

Abstract
The recently sequenced genome of the honey bee (Apis mellifera) has produced 10,157 predicted protein sequences, calling for a computational effort to extract biological insights from them. We have applied an unsupervised hierarchical protein-clustering method, which was previously used in the ProtoNet system, to nearly 200,000 proteins consisting of the predicted honey bee proteins, the SWISS-PROT protein database, and the complete set of proteins of the mouse (Mus musculus) and the fruit fly (Drosophila melanogaster). The hierarchy produced by this method has been entitled ProtoBee. In ProtoBee, the proteins are hierarchically organized into 18,936 separate tree hierarchies, each representing a protein functional family. By using the mouse and Drosophila complete proteomes as reference, we are able to highlight functional groups of putative gene-loss events, putative novel proteins of unique functionality, and bee-specific paralogs. We have studied some of the ProtoBee findings and suggest their biological relevance. Examples include novel opsin genes and intriguing nuclear matches of mitochondrial genes. The organization of bee sequences into functional clusters suggests a natural way of automatically inferring functional annotation. Following this notion, we were able to assign functional annotation to about 70% of the sequences. ProtoBee is available at http://www.protobee.cs.huji.ac.il
Comparative genomics are heavily based on computational methods. These methods provide not only automation for handling the immense amount of data held within whole genomes, but are also a means of highlighting biologically interesting differences between genomes. The recently sequenced genome of the honey bee Apis mellifera (The Honey Bee Genome Sequencing Consortium 2006) poses an excellent instance for comparative computational analysis in order to identify unique bee phenomena at the genomic and proteomic levels.
ProtoNet is a hierarchical organization of over 1,000,000 protein sequences (Kaplan et al. 2005). The hierarchy is based on an automatic unsupervised clustering method, which groups proteins according to their sequence similarity to each other. The resulting hierarchy consists of protein clusters that are arranged into several trees. Each such tree represents a protein family at various functional levels, from the level of very general super-families (represented by the roots of the trees) to the level of very specialized subfamilies (represented by the leaves). This method has been shown previously to produce both hierarchies and clusters that are highly coherent with several impartial biological data sources (Kaplan et al. 2004).
We have applied the method used in ProtoNet to 199,343 proteins consisting of the GLEAN3 set of predicted bee proteins (The Honey Bee Genome Sequencing Consortium 2006), the SWISS-PROT protein database (Bairoch et al. 2005), and the complete set of proteins of the mouse Mus musculus and the fruit fly Drosophila melanogaster from TrEMBL complementary database. The SWISS-PROT database acts as a high-quality scaffold of the protein sequence space, spanning several different taxonomical and functional areas. While the SWISS-PROT database is manually validated and thus extremely reliable, it does not contain whole genomes of complex eukaryotes. Thus, the additional mouse and Drosophila TrEMBL proteins (together with the mouse and Drosophila proteins in SWISS-PROT) act as reference genomes of multicellular eukaryotes. By combining these, one can both achieve a global overview of the bee proteome and highlight unique aspects of the bee proteome with relative ease. Specifically, we show how one can identify clusters that suggest instances of either proteins of unique bee functionality, potential gene-loss events, and bee-specific paralogs.
One key computational task for a newly sequenced genome is the automatic assignment of functional annotation to its predicted coding sequences (see Discussion in Sasson et al. 2006). By annotation we are referring to biological terms describing functional aspects of proteins, which are obtained from a standardized vocabulary such as the Gene Ontology (GO) (Camon et al. 2004; Harris et al. 2004), UniProt (Bairoch et al. 2005) keywords, and InterPro (Mulder et al. 2005) domains. Given that the hierarchies provided by the clustering method are biologically valid to a large extent (as previously demonstrated in Kaplan et al. [2004]), it is quite straightforward to exploit these hierarchies in order to infer protein annotation. This is done by using existing high-quality annotation on the UniProt proteins from several different sources. First, each cluster is assigned the annotations that represent its proteins. Next, each bee protein sequence receives the annotations of the cluster to which it belongs, and the annotations of all of the cluster's parent clusters. By providing automatic annotation for the bee sequences, we are able to complement the comparative view of the protein families.
A Web site that enables downloading, browsing, and analysis of the ProtoBee hierarchy and classification is available at http://www.protobee.cs.huji.ac.il.
Results
ProtoBee hierarchy
The resulting hierarchy of the ∼200,000 protein sequences contains 85,579 clusters that are organized into 18,936 separate trees. Each such tree is conjectured to represent a family of proteins that are functionally related. The proteins of each tree are all contained in its root cluster; therefore, the terms “tree” and “root” will be used interchangeably. Before proceeding, it is crucial to stress that the bee sequences are based on computational prediction. This means that some of the predicted coding sequences may be either partially or even fully incorrect. Furthermore, it is plausible that some proteins could be missing from the predicted set. In addition, the clustering and annotation methods are also expected to possess some degree of error as expected of any automatic computational method. Although in order to properly distinguish between these possibilities each cluster has to be inspected manually, in some instances it is possible to systematically pinpoint clusters that are more likely to possess unique bee features. This is the approach by which we proceed. In order to gain a global taxonomic view of the bee proteome, we look at two different perspectives. (1) Protein-based view. Each one of the 10,157 predicted bee sequences belongs to one of the roots. Other proteins assigned to the same root are considered to be putative homologs, belonging to the same functional family. For each protein, we check whether it has homologs from the mouse, fly, or other organisms. (2) Root-based view. There are 5095 roots that contain at least one of the 10,157 bee proteins. For each such root, we check whether it contains proteins from the mouse, fly, or other organisms in addition to the bee proteins. Figure 1 shows the summary of these results in a Venn diagram. As expected, a large majority (67%) of proteins have putative homologs both in mouse, fly, and other organisms. However, in terms of roots, these proteins are contained in 2539 roots, which represent only 50% of the total amount of roots. This suggests that several of these roots represent families that possess some functional divergence in the form of paralogs. A total of 87% of the proteins have putative fly homologs and 82% of the proteins have putative mouse homologs.
