
| MYC | V–myc Avian Myelocytomatosis Viral Oncogene Homolog | 130 | 17.23 | 17.22 | 0.63 | 2.38 | 0.0685 | 0.0705 |
| NFKB1 | Nuclear Factor of κ Light Polypeptide Gene Enhancer in B Cells 1 (p105) | 131 | 17.38 | 17.41 | 0.57 | 2.20 | 0.0178 | 0.0076 |
| PLA2G7 | Phospholipase A2, Group VII | 126 | 19.36 | 19.36 | 0.69 | 2.60 | 0.1485 | 0.5492 |
| PLAUR | Plasminogen Activator, Urokinase Receptor | 131 | 15.12 | 15.15 | 0.58 | 2.25 | 0.0275 | 0.0190 |
| PTGS2 | Prostaglandin–Endoperoxide Synthase 2 (COX–2) | 126 | 16.72 | 16.75 | 0.61 | 2.33 | 0.0505 | 0.0309 |
| PTPRC | Protein Tyrosine Phosphatase Receptor, 127 Type C (CD45) | 11.91 | 11.96 | 0.52 | 2.07 | 0.0410 | 0.0165 | |
| SERPINA1 | Serine (or Cysteine) Proteinase Inhibitor, Clade A, Member 1 (Alpha 1 Anti–Trypsin) | 131 | 13.26 | 13.27 | 0.66 | 2.50 | 0.0100 | 0.0092 |
| SERPINE1 | Serine (or Cysteine) Proteinase Inhibitor, Clade E (Ovalbumin), Member 1 (Plasminogen Activator Inhibitor Type 1) | 101 | 22.38 | 22.44 | 0.89 | 3.43 | 0.0014 | 0.0015 |
| SERPING1 | Serine (or Cysteine) Proteinase Inhibitor, Clade G (C1 Inhibitor), Member 1 (Angioedema, Hereditary) | 130 | 19.20 | 19.29 | 1.20 | 5.25 | 3.2E-04 | 6.0E-05 |
| TGFB1 | Transforming Growth Factor β 1 | 130 | 13.14 | 13.16 | 0.44 | 1.83 | 0.0115 | 0.0141 |
| TIMP1 | Tissue Inhibitor of Matrix Metalloproteinase 1 | 131 | 15.02 | 15.09 | 0.57 | 2.19 | 1.1E-05 | 4.2E-06 |
| TLR2 | Toll–Like Receptor 2 | 130 | 16.07 | 16.13 | 0.63 | 2.38 | 0.0058 | 0.0015 |
| TNF | Tumor Necrosis Factor | 124 | 20.67 | 20.55 | 0.98 | 3.90 | 1.1E-05 | 3.4E-04 |
| TNFSF5 | Tumor Necrosis Factor (Ligand) Superfamily, Member 5 (CD40 Ligand) | 131 | 17.69 | 17.67 | 0.63 | 2.38 | 1.5E-14 | 1.5E-10 |
| TNFSF6 | Tumor Necrosis Factor (Ligand) Superfamily, Member 6 (Fas Ligand) | 126 | 20.41 | 20.35 | 0.74 | 2.80 | 4.7E-04 | 0.0021 |
n is the number of samples having detectable expression for the gene in at least 3 of 4 replicate RT–PCR reactions. Mean and SD are estimated by maximum likelihood for genes where any replicates fall below the detection threshold (CT > 37).
Figure 1.

Gene distribution across 131 healthy donors. Range of CT values for each gene targeted by the panel of 48 primer sets, across 131 single-time samples. Bars span the range from the 5th to the 95th percentile of CT values for each gene.
Of the 48 genes profiled in this study, 2 important signals of inflammation, IL6 and CXCL2, lacked detectable expression in most of the apparently healthy subjects, and their CT values were at or greater than 37. Dynamic ranges and variance components were not computed for these genes. For the remaining 46 genes, the estimated SD of the ΔCT values ranged from 0.44 to 1.46 and were below 0.792 for 36 of the 46 genes, as shown in Table 1. Thus, the dynamic range of expression extending 2 SD in either direction from the geometric mean was less than 22 * °.792 or a 3-fold change (32). For normally distributed ΔCT values, this range covers 95.4% of the sample measurements. The distribution of dynamic ranges corresponding to a ±2 SD span is shown in Figure 2. The highest dynamic range observed was 7.53-fold change units for IL-8. The SDs of ΔCT values were independent of the mean Δ CT, indicating that the dynamic ranges did not depend on a gene’s expression level.
Figure 2.

Histogram of dynamic ranges of expression values, expressed as fold changes spanning 2 standard deviations of each gene’s ΔCT values (that is, 2−2SD(ΔCT)).
