

Abstract
The expression of telencephalic reelin (Reln) and glutamic acid decarboxylase mRNAs and their respective cognate proteins is down-regulated in postmortem brains of schizophrenia and bipolar disorder patients. To interpret the pathophysiological significance of this finding, immunoelectron microscopic experiments are required, but these cannot be carried out in postmortem human brains. As an alternative, we carried out such experiments in the cortex of rats and nonhuman primates. We found that Reln is expressed predominantly in layer I of both cortices and is localized to bitufted (double-bouquet), horizontal, and multipolar γ-aminobutyric acid-ergic interneurons, which secrete Reln into extracellular matrix. Reln secretion is mediated by a constitutive mechanism that depends on the expression of a specific signal peptide present in the Reln carboxy-terminal domain. Extracellular matrix Reln is found to aggregate in proximity of postsynaptic densities expressed in apical dendrite spines, which include also the α3 subunit of integrin receptors. Most pyramidal neurons of various cortical layers express the mouse-disabled 1 (Dab1) protein, which, after phosphorylation by a soluble tyrosine kinase, functions as an adapter protein, probably mediating a modulation of cytoskeleton protein expression. We hypothesize that the decrease of neuropil and dendritic spine density reported to exist in the neocortex of psychiatric patients may be related to a down-regulation of Reln–integrin interactions and the consequent decrease of cytoskeleton protein turnover.
Keywords: glutamic acid decarboxylase, schizophrenia, bipolar disorders, long-term potentiation
Reelin (Reln) is an ≈400-kDa neuronal glycoprotein secreted in the extracellular matrix (ECM) that is conserved in several vertebrate species. Its gene structure has been reviewed in current publications (1–4). Reln possesses some of the structural properties of an ECM protein: (i) a cleavable signal peptide and a region similar to F-spondin in proximity of the amino terminus domain (5), (ii) eight internal repeats of 350–390 aa, each repeat containing two related subdomains flanking a pattern of conserved cysteine residues related to the epidermal growth factor-like motif, and (iii) a short region of positively charged amino acids near the carboxy terminus that is required for secretion (4, 5).
The Reln gene of human and nonhuman primates, but not that of mice (4), contains a trinucleotide (CGG) repeat, which is located in the 5′ untranslated region of exon 1, just upstream from the open reading frame starting codon. In humans, this CGG trinucleotide repeat expands from 4 to 15, forming an array of polymorphic homozygous or heterozygous allelic variants.
In postmortem brains of schizophrenia or bipolar disorder patients with psychosis, the expression of the Reln (6) and glutamic acid decarboxylase 67 (GAD67) mRNAs and their respective translated products (6, 7) are down-regulated by at least 50%. These changes are caused neither by drugs used to treat the symptoms of the disease nor by the effect of concurrent terminal diseases overlapping with the diagnosed psychiatric illness. This raises the possibility that Reln and GAD67 down-regulation found in schizophrenia and bipolar disorder psychotic patients may be genetically determined. For example, they could be related, either to a gene-dosage insufficiency or to a decrease in the promoter function caused by either the expansion of the trinucleotide repeats or other defects of the promoter function.
In previous studies, we have found a strong preferential location of Reln in γ-aminobutyric acid (GABA)ergic cells of adult rat cortex and hippocampus and glutamatergic granule cells of cerebellum (8, 9). From these neurons in culture, Reln is secreted by a constitutive mechanism that is independent from storage in synaptic vesicles, does not require neurotransmitters or Ca2+ (10), but is dependent on a specific sequence of positively charged amino acids expressed in the Reln carboxy-terminus domain (4). Thus, here we have studied the cellular and ECM location of Reln in the neuropil of the three cortical areas of three species of nonhuman primates. We have investigated whether Reln colocalizes with GAD67 in the cortical areas of the three nonhuman primate species, and whether, after the secretion into ECM, Reln tends to be located in proximity of integrin receptor assemblies, including the α3 subunits, expressed on apical dendritic spines, as previously suggested (9).