Figure 1.

Venn diagram describing the taxonomical distribution of 10,157 bee proteins and 5095 roots with respect to fly, mouse, and all other species. Bold numbers indicate the amount of proteins in each partition. Numbers in parentheses are numbers of roots. Note 159 proteins that were not clustered with proteins from other species and are thus labeled “unique.”
One of the most interesting subset of proteins is the group of 159 proteins that do not have homologs from any organism in our database. Since these proteins appear in 143 roots, most of them consist of only one bee protein. We expect these to be either bee proteins that have a unique functionality, highly diverged bee orthologs, gene prediction mistakes, or sequences that could not be properly classified by ProtoBee. An interesting subset of these 159 proteins is the subset of proteins that belong to nonsingleton clusters (i.e., consisting of more than one protein). The reason that these are especially interesting is that the chance of them being gene-prediction mistakes is significantly reduced. Such clusters are conjectured to consist of unique bee paralogs, created by gene-duplication events that are unique to the bee. Table 1 shows a list of the nine nonsingleton clusters that contain only bee proteins.
Table 1.
Nine nonsingleton clusters containing only bee proteins

Following this comparative overview and the identification of putative bee sequences that possess a unique functionality, we would like to focus on gene-loss events in the bee. A careful testing of individual genes has previously shown cases of possible gene loss in the bee genome (Whitfield et al. 2002). The root clusters are used as our starting points. The 199,343 proteins in the database are contained in 18,936 roots. From these, 2598 roots contain fly proteins, but do not contain bee proteins. In the resulting list, it is difficult to separate these into putative bee gene-loss events and unique fly proteins. A third high-quality annotated insect genome would be helpful as a reference for separating these cases, but currently there is no such genome available (the Anopheles genome [Holt et al. 2002] is currently not sufficient). Therefore, there are two possible approaches. The first is to use the mouse genome as a reference and look at the subset of roots that contain both fly and mouse proteins but do not contain bee proteins (marked fly+/mouse+/bee−). There are 1225 such roots (a list is available on the ProtoBee Web site). While this approach would eliminate the cases of mistaking bee gene-loss events for unique Drosophila functionality, it would miss protein families that are unique in insects. Alternatively, a different approach would be to use the other insect proteins that exist in the SWISS-PROT database as a reference (there are 3465 such proteins). In this approach, we focus on clusters that do not contain bee proteins but contain at least one fly protein and at least one additional protein from a different insect (marked fly+/insect+/bee−). While this approach will certainly miss several cases of gene-loss events due to the lack of coverage of the insect proteins, it focuses on insect functionality. Still, this approach can mistake some instances of unique Drosophila functionality for bee gene-loss events (in cases where the other insect species in the cluster are evolutionarily very close to Drosophila). There are 67 such roots that do not contain bee proteins but do contain at least one fly protein and at least one protein from another insect. A list of these fly+/insect+/bee− clusters is shown in Table 2.
Table 2.
Fifty-two fly+/insect+/bee− root clusters

So far we have focused on two sets of proteins that are of special interest in a comparative study of the bee genome— proteins whose function is bee specific and proteins that are missing in the bee due to gene-loss events. One other interesting case is that of paralog enrichment. In the case of paralogs, we would like to focus on protein families that are taxonomically imbalanced. Specifically, roots that contain a high ratio of bee:fly and bee:mouse proteins may suggest that there exist several paralogs in the bee that do not exist in the fly and mouse. In order to highlight taxonomically imbalanced clusters, we use a taxonomical balance score (TB score):
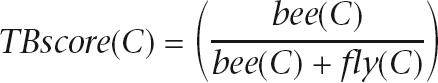 |
where bee(C) is the number of bee proteins in cluster C and fly(C) is the number of fly proteins in C. The score ranges from 1 (only bee proteins, no fly proteins) to 0 (no bee proteins, only fly proteins), 0.5 indicating an equal amount of fly and bee proteins. A score for bee:mouse ratio is derived in a similar manner. The TB score for each cluster is available through the ProtoBee Web site.
Following the procedure described in Methods, 7131 of 10,157 (70%) bee sequences were assigned annotation. While in terms of coverage this is comparable with supervised methods, the fact that the annotation sources used (see Methods) are varied in terms of scope provides viewpoints at several levels of functionality. Namely, we are able to assign a wide range of annotations from very general properties (e.g., signal transduction, metabolism) to very specific properties (e.g., glucose-6-phosphate isomerase). However, the main goal of the automatic annotation effort is to complement the view of the individual protein families. For example, suppose that by using the comparative approach previously described we find that a protein cluster of polymerases does not contain bee orthologs. A natural question would be whether bee polymerases can be found in other clusters. This can be easily examined by checking which bee proteins were annotated as polymerases. Figure 2 shows the distribution of annotated proteins into GO functional categories. A list summarizing the amount of proteins per annotation is available on the ProtoBee Web site.
Figure 2.

Protein annotation summary for several Gene Ontology categories. A full list is available on the ProtoBee Web site.
Manual evaluation of the results
It is obvious that the full extent of the biological relevance of the results that are produced by this computational approach cannot be assessed without manual inspection of each and every prediction. Therefore, we proceed by providing an in-depth biological analysis of only some of the results.