The Majority of Genes Have Expression Values following Log-Normal Distributions
Commonly used parametric tests for differential gene expression between groups of samples, such as t tests and analysis of variance, are based partly on the assumption that the values being compared are sampled from normal distributions. Although it is commonly asserted that transcription levels of many genes are log-normally distributed (28,29), it is important to test this assumption to use such tests for disease diagnosis and detection. The majority of expressed transcripts followed approximately log-normal distributions, according to the Anderson-Darling and Shapiro-Wilk tests (Table 1, Figure 3). The gene most closely following a normal distribution of ΔCT values was IL1R1 (Figure 3A), with an Anderson-Darling P value of 0.945. Among the 46 genes tested, 34 had P values greater than 0.001. All genes had unimodal distributions; the deviations from normality involved moderate degrees of left or right skewness, and/or heavy or light tails. Although these departures were not dramatic, they will need to be incorporated into the predicted error rates for diagnostic tests based on expression of these genes.
Figure 3.

Q-Q normal plots and histograms of ΔCT values for the genes deviating least and most from a normal distribution (IL1R1 in Figure 3A and TNFSF5 in Figure 3B, respectively), according to the Anderson-Darling test. Unit diagonals and normal density curves are drawn on the Q-Q normal plots and histograms, respectively, for comparison with a normal distribution with the same mean and variance as observed. P values computed by the Anderson-Darling normality test are shown above each histogram.
Of the 48 genes shown in Table 1, the gene deviating most from a normal distribution of ΔCT values was TNFSF5 (CD40 ligand, Figure 3B), with an Anderson-Darling P value of 1.52 × 10−10. The observed distribution is characterized by a heavy tail and large ΔCT, suggesting the presence of a subpopulation with an unusually low expression level of this gene.
Minor Variations in Expression May Be Based on Sex, Ethnicity, and Age
Table 2 shows the contributions of sex, age, and ethnicity on interindividual variation estimated by the LME model (equation 2). For the 43 genes examined, the observed effects of sex, ethnicity, and age were small. Only 10 genes had contributions from these effects, explaining more than 20% of the intersubject variance; the maximum contribution was only 27.9% for NFKB1. For most genes, sex effects accounted for most of this contribution. Fifteen genes showed significant sex differences (unadjusted P value < 0.05), but the largest fold change from women to men was only 1.62 for TNFSF6. Likewise, only moderate ethnicity effects were observed. Five genes (MPO, MYC, TNFSF6, ELA2, and HMGB1) showed significant differential expression between white (non-Hispanic) and Hispanic subjects, with the largest change being a 2.5-fold overexpression of ELA2 in Hispanic women relative to white women.
Table 2.
Sex, age, and ethnicity (fixed effect) contributions to intersubject variation for 43 genes, in decreasing order of percentage of variance explained (equation 3).
|
P Values for Effect
|
Fold Change Corresponding to Effect
|