Integrins are a family of surface receptors composed of two subunits, each selected from one subunit family (α and β). The integrin receptor interaction with ECM proteins by means of an arginine-glycine-aspartic acid (RGD) binding motif promotes integrin receptor clustering and triggers activation of certain cytosolic tyrosine kinases, such as focal adhesion kinases (Fak) or tyrosine kinases of the Src family (e.g., Fyn) (11). These kinases are involved in the signaling transduction pathways that control cytoskeleton protein turnover and organization (11). Migrating cortical neurons in embryos express integrin receptors containing α3β1 subunits, and the disruption of the α3 integrin subunit results in a reeler-like phenotype (12). In the present study, the mouse-disabled homologue protein (Dab1) (13, 14) is considered an essential component of any Reln signaling pathway. Once Dab1 adapter function is activated by tyrosine phosphorylation mediated by the Reln interaction with integrin receptors, an activation of intracellular microtubule network enzymes takes place (13–16). Because Dab1 needs to be phosphorylated to participate in the Reln signaling pathway (17), it is possible that Reln, binding to an extracellular integrin receptor domain (8, 12), not only promotes integrin receptor clustering, but also activates cytosolic Fyn kinase that in turn phosphorylates Dab1. Cadherin-related neuronal receptors (CNRs) also have been considered as candidate receptors for Reln (18). CNRs, like the integrin receptors, possess on the cytoplasmic domain a binding consensus for Fyn kinase and on the extracellular domain a Reln binding sequence with a RGD motif that is identical to the motif that function in ECM protein–integrin receptor interaction. Other signaling mechanisms, such as very low density lipoprotein receptor (VLDLR) and apolipoprotein E receptor 2 (ApoER2), anchoring Dab1 to their cytoplasmic domain (19, 20), have been suggested to be operative in the actions of Reln. However, VLDLR and ApoER2 do not express a cytoplasmic domain that activates specifically tyrosine kinases. Therefore, a hypothetical model for Reln signaling involves a direct Reln interaction with integrin or integrin-related receptors on the neuronal surface (9). This results in activation of cytoplasmic kinases and promotes phosphorylation of Dab1.
Materials and Methods
Subjects.
Subjects were three female Fisher rats (300 g body weight), one ≈55-yr-old female patas (Erythrocebus pata), one 20-yr-old female macaque (Macaca artoides), and two ≈8-yr-old female baboons (Papio papio). The animals were euthanized with ketamine (10 mg/kg i.m.) and pentobarbital (20–30 mg/kg i.v.), and perfused through the internal carotid artery (monkeys) or intracardially (rats) with PBS followed by 4% paraformaldehyde in PBS. All brains were placed in fixative at 4°C for 24 hr, then transferred to PBS for 6 hr, and embedded in 30% sucrose in PBS.
Dissection of Brain Areas Selected.
Coronal blocks of 0.5 cm were taken through the following areas of the nonhuman primate brains by using the Rhesus monkey brain atlas of Paxinos et al. (21) as a guide: anterior cingulate cortex (CingC; area 24, figure 14) prefrontal cortex (PFC; area 9/L, figure 14), and primary visual cortex (VC; area V1, figure 115).
Antibodies.
The mouse mAb Reln 142 (1:500), which was raised against the N-terminal region (amino acid residues 40–189), was a generous gift from A. Goffinet (Fac Univ Notre-Dames de la Paix School of Medicine, Namur, Belgium); the rabbit polyclonal antibody DAB1 B3 (1:500) was a generous gift from B.W. Howell (National Institutes of Health, Bethesda, MD); and the sheep antiserum 1440 to GAD67 (1:1500) was provided by the National Institutes of Health, developed by I.J. Kopin, W. Oertel, D.E. Schmechel, and M. Tappaz. Mouse anti-neuronal nuclei (NeuN, 1:500), and rabbit antiserum raised against a peptide corresponding to the cytosolic loop of the C-terminal domain of integrin α3-subunit (1:250) were purchased from Chemicon.
Immunolabeling for Light Microscopy.
Reln immunostaining.
For Reln immunostaining, sections were first mounted on superfrost slides, air-dried, microwaved (2 min in citrate buffer, pH 6.0), and immunostained as described (8).
NeuN immunostaining.
Pilot studies were conducted in which section thickness and incubation times were varied to establish the maximum thickness of section that could accurately be used for neuronal quantification. Counterstaining with the neutral red cell stain revealed that, at a maximum of 40 μm and a primary incubation time of 3 days, all neurons are labeled positive for the neuron-specific nuclear protein. All subsequent steps were performed as described (8).
Integrin α3-subunit immunostaining.
All steps were performed as described (8), except that a 2-day incubation time was necessary to obtain immunolabeling for the α3 subunit of integrin.
Immunolabeling for Electron Microscopy.
For single Reln labeling, 40-μm sections were cut with a cryostat and immunolabeled for Reln following our previously described method, except that for electron microscopy, a higher concentration of Reln 142-antibody (1:250), but neither antigen-retrieval technique nor detergent were used. For double-labeling of integrin α3 and Reln, sections first were labeled for integrin α3 by ImmunoGold, and then labeled for Reln with diaminobenzidine. In this case, sections were ImmunoGold-labeled following the method described (22), except that the gold particles were enhanced only for 5–6 min. Sections then underwent Reln-immunostaining as described (8). After immunostaining, sections were postfixed for 1 hr in 1% osmium tetroxide in 0.1 M cacodylate buffer, dehydrated, and flat-embedded in Epon. Ultrathin sections (60–80 nm) from the surface of the embedded cryostat section were mounted onto copper mesh grids, dried, and examined and photographed without any counterstaining by using a Zeiss 902 electron microscope.
Double-Immunofluorescent Labeling of Reln and GAD67 for Confocal Microscopy.