We start by examining the set of fly+/insect+/bee− clusters (Table 2). Fifteen of these clusters contain multiple biological groups that are apparently unrelated (note that according to our annotation-inference method, such clusters will not be used to infer annotations). However, it is apparent that in some instances these predictions are meaningful, considering the fact that they suggest specific functionally related groups of proteins to be missing (such as mitochondrial proteins, chorion proteins, vision proteins, and developmental proteins). Still, it is crucial to note that not all biologically coherent clusters necessarily indicate gene-loss events. For example, in the case of glucose-6-phosphate isomerase (G6PI), the protein seems to be missing, as it does not appear clustered with the Drosophila protein. G6PI is conserved amongst several species and is crucial for glycolysis, so it is highly unlikely that it does not have a bee homolog. Browsing the ProtoBee annotations, we find that ProtoBee annotates one of the bee proteins as G6PI, and this protein indeed seems to show very high similarity to G6PI proteins. Thus, we suggest that in order to determine whether a fly+/insect+/bee− cluster is indicative of a putative gene loss, one should complement the study of each such cluster with an examination of the corresponding annotations.
Mitochondrial proteins
The bee mitochondrion has been sequenced and is known to contain 13 genes (Crozier and Crozier 1993), all involved in oxidative phosphorylation, i.e., Cytochrome c oxidase (COX) (subunits 1, 2, and 3), Cytochrome b, ATP synthase (subunits 6 and 8), and NADH dehydrogenase (ND) (subunits 1, 2, 3, 4, 4L, 5, and 6). The 10,157 predicted protein sequences in ProtoBee consisted only of nuclear DNA. Therefore, we would expect these proteins to appear to be missing and can use this group of proteins in order to evaluate the biological predictions made by ProtoBee. Table 1 shows that 10 of the 13 genes are indeed predicted to be missing in bee nuclear proteome. However, COX1, COX3, and ND1 do not appear in this list, indicating that either they have bee homologs in the nuclear DNA or that ProtoBee was unable to group these protein families correctly. Further inspection shows that the former is the case. In all three cases, bee sequences with significant similarity are found.
In the case of COX1, ProtoBee is able to correctly cluster the COX1 protein family into a unique tree. However, one of the clusters in this tree also contains GB17755, a 30 amino acid bee protein. GB17755 shows a high level of similarity (59% identity spanning all 30 amino acids) to COX1 from various organisms. Returning to the genome, we find that the sequence of GB17755 is not part of a full-length COX1 homolog in the genome. No evidence of expression or mitochondrial targeting signal was found. In the case of ND1, we find that the bee sequence GB12194 was clustered in a cluster of ND1 orthologs. A BLAST search using GB12194 as the query shows that the best matching sequence in UniProt is the sequence of the bee mitochondrial ND1 (84% identity on a region of over 60 amino acids). Searching the genome showed that this sequence is not part of a full-length nuclear homolog of ND1. Therefore, we do not expect this to be an instance in which a high-similarity homolog was missed in the gene prediction process. Once again, no evidence of EST expression or mitochondrial targeting was found. In light of this, the most likely explanation for the appearance of these sequences in the nuclear DNA is the migration of the mitochondrial sequence to the nuclear DNA, creating NUMTs (nuclear mitochondrial DNA).
In order to further investigate whether these sequences are indeed NUMTs (Richly and Leister 2004), we used the full-length nucleotide sequences of bee ND1 and COX1 in order to search for additional similarities within the bee genome. Figure 3 depicts several nuclear matches that were found for both ND1 and COX1. Additional comparisons of other mitochondrial genes indicate that this phenomenon is indeed widespread in the bee genome (data not shown).
Figure 3.

Nuclear matches of mitochondrial genes. Striped rectangle represents the nucleotide sequence of the mitochondrial gene. Dark rectangles above and below the sequence represent high-similarity nuclear matches on the plus and minus strands, respectively. (Top) Cytochrome c oxidase subunit 1; (bottom) NADH dehydrogenase subunit 1. Display was adopted from ENSEMBL (Hubbard et al. 2005).
In the case of COX3, we find that the bee sequence GB11138 has been clustered in a cluster with bacterial COX3 and ubiquinol oxidase subunit 3 (UOX3) proteins. GB11138 shows the highest level of similarity (54% identity spanning ∼90% of the protein) to UOX3 from Escherichia coli. Furthermore, the length of the proteins (206 amino acids) matches that of prokaryotic COX3 and UOX3 proteins rather than that of eukaryotic COX3. The high similarity of this protein to prokaryotic COX3 and UOX3 suggested that this sequence may be of bacterial origin. Examining the contig in which this sequence appears, we have identified two adjacent sequences with high similarity to bacterial UOX and ferredoxin proteins. The contig is currently unlocalized within the genome. No evidence of expression or mitochondrial targeting signal was found. We suggest that GB11138 and its contig are either the result of a recent lateral gene-transfer event or of a contamination within the genome sequence. In the cases of all three sequences it is apparent that although these sequences probably do not code for proteins, the classifications made by ProtoBee in each of these instances were justifiable.