Fold Change to Age Difference (69 vs. 23 years)Corresponding
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Percent of Variance Explained by Sex, Age, and Ethnicity | Sex | Ethnicity | Sex + Ethnicity | Age | Sex + Age | Ethnicity + Age | Sex + Ethnicity + Age | WM vs. WF | HF vs. WF | HM vs. WF | WF | WM | HF | HM |
| NFKB1 | 27.90 | 0.0022 | 0.7083 | 0.1098 | 0.3335 | 0.0553 | 0.1896 | 0.2572 | 1.35 | −1.04 | −1.03 | −1.29 | −2.48 | 1.32 | 1.43 |
| MPO | 27.74 | 0.0128 | 0.0005 | 0.0116 | 0.2260 | 0.3535 | 0.8682 | 0.0909 | 1.42 | 1.82 | 1.31 | 1.57 | 1.00 | 1.43 | 4.48 |
| IL18 | 27.46 | 0.0220 | 0.6228 | 0.9333 | 0.0119 | 0.0468 | 0.9150 | 0.1023 | 1.25 | 1.06 | 1.30 | 1.97 | −1.02 | 1.88 | 2.77 |
| MYC | 26.91 | 0.0180 | 0.0132 | 0.7298 | 0.5240 | 0.1344 | 0.9658 | 0.1843 | 1.31 | −1.42 | −1.17 | −1.21 | −2.19 | −1.24 | 1.23 |
| TGFB1 | 25.88 | 0.0121 | 0.0726 | 0.5024 | 0.5696 | 0.0809 | 0.3451 | 0.1967 | 1.22 | −1.18 | −1.07 | −1.13 | −1.84 | 1.21 | 1.47 |
| TNFSF6 | 23.90 | 0.0008 | 0.0344 | 0.0123 | 0.1039 | 0.0892 | 0.8499 | 0.4898 | 1.62 | 1.39 | 1.17 | 1.78 | −1.28 | 1.61 | 1.32 |
| LTA | 23.87 | 0.0179 | 0.8223 | 0.0131 | 0.4992 | 0.1109 | 0.9253 | 0.4470 | 1.32 | −1.03 | −1.33 | −1.22 | −2.32 | −1.17 | −1.27 |
| ELA2 | 23.65 | 0.3536 | 0.0013 | 0.0998 | 0.0262 | 0.2949 | 0.1695 | 0.0889 | 1.24 | 2.53 | 1.50 | 4.21 | 1.78 | 1.09 | 6.94 |
| CD86 | 21.03 | 0.0289 | 0.2781 | 0.4782 | 0.1418 | 0.1509 | 0.3672 | 0.3403 | 1.24 | 1.13 | 1.23 | 1.47 | −1.11 | 2.12 | 2.42 |
| CD14 | 20.75 | 0.0066 | 0.9157 | 0.1150 | 0.1484 | 0.4972 | 0.0631 | 0.6869 | 1.42 | −1.02 | −1.05 | −1.65 | −2.24 | 1.64 | 1.71 |
| C1QA | 19.11 | 0.9650 | 0.2648 | 0.7856 | 0.0458 | 0.1489 | 0.0286 | 0.0015 | 1.01 | 1.25 | 1.15 | 2.51 | 1.07 | −1.89 | 9.27 |
| GCLC | 19.04 | 0.1281 | 0.0510 | 0.4470 | 0.1961 | 0.5864 | 0.4943 | 0.1936 | 1.19 | 1.31 | 1.33 | 1.51 | 1.21 | 2.10 | −1.66 |
| HSPA1A | 18.91 | 0.0208 | 0.3329 | 0.8448 | 0.2111 | 0.5560 | 0.1404 | 0.3747 | 1.37 | −1.17 | 1.12 | −1.58 | −2.09 | 1.46 | 2.47 |
| TNF | 18.22 | 0.0489 | 0.6451 | 0.2638 | 0.6774 | 0.8716 | 0.0660 | 0.4877 | 1.44 | −1.11 | −1.14 | −1.23 | −1.11 | 3.34 | 1.59 |
| HMGB1 | 17.72 | 0.0630 | 0.0047 | 0.1891 | 0.2164 | 0.3968 | 0.6109 | 0.9320 | 1.24 | 1.50 | 1.39 | 1.48 | 1.05 | 1.16 | −1.31 |
| CYBB | 17.34 | 0.0223 | 0.9489 | 0.3272 | 0.9352 | 0.2462 | 0.1217 | 0.5252 | 1.31 | −1.01 | 1.05 | 1.03 | −1.56 | 2.17 | 2.22 |
| SERPINA1 | 17.27 | 0.0730 | 0.2471 | 0.9965 | 0.2167 | 0.5933 | 0.0938 | 0.5365 | 1.28 | −1.21 | 1.06 | −1.59 | −2.06 | 1.66 | 2.28 |
| MMP9 | 16.14 | 0.1858 | 0.1057 | 0.3289 | 0.1730 | 0.6537 | 0.5372 | 0.3199 | 1.36 | −1.57 | 1.34 | −2.39 | −3.46 | −1.31 | 2.56 |
| CXCL1 | 15.91 | 0.1965 | 0.1305 | 0.5228 | 0.3722 | 0.4229 | 0.1961 | 0.4267 | 1.18 | −1.26 | 1.09 | −1.36 | −1.94 | 1.46 | 2.02 |
| EGR1 | 15.53 | 0.0263 | 0.3387 | 0.4572 | 0.2550 | 0.0909 | 0.6418 | 0.2217 | 1.33 | −1.15 | −1.04 | 1.48 | −1.44 | 1.16 | 1.54 |
| IL15 | 15.44 | 0.4922 | 0.0501 | 0.0862 | 0.3364 | 0.9222 | 0.3302 | 0.2744 | 1.10 | 1.37 | −1.03 | 1.42 | 1.36 | 2.45 | −1.15 |
| DPP4 | 15.00 | 0.2114 | 0.4354 | 0.4450 | 0.1223 | 0.6509 | 0.1781 | 0.5079 | 1.15 | −1.11 | −1.14 | −1.63 | −1.95 | 1.18 | −1.70 |
| CD4 | 14.86 | 0.0816 | 0.4197 | 0.3359 | 0.9445 | 0.0935 | 0.4285 | 0.4223 | 1.19 | −1.10 | −1.11 | 1.02 | −1.77 | 1.42 | 1.34 |
| HMOX1 | 14.81 | 0.0587 | 0.8654 | 0.6194 | 0.2167 | 0.0801 | 0.9441 | 0.0954 | 1.27 | −1.03 | 1.10 | 1.53 | −1.44 | 1.59 | 3.05 |
| PLA2G7 | 14.20 | 0.0865 | 0.9282 | 0.7051 | 0.4431 | 0.8269 | 0.0575 | 0.6794 | 1.27 | 1.02 | 1.17 | −1.34 | −1.49 | 2.31 | 3.07 |
| PLAUR | 13.46 | 0.2046 | 0.4947 | 0.9590 | 0.4582 | 0.2254 | 0.6593 | 0.1645 | 1.17 | −1.10 | 1.07 | −1.28 | −2.15 | −1.02 | 1.83 |
| TIMP1 | 13.33 | 0.0658 | 0.5106 | 0.2000 | 0.4669 | 0.1399 | 0.8392 | 0.3714 | 1.23 | −1.09 | −1.17 | 1.25 | −1.44 | 1.37 | 1.51 |
| CD8A | 13.23 | 0.0412 | 0.1604 | 0.8832 | 0.2939 | 0.7299 | 0.4394 | 0.8534 | 1.30 | 1.24 | 1.56 | −1.44 | −1.24 | 1.05 | 1.44 |
| ADAM17 | 12.71 | 0.1919 | 0.8616 | 0.6441 | 0.1633 | 0.2687 | 0.7874 | 0.7724 | 1.15 | 1.02 | 1.29 | 1.49 | −1.01 | 1.68 | 1.37 |
| PTPRC | 12.65 | 0.0279 | 0.7490 | 0.7072 | 0.5853 | 0.4100 | 0.2291 | 0.5759 | 1.24 | 1.04 | 1.20 | −1.15 | −1.53 | 1.41 | 1.53 |
| PTGS2 | 12.55 | 0.0630 | 0.9846 | 0.6788 | 0.0812 | 0.9289 | 0.1552 | 0.9724 | 1.28 | −1.00 | 1.15 | −1.87 | −1.79 | 1.17 | 1.18 |
| IL1RN | 12.41 | 0.3848 | 0.0842 | 0.1495 | 0.1451 | 0.6460 | 0.1061 | 0.8547 | −1.13 | −1.33 | −1.03 | −1.73 | −1.38 | 1.48 | 2.18 |
| ICAM1 | 11.99 | 0.2755 | 0.2793 | 0.7173 | 0.4375 | 0.4882 | 0.2839 | 0.3328 | 1.13 | −1.16 | 1.06 | −1.27 | −1.68 | 1.31 | 2.10 |
| APAF1 | 11.91 | 0.0852 | 0.8732 | 0.8466 | 0.8508 | 0.4636 | 0.0957 | 0.6159 | 1.20 | −1.02 | 1.13 | −1.05 | −1.38 | 1.96 | 1.06 |
| MNDA | 11.64 | 0.0662 | 0.9441 | 0.3044 | 0.5092 | 0.6863 | 0.3526 | 0.3483 | 1.24 | −1.01 | −1.03 | −1.23 | −1.46 | 1.28 | 2.29 |
| IL18BP | 10.94 | 0.1220 | 0.0913 | 0.1308 | 0.6556 | 0.3362 | 0.4393 | 0.4857 | 1.14 | 1.18 | 1.06 | 1.11 | −1.20 | 1.45 | 1.62 |
| SERPING1 | 10.70 | 0.4339 | 0.2313 | 0.9868 | 0.5224 | 0.9825 | 0.7008 | 0.2993 | −1.21 | −1.41 | −1.69 | −1.52 | −1.49 | −1.03 | 5.36 |
| IL1R1 | 10.49 | 0.1039 | 0.7632 | 0.9654 | 0.7602 | 0.3975 | 0.2078 | 0.9550 | 1.34 | −1.07 | 1.28 | −1.16 | −2.00 | 2.25 | 1.40 |
| IL1B | 10.09 | 0.6746 | 0.3584 | 0.4113 | 0.3254 | 0.7338 | 0.1561 | 0.9919 | −1.07 | −1.19 | 1.01 | −1.52 | −1.84 | 1.68 | 1.40 |
| TLR2 | 9.82 | 0.2787 | 0.6278 | 0.5153 | 0.7197 | 0.1332 | 0.2841 | 0.8863 | 1.15 | 1.08 | 1.06 | 1.14 | −1.77 | 2.05 | 1.16 |
| TNFSF5 | 9.48 | 0.3867 | 0.3648 | 0.1478 | 0.0599 | 0.4390 | 0.3545 | 0.6951 | 1.12 | 1.16 | −1.12 | −2.01 | −1.39 | −1.19 | −1.18 |
| IFI16 | 6.46 | 0.5860 | 0.6442 | 0.5085 | 0.2437 | 0.8924 | 0.4439 | 0.9634 | 1.09 | 1.09 | −1.02 | −1.62 | −1.74 | 1.00 | −1.02 |
| CD19 | 5.24 | 0.1827 | 0.4304 | 0.1925 | 0.3513 | 0.1437 | 0.4404 | 0.5098 | 1.23 | 1.16 | −1.03 | 1.48 | −1.51 | −1.11 | −1.25 |
Values were computed only for white and Hispanic subjects for whom sex and age were recorded (n = 68). Unadjusted P values are shown for each effect, including interaction terms, and underlined (together with corresponding fold changes) when < 0.05. Fold changes for sex and ethnicity effects are computed by raising 2 to the power of the corresponding ΔCT effect terms; for age effects, they are computed by multiplying the corresponding slope effect by the range of ages in the sample (69 - 23) and then exponentiating. HF indicates Hispanic female; HM, Hispanic male; WF, white female; WM, white male.