Opsins
Opsins are rhodopsin-like G-protein coupled receptors that act as photoreceptors. It has been recently shown that some opsins are not involved in vision but are involved in photic entrainment of the circadian rhythm (Thresher et al. 1998). From Table 2 it seems that the bee opsin family is significantly different from that of the fly. Of the six known opsins in fly (Rh1–Rh6), three seem to lack bee homologs. In order to search for alternative opsins in the bee, we look at bee proteins that were automatically assigned the annotation “opsin” in ProtoBee. Altogether there are six such proteins (Table 3). Three of the proteins (GB18171, GB13493, and GB19657) match the three known bee opsins (ultraviolet-sensitive opsin [UVOP], blue-sensitive opsin [BLOP], and long-wavelength opsin [LWOP], respectively). UVOP and BLOP are clustered with fly opsins Rh3 and Rh5. LWOP is found clustered with various arthropod rhodopsins, but without any fly opsins. Surprisingly, three other proteins appear to be annotated as opsins. Manual inspection shows that one of these seems to be incorrectly assigned this annotation, while the other two show significant levels of similarity to opsins. GB19336 is clustered with fly Rh6 opsin, but also possesseses a strong similarity to LWOP (64% identity by global alignment). Furthermore, it is located <1 kb away from LWOP, but appears on the opposite strand. This evidence strongly suggests that this protein is a LWOP paralog that has been created by a duplication event. Note that the existence of such a paralog in several insects has been suggested (Spaethe and Briscoe 2004). GB12200 is found to be clustered with several different rhodoposin proteins. While the similarity of this sequence to rhodopsins is low, but statistically significant (32% identity, e-value 10−40), its function as a rhodopsin is also supported by other search methods (i.e., InterPro-Scan). Interestingly, the proteins that were found to be most similar to this sequence are encephalopsins and pineal opsins. Encephalopsins are opsins that were previously found to be specifically expressed in the mammalian brain and suggested to be nonvisual opsins involved in encephalic photoreception and in photic entrainment of the circadian rhythm (Blackshaw and Snyder 1999). Pineal opsins are nonvisual opsins expressed in the pineal of several species (Max et al. 1995). In order to test whether GB12200 might have a similar function as a nonvisual opsin, we searched the EST database using the untranslated coding sequence of GB12200. Two matching ESTs were found (BQ103783 and BI509943), both expressed in the bee brain. Thus, we suggest that GB12200 might indeed function as a non-visual opsin and might provide input for the circadian rhythm. After the completion of this study, GB12200 was independently discovered as an opsin and shown experimentally to be expressed in the bee brain but not in the eye (Velarde et al. 2005).
Table 3.
Protein sequences that were automatically assigned the annotation "opsin by ProtoBee

Pigment Dispersal Hormone
Another protein that surprisingly seems to be missing is the Pigment Dispersal Hormone (PDH). PDH has been suggested to be involved both in vision and the circadian rhythm (Park and Hall 1998). However, it is also suggested to exist in the bee and be highly conserved amongst insects (Bloch et al. 2003). Running a BLAST search of PDH against the entire 10,157 proteins set using the fly PDH preprotein as query finds no matching sequences. Since the experimental evidence suggested that this protein does exist, we independently searched the bee genome for a homolog of PDH, using the fly PDH preprotein. One matching sequence was found, displaying an extremely high degree of conservation (identical in all but two amino acids), which is restricted to the part of the preprotein that codes for the PDH peptide. Apart from this region of similarity, the rest of the preprotein sequence does not have matches in the genome. In light of this strong evidence, we suggest that a homolog of PDH does exist in the bee, but was missed by the computational gene prediction.
Unique bee paralogs
We proceed by examining the nine nonsingleton bee-specific clusters (Table 1). Four of these nine clusters seemed to be grouped due to very weak similarity and will not be discussed further. The five other clusters possess high inner similarity and seem to be true instances of paralogs. In three of the five clusters, we have found experimental expression evidence for at least one protein. In three of these clusters, the genes are also localized to the same locus, strengthening the notion of gene-duplication events. Cluster 345,857 consists of a group of three proteins that are localized to the same locus (separated by ∼1.5–2.5 kb) and are predicted by InterProScan to be olfactory receptors.
Cluster 391,502 consists of Apamin and Mast Cell Degranulating Protein (MCDP), both constituents of the bee venom. Apamin and MCDP were previously shown to share their 3′ exon (Gmachl and Kreil 1995). Cluster 388,340 consists of four proteins sharing a region of 44 amino acids that is extremely conserved (see Fig. 3). Three of these sequences (GB16868, GB10213, and GB11167) are located on one contig with ∼30–40 kb separating them from one another, and the fourth (GB19685) is found on a separate contig. Although InterPro detects no known domains on these proteins, one of the proteins seems to be coded by the recently discovered csd gene (Beye et al. 2003). The csd gene has been discovered by Beye et al. via positional cloning and was shown, by means of RNAi gene silencing, to be directly responsible for complementary sex determination. While all of the four sequences found are shorter then the sequence presented by Beye et al., we attribute this to inaccurate gene prediction. Following the gene-silencing of csd by RNAi performed in (Beye et al. 2003), the csd gene has been suggested to be solely responsible for bee sex determination. In addition, csd was found to have several allelic variants, which were thought to govern this process (Hasselmann and Beye 2004). Allelic variants were detected in the sequences as they assemble to separate chromosomes. Signatures of duplication were also detected in this region that could possibly harbor functional genes and pseudogenes (M. Beye, pers. comm.). Nonetheless, the identification of these sequences demonstrates the ability of our approach to easily detect unique bee-specific functional groups.