Age effects were difficult to measure in this data set, due to the markedly different age distributions between the female and male blood donors. Male blood donors had a median age of 53 years, compared with 43 years for females. Therefore, sex and age effects are potentially confounded. The LME model defined in equation 2 addresses the confounding factors by fitting the ΔCT versus age data to different slopes for each sex/ethnicity combination. According to the LME model, 3 genes (IL18, ELA2, and C1QA) had significant age effects for at least 1 sex/ethnicity combination. For all 3 of these genes, the fitted slopes were markedly different between sexes. For example, age had virtually no effect on IL18 expression in white men, whereas in white women the slope corresponded to a 2-fold increase from age 23 to age 69. Similarly, the fitted slopes suggest dramatic differences in age effects among ethnicities. Overall, the size of the sample is too small to reliably estimate ethnic differences.
Variation of Expression within Subjects Over Time Is Limited
To compare the contributions of intersubject, temporal, and technical components to the overall variation in gene expression, we fitted the LME model (equation 1) to the longitudinal set of measurements described in “Materials and Methods.” For this data set, we fitted the model for each of 29 genes with detectable expression in at least 90% of the samples to obtain, for each gene, a set of variance parameters σ2S, σ2T, and σ2R. These are approximate estimates of the contributions to the total variance from intersubject variation, variation among samples taken at different times from each subject, and residual variation between replicate reactions, respectively.
The results of the initial LME model analysis are summarized in Figure 4, which shows the fitted standard error parameters σ2S, σ2T, and σ2R for each gene. For 6 of the 29 genes examined (CD19, TNFSF13B, HMOX1, C1QA, CD8A, and CD4), intersubject variation comprised more than 50% of the total variance of ΔCT values. For the remaining 23 genes, variation between samples taken at different times was the largest component. However, the magnitude of the temporal variation was limited; the parameter σT ranged from 0.36 ΔCT units for the gene PTPRC to 0.72 ΔCT units for MMP9. The dynamic ranges corresponding to 2σT ranged from 1.66- to 2.72-fold change units. Because measurements from samples taken over a period of 8 months may be subject to several sources of technical variation (for example, instrument calibration, reagent lots, and variations in sample handling), these ranges can be considered upper limits on the true temporal variation of expression for the genes analyzed.
Figure 4.

Source of variance in gene expression. (A) Variance components estimated from mixed-effect models, representing variation between subjects (dark grey), between longitudinal samples from same subject (grey), and between replicate RT-PCR reactions for same sample (white). Systematic variations affecting all samples drawn on same date have been subtracted before estimating variance components. (B) Variance components expressed as percentages relative to sum of components.
LPS Stimulation Induces Transient Gene Expression Changes in Excess of Natural Variation
To demonstrate that changes marked beyond the normal reference range occur, gene expression was measured in blood collected from healthy subjects injected with LPS. Healthy subjects who receive an injection of LPS experience mild fever and flu-like symptoms that subside within 24 h (35). Figure 5 shows the expression of a subset of genes with significant changes at any time point after LPS injection. Reference ranges (mean ± 2 SD) for healthy subjects are indicated by dashed lines. The plotted ΔΔCT values are computed relative to the mean ΔCT for the apparently healthy blood donors. Individual time courses are shown for each subject. Twenty-seven genes had significant changes in expression in LPS-injected subjects at any time postinfusion relative to apparently healthy blood donors, with adjusted false discovery rates of less than 5%. Each of these genes had pre-injection expression levels within the normal reference range for apparently healthy blood donors; each showed increased or decreased expression at 2 and/or 5 h postinfusion; and most returned to the normal expression range by 21 h after infusion. Fifteen genes increased or decreased expression by a factor greater than 10-fold, and 2 (MMP9 and IL1RN) increased more than 90-fold (Figure 5). Because the innate immune system’s immediate response to LPS infusion is the production of inflammatory mediators by monocytes, it is not surprising that the genes showing substantial increases in expression include cytokines and chemokines associated with the monocyte/macrophage lineage, such as TNF, IL1B, CXCL1, and IL18. Key cell-surface markers (ICAM1, CD14) and signaling molecules (PTGS2/COX-2) also respond. Interestingly, the anti-inflammatory regulator IL1RN, which blocks the binding of IL1 to its receptor, was 1 of the 2 most overexpressed genes. This fits with the premise that inflammatory processes are tightly regulated by coordinated expression of pro-inflammatory and anti-inflammatory factors. These include genes with significant decreases in expression such as PLA2G7 and TNFSF5 (CD40 ligand) (see Figure 5).
Figure 5.

Time course of expression for 12 genes with significant responses to LPS infusion in 3 healthy male subjects. Whole blood was sampled at pre-LPS (0 h) and 2, 5 and 21 h post-LPS infusion. Gene expression is plotted as ΔCT values relative to mean ΔCT for healthy blood donors, with points and lines colored by subject. Mean and mean ± 2 SD are indicated by horizontal dashed lines. ΔCT scale is inverted, so upward direction corresponds to increasing expression.
DISCUSSION
The studies reported here are an initial step toward establishing normal reference ranges for the expression of genes related to inflammation and immunity. Several key observations emerged. First, the dynamic range of expression of most immune response genes is relatively limited among apparently healthy subjects. Second, expression levels for most genes analyzed are approximately log-normally distributed. Third, individuals exposed to bacterial endotoxin have gene expression profiles that are easily (albeit transiently) distinguished from those of an apparently healthy population. In developing the methods for these studies, it was also observed that multiple technical factors, including sample handling procedures, PCR reagents, and instrument calibration, contribute to the overall variation, which must be carefully controlled. Taken together, these observations support both the usefulness and practicality of establishing normal reference ranges for gene expression assays related to immune system function.
A variety of biological factors may contribute to the variation of expression observed in apparently healthy subjects (18). In general, these factors can be divided into intrinsic (for example, age, sex, genetics) and extrinsic (for example, inflammatory, autoimmune disease, cancer, infections, and metabolism) factors. The apparently healthy blood donor population studied here may have included individuals with subacute illnesses or chronic conditions that contributed to the variability in expression of some immune response genes. Many chronic inflammatory and atopic diseases, such as arthritis, asthma, ulcers, gastritis, and allergies, are highly prevalent in the U.S. adult population, with frequencies ranging from 7% to 27% (36). Nonetheless, individuals with these conditions are deemed “healthy” and permitted to donate blood, provided these “chronic conditions are bring treated and the condition is under control,” and they “feel well and are able to perform normal activities” (30).
Atherosclerosis is another highly prevalent condition which develops over several years and is asymptomatic in its early or even late stages. Several studies have demonstrated an elevation of C-reactive protein and other markers of inflammation in early stages of cardiovascular disease (37,38). Chronic infections with viruses (cytomegalovirus, Epstein-Barr virus, genital herpes, and human papillomavirus), bacteria (Helicobacter pylori), and protozoans (Toxoplasma gondii ) also are common in the U.S. population, but do not consistently produce symptoms in immunocompetent persons. Periodic reactivation and suppression of these infections may account for some of the background variation in immune response gene expression. Dietary influences on immune system gene expression may include consumption of omega-3 fatty acids, arginine, and other nutrients as well as vegetarian diets (39,40).
Age, sex, and ethnicity also may contribute to the intersubject variation observed for several transcripts. However, the contributions of these factors appeared to be modest in the present study. Variations associated with age and sex have been previously reported (18,41,42), with some sex differences being directly attributable to differences in sex chromosomes (18). Several studies (18,42) have observed individual differences in interferon-responsive genes among individuals, suggesting further stratification in an apparently normal healthy subject group. Larger studies specifically targeting some of these factors are needed to elucidate the effects so that populations can be stratified for more precise diagnostic resolution.
Intrinsic and extrinsic factors can also alter the proportions of blood cell types such as neutrophils, monocytes, and lymphocytes, as well as the relative expression of individual transcripts within each cell type. These effects combine to produce the observed variation in transcript abundances in whole blood. The individual contributions of cell populations and gene regulation within cell types could be examined using flow cytometry combined with QRT-PCR, and deserve further study.
Given the variety of factors that can affect the expression of immune response genes in a blood donor population, it is remarkable that the overall dynamic range of expression is not wider than observed in the present study, whereas larger, up to 90-fold, but transient changes can be induced by the severe acute inflammatory stimulus LPS. In other diseases, such as rheumatoid arthritis and lupus, differences in gene expression from apparently healthy normals are more modest, 2- to 5-fold (43). These observations support the view that expression of these genes is maintained within limits by regulatory mechanisms, possibly to reduce the danger of tissue damage from constant activation of immune responses, while allowing appropriate responses to infectious threats. The limited dynamic range observed supports the development of expression-based diagnostics, allowing expression outside the normal reference range to indicate the presence of infections, cancer, or indolent autoimmune diseases.
Molecular diagnostics, including those based on gene expression, are increasingly being applied in the clinic (44,45). These tests have improved the selection of therapies, as well as dosage and treatment schedule. In addition, “treat-to-normal” strategies are routinely used in major diseases such as hypertension and diabetes. Assays based on precise, quantitative measurements of immune system gene expression offer the promise of effective clinical monitors in infection, autoimmune diseases, and other immune-related conditions, such as transplant rejection and drug- or virus-induced immunosuppression, as well as cancer. A better understanding of the relevant factors that contribute to the individuality of gene expression in the human will help to establish the most appropriate normal reference values in the clinic and will serve as an essential step in the development of effective molecular diagnostics for these and other inflammatory and immunologic diseases.
ACKNOWLEDGMENTS
The authors would like to thank C. Edwards, C. Dinarello, A. Rasley, D. Nelson, M. Ascher, and C.T. Rigl for helpful comments, review, and discussion. This work was performed under the auspices of the Lawrence Livermore National Laboratory and was supported with funds from the Laboratory Directed Research and Development (LDRD) Program.