Discussion
Once a new genome is sequenced, there are several computational tasks that may be performed on it in order to learn about its biology. These include gene prediction, automatic annotation, and comparative analyses. For each of these tasks there are several different approaches. In this work we present a novel method that combines both the tasks of comparative analysis and automatic annotation. One unique aspect of the clustering method used by ProtoBee is the fact that it is an unsupervised method. In the supervised approach, the algorithm is typically provided with a training set of proteins known to belong to the same family, and then learns common features in order to detect new members of this family. This is the most commonly used approach for machine learning of protein families. While this approach delivers extremely high performance in terms of sensitivity and specificity, it creates a heavy bias toward the detection of only that which is known and cannot detect novel protein families. In the unsupervised approach, on the other hand, the method looks for intrinsic features of the data in order to organize it, rather than being guided externally. Using an unsupervised clustering method, ProtoBee is expected to be inferior to supervised methods such as InterProScan in terms of sensitivity/specificity. Thus, we suggest using our annotation method in conjunction with supervised methods in order to provide maximal coverage and specificity. However, the method makes up for this inferiority by its ability to detect novel protein families (e.g., nonsingleton clusters that are unique to bee) and provide a hierarchical comparative view.
A genomic view that is based on the comparison of a genome to only two other genomes may be somewhat biased. However, since the computation required by this method is demanding (nearly 4 × 1010 sequence comparisons), a three-way comparison seems to be a reasonable compromise between biological accuracy and computational feasibility.
Testing our method, we have discovered that the phenomenon of NUMTs is extensive in the honey bee genome. The significant appearance of NUMTs in the bee genome is quite surprising considering that this phenomenon in nearly absent both in Anopheles gambiae and in Drosophila melanogaster (Richly and Leister 2004).
In contrast to the previous application of this method in ProtoNet, the focus in ProtoBee is on a whole-genome comparative view. The ability to divide the proteins into functional groups and view each group in light of three whole proteomes provides a unique view of the functional organization of the bee proteome in light of two other metazoan proteomes. This led us to highlight interesting groups of proteins that may be able to account for unique biological characteristics of the bee. It is important to recognize that the predictions made by this method may be, in some cases, lacking or mistaken. However, our goal in highlighting potentially interesting clusters is not to provide a finalized comprehensive list of gene-loss and function-gain events, but merely to select a subset of clusters that suggest further in-depth examination. By studying a few examples of such clusters it is evident that some of these are genuinely interesting. The purpose of the examples that we provide is to demonstrate the ability of the ProtoBee method to pinpoint interesting and often surprising biology in the genome. Obviously, these biological findings require further research in order to evaluate their significance. We expect the lists of putative gene losses, unique function proteins, and bee-specific paralogs to conceal within them many more exciting biological stories.
Methods
Sources and tools
The protein database that was clustered consisted of the SWISS-PROT database version 41.21 (133,312 proteins), additional mouse and fly proteins from TrEMBL version 24.8 (20,730 Drosophila proteins and 35,199 mouse proteins), and the GLEAN3 set of predicted proteins (http://www.protobee.cs.huji.il and http://www.hgsc.bcm.tmc.edu/projects/honeybee) from release v3.0 of the Apis mellifera genome (10,157 proteins). Fifty-five previously known bee proteins that appeared in SWISS-PROT were removed from the database in order to avoid duplicate instances of the proteins, leaving our protein database at a total size of 199,343 protein sequences.
For sequence comparison, NCBI BLAST (Altschul et al. 1997) was used for local alignment and the EMBOSS Align (Olson 2002) implementation of the Needleman-Wunsch algorithm (Needleman and Wunsch 1970) was used for global alignment. Genomic searches were performed using ENSEMBL genomic BLAST (Hubbard et al. 2005). EST searches were performed using NCBI BLAST against dbEST (Boguski et al. 1993). Multiple sequence alignment was performed by CLUSTALW (Thompson et al. 1994). Phylogenetic analysis was performed using the PHYLIP package v3.65 (Felsenstein 1988) with the neighbor-joining algorithm for tree construction. Subcellular localization and mitochondrial targeting were predicted using TargetP (Emanuelsson et al. 2000) and WoLF PSORT (Nakai and Horton 1999). InterPro domain detection was performed by InterProScan (Quevillon et al. 2005).
Protein clustering
The organization of the proteins into a set of trees is composed of four steps.
1. All-against-all BLAST. NCBI BLAST is run on all pairs of proteins, using BLOSUM62. All E-values lower than 100 are kept in a matrix. E-values higher than 100 are considered to be 100.
2. Hierarchical clustering. An agglomerative clustering procedure is applied, in which all clusters start as singletons, and at each step the two clusters that have the lowest score are merged into a new cluster. The score between two clusters is defined as the arithmetic mean of the E-values from all intercluster pairs of proteins:
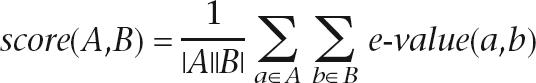 |
3. Cutoff. All clusters that are created at ProtoLevel 80 or later are eliminated in order to increase biological validity.
4. Pruning. Following the pruning method presented in Kaplan et al. (2004), all clusters with a “lifetime” of <1 are eliminated. The rational and biological justification for pruning is discussed in Kaplan et al. (2004). Following this step, 85,579 clusters remain, organized into 18,936 trees.
Protein annotation
The annotation of the bee sequences was performed in the following manner. First, we calculate for each cluster what the annotations are that best represent its proteins. In this step, all bee proteins are ignored. For an annotation to represent a cluster we require that (1) the annotation will be shared by at least 75% of the proteins in the cluster, (2) the cluster will contain at least five proteins, and (3) the annotation will achieve a P-value smaller than 0.001 for the assumption that the annotations are distributed hypergeometrically. The P-value for a cluster C and an annotation a given the database D is calculated according to the hypergeometric distribution:
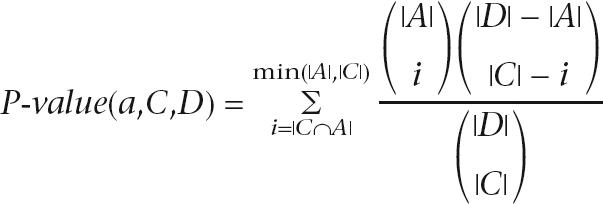 |
where A is the set of all proteins in the database that have annotation a. These relatively strict requirements ensure that clusters that are biologically incoherent do not affect the process of assigning annotations and that uninformative annotations are avoided. The annotations that are assigned to the clusters are taken from the following sources: UniProt keywords, InterPro, GO “molecular function” and “biological process” terms (including the GOA mapping), and E.C. (Enzyme Classification) numbers. Finally, each bee protein is assigned the annotations that were given to the cluster to which it belongs and the annotations that were assigned to all of the cluster's parents in the hierarchy.
Acknowledgments
We thank Ori Sasson for his effort in preparing the data for ProtoBee and Alex Savenok for development of the ProtoBee Web site. We thank the Baylor College of Medicine Human Genome Sequencing Center for making the Apis mellifera genome sequence publicly available prior to publication. This work is supported in part by the EU Framework VI, NoE BioSapiens consortium for Genome Annotation. N.K. is a fellow of the Sudarsky Center for Computational Biology of the Hebrew University.
Footnotes
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.4916306.
References
- Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J., Zhang J., Zhang Z., Miller W., Lipman D.J., Zhang Z., Miller W., Lipman D.J., Miller W., Lipman D.J., Lipman D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A., Apweiler R., Wu C.H., Barker W.C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Apweiler R., Wu C.H., Barker W.C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Wu C.H., Barker W.C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Barker W.C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Gasteiger E., Huang H., Lopez R., Magrane M., Huang H., Lopez R., Magrane M., Lopez R., Magrane M., Magrane M., et al. The universal protein resource (UniProt) Nucleic Acids Res. 2005;33:D154–D159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beye M., Hasselmann M., Fondrk M.K., Page R.E., Omholt S.W., Hasselmann M., Fondrk M.K., Page R.E., Omholt S.W., Fondrk M.K., Page R.E., Omholt S.W., Page R.E., Omholt S.W., Omholt S.W. The gene csd is the primary signal for sexual development in the honeybee and encodes an SR-type protein. Cell. 2003;114:397–398. doi: 10.1016/s0092-8674(03)00606-8. [DOI] [PubMed] [Google Scholar]
- Blackshaw S., Snyder S.H., Snyder S.H. Encephalopsin: A novel mammalian extraretinal opsin discretely localized in the brain. J. Neurosci. 1999;19:3681–3690. doi: 10.1523/JNEUROSCI.19-10-03681.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch G., Solomon S.M., Robinson G.E., Fahrbach S.E., Solomon S.M., Robinson G.E., Fahrbach S.E., Robinson G.E., Fahrbach S.E., Fahrbach S.E. Patterns of PERIOD and pigment-dispersing hormone immunoreactivity in the brain of the European honeybee (Apis mellifera): Age- and time-related plasticity. J. Comp. Neurol. 2003;464:269–284. doi: 10.1002/cne.10778. [DOI] [PubMed] [Google Scholar]
- Boguski M.S., Lowe T.M., Tolstoshev C.M., Lowe T.M., Tolstoshev C.M., Tolstoshev C.M. dbEST–database for “expressed sequence tags. Nat. Genet. 1993;4:332–333. doi: 10.1038/ng0893-332. [DOI] [PubMed] [Google Scholar]
- Camon E., Magrane M., Barrell D., Lee V., Dimmer E., Maslen J., Binns D., Harte N., Lopez R., Apweiler R., Magrane M., Barrell D., Lee V., Dimmer E., Maslen J., Binns D., Harte N., Lopez R., Apweiler R., Barrell D., Lee V., Dimmer E., Maslen J., Binns D., Harte N., Lopez R., Apweiler R., Lee V., Dimmer E., Maslen J., Binns D., Harte N., Lopez R., Apweiler R., Dimmer E., Maslen J., Binns D., Harte N., Lopez R., Apweiler R., Maslen J., Binns D., Harte N., Lopez R., Apweiler R., Binns D., Harte N., Lopez R., Apweiler R., Harte N., Lopez R., Apweiler R., Lopez R., Apweiler R., Apweiler R. The Gene Ontology Annotation (GOA) Database: Sharing knowledge in UniProt with Gene Ontology. Nucleic Acids Res. 2004;32:D262–D266. doi: 10.1093/nar/gkh021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozier R.H., Crozier Y.C., Crozier Y.C. The mitochondrial genome of the honeybee Apis mellifera: Complete sequence and genome organization. Genetics. 1993;133:97–117. doi: 10.1093/genetics/133.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson O., Nielsen H., Brunak S., von Heijne G., Nielsen H., Brunak S., von Heijne G., Brunak S., von Heijne G., von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Phylogenies from molecular sequences: Inference and reliability. Annu. Rev. Genet. 1988;22:521–565. doi: 10.1146/annurev.ge.22.120188.002513. [DOI] [PubMed] [Google Scholar]