Footnotes
Online address: http://www.molmed.org
REFERENCES
- 1.Bild AH, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:274–5. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 2.Gladkevich A, Nelemans SA, Kauffman HF, Korf J. Microarray profiling of lymphocytes in internal diseases with an altered immune response: potential and methodology. Mediators Inflamm. 2005;2005:317–30. doi: 10.1155/MI.2005.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Han D, Leith J, Alejandro R, Bolton W, Ricordi C, Kenyon NS. Peripheral blood cytotoxic lymphocyte gene transcript levels differ in patients with long-term type 1 diabetes compared to normal controls. Cell Transplant. 2005;14:403–9. doi: 10.3727/000000005783982972. [DOI] [PubMed] [Google Scholar]
- 4.Perez EA, Pusztai L, Van de Vijver M. Improving patient care through molecular diagnostics. Semin Oncol. 2004;31(5 Suppl 10):14–20. doi: 10.1053/j.seminoncol.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 5.Baechler EC, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100:2610–5. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rus V, Chen H, Zernetkina V, Magder LS, Mathai S, Hochberg MC, Via CS. Gene expression profiling in peripheral blood mononuclear cells from lupus patients with active and inactive disease. Clin Immunol. 2004;112:231–4. doi: 10.1016/j.clim.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 7.Chen X, et al. Gene expression patterns in human liver cancers. Mol Biol Cell. 2002;13:1929–39. doi: 10.1091/mbc.02-02-0023.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gang J, et al. Discovery and analysis of pancreatic adenocarcinoma genes using DNA microarrays. World J Gastroenterol. 2005;11:6543–8. doi: 10.3748/wjg.v11.i41.6543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang JC, et al. Gene expression profiling for the prediction of therapeutic response to docetaxel in patients with breast cancer. Lancet. 2003;362:362–9. doi: 10.1016/S0140-6736(03)14023-8. [DOI] [PubMed] [Google Scholar]
- 10.US patent no. 6,960,439: Identification, monitoring and treatment of disease and characterization of biological condition using gene expression profiles, covering the use of a Healthy Normals Reference Dataset, issued to Source MDx, Nov. 4, 2005.
- 11.Stitt JT. Fever versus hyperthermia. Fed Proc. 1979;38:39–43. [PubMed] [Google Scholar]
- 12.Conti B, Tabarean I, Andrei C, Bartfai T. Cytokines and fever. Front Biosci. 2004;9:1433–49. doi: 10.2741/1341. [DOI] [PubMed] [Google Scholar]
- 13.Dinarello CA. Infection, fever, and exogenous and endogenous pyrogens: some concepts have changed. J Endotoxin Res. 2004;10:201–22. doi: 10.1179/096805104225006129. [DOI] [PubMed] [Google Scholar]
- 14.Jiang H, Chess L. An integrated view of suppressor T cell subsets in immunoregulation. J Clin Invest. 2004;114:1198–1208. doi: 10.1172/JCI23411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell C, Vernon SD, Karem KL, Nisenbaum R, Unger ER. Assessment of normal variability in peripheral blood gene expression. Dis Markers. 2002;18:201–6. doi: 10.1155/2002/462465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chowers I, Liu D, Farkas RH, et al. Gene expression variation in the adult human retina. Hum Mol Genet. 2003;12:2881–93. doi: 10.1093/hmg/ddg326. [DOI] [PubMed] [Google Scholar]
- 17.Cole J, Tsou R, Wallace K, Gibran N, Isik F. Comparison of normal human skin gene expression using cDNA microarrays. Wound Repair Regen. 2001;9:77–85. doi: 10.1046/j.1524-475x.2001.00077.x. [DOI] [PubMed] [Google Scholar]
- 18.Whitney AR, Diehn M, Popper SJ, Alizadeh AA, Boldrick JC, Relman DA, Brown PO. Individuality and variation in gene expression patterns in whole blood. Proc Natl Acad Sci U S A. 2003;100:1896–1901. doi: 10.1073/pnas.252784499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheung VG, Conlin LK, Weber TM, Arcaro M, Jen KY, Morley M, Spielman RS. Natural variation in human gene expression assessed in lymphoblastoid cells. Nat Genet. 2003;33:422–5. doi: 10.1038/ng1094. [DOI] [PubMed] [Google Scholar]
- 20.Baechler EC, Batliwalla FM, Karypis G, et al. Expression levels for many genes in human peripheral blood cells are highly sensitive to ex vivo incubation. Genes Immun. 2004;5:347–53. doi: 10.1038/sj.gene.6364098. [DOI] [PubMed] [Google Scholar]
- 21.Debey S, Schoenbeck U, Hellmich M, Gathof BS, Pillai R, Zander T, Schultze JL. Comparison of different isolation techniques prior gene expression profiling of blood derived cells: impact on physiological responses, on overall expression and the role of different cell types. Pharmacogenomics J. 2004;4:193–207. doi: 10.1038/sj.tpj.6500240. [DOI] [PubMed] [Google Scholar]