- Gmachl M., Kreil G., Kreil G. The precursors of the bee venom constituents apamin and MCD peptide are encoded by two genes in tandem which share the same 3′-exon. J. Biol. Chem. 1995;270:12704–12708. doi: 10.1074/jbc.270.21.12704. [DOI] [PubMed] [Google Scholar]
- Harris M.A., Clark J., Ireland A., Lomax J., Ashburner M., Foulger R., Eilbeck K., Lewis S., Marshall B., Mungall C., Clark J., Ireland A., Lomax J., Ashburner M., Foulger R., Eilbeck K., Lewis S., Marshall B., Mungall C., Ireland A., Lomax J., Ashburner M., Foulger R., Eilbeck K., Lewis S., Marshall B., Mungall C., Lomax J., Ashburner M., Foulger R., Eilbeck K., Lewis S., Marshall B., Mungall C., Ashburner M., Foulger R., Eilbeck K., Lewis S., Marshall B., Mungall C., Foulger R., Eilbeck K., Lewis S., Marshall B., Mungall C., Eilbeck K., Lewis S., Marshall B., Mungall C., Lewis S., Marshall B., Mungall C., Marshall B., Mungall C., Mungall C., et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–D261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmann M., Beye M., Beye M. Signatures of selection among sex-determining alleles of the honey bee. Proc. Natl. Acad. Sci. 2004;101:4888–4893. doi: 10.1073/pnas.0307147101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt R.A., Subramanian G.M., Halpern A., Sutton G.G., Charlab R., Nusskern D.R., Wincker P., Clark A.G., Ribeiro J.M., Wides R., Subramanian G.M., Halpern A., Sutton G.G., Charlab R., Nusskern D.R., Wincker P., Clark A.G., Ribeiro J.M., Wides R., Halpern A., Sutton G.G., Charlab R., Nusskern D.R., Wincker P., Clark A.G., Ribeiro J.M., Wides R., Sutton G.G., Charlab R., Nusskern D.R., Wincker P., Clark A.G., Ribeiro J.M., Wides R., Charlab R., Nusskern D.R., Wincker P., Clark A.G., Ribeiro J.M., Wides R., Nusskern D.R., Wincker P., Clark A.G., Ribeiro J.M., Wides R., Wincker P., Clark A.G., Ribeiro J.M., Wides R., Clark A.G., Ribeiro J.M., Wides R., Ribeiro J.M., Wides R., Wides R., et al. The genome sequence of the malaria mosquito Anopheles gambiae . Science. 2002;298:129–149. doi: 10.1126/science.1076181. [DOI] [PubMed] [Google Scholar]
- The Honey Bee Genome Sequencing Consortium. Insights into social insects from the genome of the honey bee Apis mellifera. Nature. 2006;((in press)) [Google Scholar]
- Hubbard T., Andrews D., Caccamo M., Cameron G., Chen Y., Clamp M., Clarke L., Coates G., Cox T., Cunningham F., Andrews D., Caccamo M., Cameron G., Chen Y., Clamp M., Clarke L., Coates G., Cox T., Cunningham F., Caccamo M., Cameron G., Chen Y., Clamp M., Clarke L., Coates G., Cox T., Cunningham F., Cameron G., Chen Y., Clamp M., Clarke L., Coates G., Cox T., Cunningham F., Chen Y., Clamp M., Clarke L., Coates G., Cox T., Cunningham F., Clamp M., Clarke L., Coates G., Cox T., Cunningham F., Clarke L., Coates G., Cox T., Cunningham F., Coates G., Cox T., Cunningham F., Cox T., Cunningham F., Cunningham F., et al. Ensembl 2005. Nucleic Acids Res. 2005;33:D447–D453. doi: 10.1093/nar/gki138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N., Friedlich M., Fromer M., Linial M., Friedlich M., Fromer M., Linial M., Fromer M., Linial M., Linial M. A functional hierarchical organization of the protein sequence space. BMC Bioinformatics. 2004;5:196. doi: 10.1186/1471-2105-5-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N., Sasson O., Inbar U., Friedlich M., Fromer M., Fleischer H., Portugaly E., Linial N., Linial M., Sasson O., Inbar U., Friedlich M., Fromer M., Fleischer H., Portugaly E., Linial N., Linial M., Inbar U., Friedlich M., Fromer M., Fleischer H., Portugaly E., Linial N., Linial M., Friedlich M., Fromer M., Fleischer H., Portugaly E., Linial N., Linial M., Fromer M., Fleischer H., Portugaly E., Linial N., Linial M., Fleischer H., Portugaly E., Linial N., Linial M., Portugaly E., Linial N., Linial M., Linial N., Linial M., Linial M. ProtoNet 4.0: A hierarchical classification of one million protein sequences. Nucleic Acids Res. 2005;33:D216–D218. doi: 10.1093/nar/gki007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Max M., McKinnon P.J., Seidenman K.J., Barrett R.K., Applebury M.L., Takahashi J.S., Margolskee R.F., McKinnon P.J., Seidenman K.J., Barrett R.K., Applebury M.L., Takahashi J.S., Margolskee R.F., Seidenman K.J., Barrett R.K., Applebury M.L., Takahashi J.S., Margolskee R.F., Barrett R.K., Applebury M.L., Takahashi J.S., Margolskee R.F., Applebury M.L., Takahashi J.S., Margolskee R.F., Takahashi J.S., Margolskee R.F., Margolskee R.F. Pineal opsin: A nonvisual opsin expressed in chick pineal. Science. 1995;267:1502–1506. doi: 10.1126/science.7878470. [DOI] [PubMed] [Google Scholar]
- Mulder N.J., Apweiler R., Attwood T.K., Bairoch A., Bateman A., Binns D., Bradley P., Bork P., Bucher P., Cerutti L., Apweiler R., Attwood T.K., Bairoch A., Bateman A., Binns D., Bradley P., Bork P., Bucher P., Cerutti L., Attwood T.K., Bairoch A., Bateman A., Binns D., Bradley P., Bork P., Bucher P., Cerutti L., Bairoch A., Bateman A., Binns D., Bradley P., Bork P., Bucher P., Cerutti L., Bateman A., Binns D., Bradley P., Bork P., Bucher P., Cerutti L., Binns D., Bradley P., Bork P., Bucher P., Cerutti L., Bradley P., Bork P., Bucher P., Cerutti L., Bork P., Bucher P., Cerutti L., Bucher P., Cerutti L., Cerutti L., et al. InterPro, progress and status in 2005. Nucleic Acids Res. 2005;33:D201–D205. doi: 10.1093/nar/gki106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai K., Horton P., Horton P. PSORT: A program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem. Sci. 1999;24:34–36. doi: 10.1016/s0968-0004(98)01336-x. [DOI] [PubMed] [Google Scholar]
- Needleman S.B., Wunsch C.D., Wunsch C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Olson S.A. EMBOSS opens up sequence analysis. European Molecular Biology Open Software Suite. Brief. Bioinform. 2002;3:87–91. doi: 10.1093/bib/3.1.87. [DOI] [PubMed] [Google Scholar]
- Park J.H., Hall J.C., Hall J.C. Isolation and chronobiological analysis of a neuropeptide pigment-dispersing factor gene in Drosophila melanogaster . J. Biol. Rhythms. 1998;13:219–228. doi: 10.1177/074873098129000066. [DOI] [PubMed] [Google Scholar]
- Quevillon E., Silventoinen V., Pillai S., Harte N., Mulder N., Apweiler R., Lopez R., Silventoinen V., Pillai S., Harte N., Mulder N., Apweiler R., Lopez R., Pillai S., Harte N., Mulder N., Apweiler R., Lopez R., Harte N., Mulder N., Apweiler R., Lopez R., Mulder N., Apweiler R., Lopez R., Apweiler R., Lopez R., Lopez R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005;33:W116–W120. doi: 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richly E., Leister D., Leister D. NUMTs in sequenced eukaryotic genomes. Mol. Biol. Evol. 2004;21:1081–1084. doi: 10.1093/molbev/msh110. [DOI] [PubMed] [Google Scholar]
- Sasson O., Kaplan N., Linial M., Kaplan N., Linial M., Linial M. Functional annotation prediction: All for one and one for all. Protein Sci. 2006;15((in press)) doi: 10.1110/ps.062185706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaethe J., Briscoe A.D., Briscoe A.D. Early duplication and functional diversification of the opsin gene family in insects. Mol. Biol. Evol. 2004;21:1583–1594. doi: 10.1093/molbev/msh162. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G., Gibson T.J., Higgins D.G., Gibson T.J., Gibson T.J. ClustalW: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thresher R.J., Vitaterna M.H., Miyamoto Y., Kazantsev A., Hsu D.S., Petit C., Selby C.P., Dawut L., Smithies O., Takahashi J.S., Vitaterna M.H., Miyamoto Y., Kazantsev A., Hsu D.S., Petit C., Selby C.P., Dawut L., Smithies O., Takahashi J.S., Miyamoto Y., Kazantsev A., Hsu D.S., Petit C., Selby C.P., Dawut L., Smithies O., Takahashi J.S., Kazantsev A., Hsu D.S., Petit C., Selby C.P., Dawut L., Smithies O., Takahashi J.S., Hsu D.S., Petit C., Selby C.P., Dawut L., Smithies O., Takahashi J.S., Petit C., Selby C.P., Dawut L., Smithies O., Takahashi J.S., Selby C.P., Dawut L., Smithies O., Takahashi J.S., Dawut L., Smithies O., Takahashi J.S., Smithies O., Takahashi J.S., Takahashi J.S., et al. Role of mouse cryptochrome blue-light photoreceptor in circadian photoresponses. Science. 1998;282:1490–1494. doi: 10.1126/science.282.5393.1490. [DOI] [PubMed] [Google Scholar]
- Velarde R.A., Sauer C.D., Walden K.K., Fahrbach S.E., Robertson H.M., Sauer C.D., Walden K.K., Fahrbach S.E., Robertson H.M., Walden K.K., Fahrbach S.E., Robertson H.M., Fahrbach S.E., Robertson H.M., Robertson H.M. Pteropsin: A vertebrate-like non-visual opsin expressed in the honey bee brain. Insect Biochem. Mol. Biol. 2005;35:1367–1377. doi: 10.1016/j.ibmb.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Whitfield C.W., Band M.R., Bonaldo M.F., Kumar C.G., Liu L., Pardinas J.R., Robertson H.M., Soares M.B., Robinson G.E., Band M.R., Bonaldo M.F., Kumar C.G., Liu L., Pardinas J.R., Robertson H.M., Soares M.B., Robinson G.E., Bonaldo M.F., Kumar C.G., Liu L., Pardinas J.R., Robertson H.M., Soares M.B., Robinson G.E., Kumar C.G., Liu L., Pardinas J.R., Robertson H.M., Soares M.B., Robinson G.E., Liu L., Pardinas J.R., Robertson H.M., Soares M.B., Robinson G.E., Pardinas J.R., Robertson H.M., Soares M.B., Robinson G.E., Robertson H.M., Soares M.B., Robinson G.E., Soares M.B., Robinson G.E., Robinson G.E. Annotated expressed sequence tags and cDNA microarrays for studies of brain and behavior in the honey bee. Genome Res. 2002;2:555–566. doi: 10.1101/gr.5302. [DOI] [PMC free article] [PubMed] [Google Scholar]