- 22.Han ES, Wu Y, McCarter R, Nelson JF, Richardson A, Hilsenbeck SG. Reproducibility, sources of variability, pooling and sample size: important considerations for the design of high-density oligonucleotide array experiments. J Gerontol A Biol Sci Med Sci. 2004;59:306–15. doi: 10.1093/gerona/59.4.b306. [DOI] [PubMed] [Google Scholar]
- 23.Rainen L, Oelmueller U, Jurgensen S, et al. Stabilization of mRNA expression in whole blood samples. Clin Chem. 2002;48:1883–90. [PubMed] [Google Scholar]
- 24.Tan PK, Downey TJ, Spitznagel EL, et al. Evaluation of gene expression measurements from commercial microarray platforms. Nucleic Acids Res. 2003;31:5676–84. doi: 10.1093/nar/gkg763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snider JV, Wechser MA, Lossos IS. Human disease characterization: real-time quantitative PCR analysis of gene expression. Drug Discov Today. 2001;6:1062–7. doi: 10.1016/s1359-6446(01)01988-2. [DOI] [PubMed] [Google Scholar]
- 26.Liles WC, Van Voorhis WC. Nomenclature and biological significance of cytokines involved in inflammation and the host immune response. J Infect Dis. 1995;172:1573–80. doi: 10.1093/infdis/172.6.1573. [DOI] [PubMed] [Google Scholar]
- 27.Joyce DA, Steer JH, Beilharz MW, Stranger R. A system for assessment of monokine gene expression using human whole blood. Genet Anal. 1995;12:39–43. doi: 10.1016/1050-3862(95)00104-2. [DOI] [PubMed] [Google Scholar]
- 28.Inoue M, Nishimura S, Hori G, Nakahara H, Saitom M, Yoshihara Y, Amari S. Improved parameter estimation for variance-stabilizing transformation of gene-expression microarray data. J Bioinform Comput Biol. 2004;2:669–79. doi: 10.1142/s0219720004000806. [DOI] [PubMed] [Google Scholar]
- 29.Naef F, Hacker CR, Patil N, Magnasco M. Empirical characterization of the expression ratio noise structure in high-density oligonucleotide arrays. Genome Biol. 2002;Epub 2002 Mar 22;3(4) doi: 10.1186/gb-2002-3-4-research0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.American Red Cross: www.redcross.org
- 31.Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986–94. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- 32.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-??CT method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.Pinheiro J, Bates DM. (2000) Mixed-Effects Models in S and S-PLUS New York: Springer. 528 pp
- 34.R Development Core Team (2004) R Foundation for Statistical Computing, Vienna, Austria.
- 35.Martich G, Boujoukos A, Suffredini A. Response of man to endotoxin. Immunobiology. 1993;187:403–16. doi: 10.1016/S0171-2985(11)80353-0. [DOI] [PubMed] [Google Scholar]
- 36.Schiller JS, Adams PF, Nelson ZC. Summary health statistics for the US population: National health interview survey, 2003. Vital Health Stat. 2005 Apr;10(224):1–104. [PubMed] [Google Scholar]
- 37.Koenig W, et al. C-Reactive protein, a sensitive marker of inflammation, predicts future risk of coronary heart disease in initially healthy middle-aged men: results from the MONICA (Monitoring Trends and Determinants in Cardiovascular Disease) Augsburg Cohort Study, 1984 to 1992. Circulation. 1999;99:237–42. doi: 10.1161/01.cir.99.2.237. [DOI] [PubMed] [Google Scholar]
- 38.Pearson TA, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: A statement for health-care professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107:499–511. doi: 10.1161/01.cir.0000052939.59093.45. [DOI] [PubMed] [Google Scholar]
- 39.Bistrian BR. Practical recommendations for immune-enhancing diets. J Nutr. 2004;134:2868S–72S. doi: 10.1093/jn/134.10.2868S. [DOI] [PubMed] [Google Scholar]
- 40.Simopoulos AP. Omega-3 fatty acids in inflammation and autoimmune diseases. J Am Coll Nutr. 2002;21:495–505. doi: 10.1080/07315724.2002.10719248. [DOI] [PubMed] [Google Scholar]
- 41.Eady J, et al. Variation on gene expression profiles of peripheral blood mononuclear cells from healthy volunteers. Physiol Genomics. 2005;22:402–11. doi: 10.1152/physiolgenomics.00080.2005. [DOI] [PubMed] [Google Scholar]
- 42.Radich J, et al. Individual-specific variation of gene expression in peripheral blood leukocytes. Genomics. 2004;83:980–8. doi: 10.1016/j.ygeno.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 43.Tryon V et al. High-precision gene expression analysis of rheumatoid arthritis and other inflammatory diseases. Int. Assoc. Inflammation Soc. Meeting Poster Presentation, Vancouver BC, August 2003.
- 44.Ross J, et al. The HER-2/neu gene and protein in breast cancer 2003: biomarker and target of therapy. Oncologist. 2003;8:307–25. doi: 10.1634/theoncologist.8-4-307. [DOI] [PubMed] [Google Scholar]
- 45.Madhusudan S, Ganesan TS. Tyrosine kinase inhibitors in cancer therapy. Clin Biochem. 2004;37:618–35. doi: 10.1016/j.clinbiochem.2004.05.006. [DOI] [PubMed] [Google Scholar]