Twenty-micrometer sections were mounted on slides, air-dried for 3 hr, and rinsed in TBS and TBS with 0.05% Triton (TBST). Sections then were blocked with 4% normal goat serum (NGS) and 4% normal rabbit serum (NRS) in TBST for 30 min. Sections then were simultaneously incubated with both Reln 142 (1:250) and GAD1440 primary antibodies diluted in 4% NGS/NRS in TBST, overnight at room temperature. After TBS rinses, sections were incubated with biotinylated anti-sheep (1:250, Vector Laboratories) diluted in 4% NGS/NRS in TBST. Sections were rinsed (TBS) and blocked with 0.5% NEN blocking solution (TNB; NEN Life Science) for 30 min. Sections then were incubated for 1 hr in anti-mouse horseradish peroxidase (1:200, NEN Life Science) and rinsed in TBS. Sections then were incubated for 10 min in fluorescein-conjugated tyramide diluted (1:50) in amplification diluent (NEN Life Science) and rinsed in TBS followed by TNB. Finally, sections were incubated for 30 min in Texas Red-conjugated streptavidin (1:500) diluted in TNB, rinsed, and cover-slipped with Vectashield antifading agent (Vector Laboratories). Sections were examined and images were produced with a Leica TCS-NT laser confocal microscope.
Three-Dimensional Stereological Computer-Assisted Counting Method.
To quantify the number of Reln-positive cells, as well as the number of neurons in each layer (NeuN-positive cells) in the three cortical areas of the three nonhuman primate species, we used 40-μm coronal sections and a variation of the three-dimensional cell-counting method described by Rajikowska, adapted from Williams and Rakic (23). The volumetric cell counting was based on the optical dissector method using laser confocal microscopy at a magnification of ×40 that allows counting the number of cells coming into view (or alternatively disappearing from view) as one focuses through a known depth (z) of the tissue. In each of the three brain areas (PFC, CingC, VC), cell number was counted in two randomly selected columns in each of four brain sections (every fourth 40-μm section) from each animal for a total of eight columns per cortical area per animal. In each of these columns, the number of cells was counted in each cortical lamina by using three-dimensional counting boxes (100 μm × 100 μm × z μm). The position of the top and bottom sides of the box and the scan from top to bottom are determined by the laser confocal microscope. Cells located completely inside the counting box and those that cross the top, right, or back sides are counted, whereas those partially inside the box but crossing other planes are excluded. Cell counting is performed in sections from different subjects taken at the same level as ascertained by comparison with stereotaxic atlas. Confocal image stacks were analyzed with the stereo investigator confocal software program (Microbrightfield, Colchester, VT). Preliminary experiments using just a few sections allow the program to determine the number of samples that is necessary to obtain statistically reliable stereological counts.
Results
Cortical Cell Counts.
Using the computer-assisted stereological counting method described in the previous section, we evaluated neuronal density expressed in the PFC, CingC, and VC of patas, macaque, and baboon, three nonhuman primate species. Immunostaining with an antibody raised against a NeuN was used to specifically label the neurons to be counted. In every species studied, the lowest neuronal density was detected in layer I, while the highest neuronal density was detected in layers II–VI of the VC (Table 1). An example of NeuN and Reln immunostaining in baboon is reported in Fig. 1. It must be noted that two classes of Reln-immunopositive cells can be distinguished. One population is strongly labeled and consists of cells with round or elongated somata, which appear to be more abundant in layer I than in layers II and III (Fig. 1); these cells are only occasionally detected in the deeper layers (i.e., PFC of baboon). The second population of Reln-immunopositive cells have much fainter Reln immunostaining. These cells appear to be pyramidal cells, based on the shape, size, and location of their somata (see Fig. 3C). The counts of Reln-positive cells reported in Table 1 refer to the densely Reln-immunostained round and elongated somata, which appear to have the shape of bitufted (double bouquet, ref. 24), or multipolar GABAergic interneurons (see Fig. 3C). In layer I of the VC of baboons, virtually every neuron is Reln positive, whereas in the analogous layer of macaque and patas, about 70% and 76% of the neurons, respectively, are Reln positive (Table 1). The percentage of Reln-positive neurons sharply decreases in layers II and III. This decline is not only because of the sharp increase in the number of neurons in this layer, but also because of an actual decrease of the number of Reln-positive neurons in these layers (Table 1).
Table 1.
Reln-expressing neurons in three cortical areas of nonhuman primates
| Patas (neurons per mm3 × 103)
|
Macaque (neurons per mm3 × 103)
|
Baboon (neurons per mm3 × 103)
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PFC | CingC | VC | PFC | CingC | VC | PFC | CingC | VC | ||
| I | RELN | 7.8 ± 0.73 | 9.7 ± 0.87 | 5.0 ± 0.66 | 5.9 ± 0.45 | 8.1 ± 1.13 | 7.5 ± 0.47 | 6.3 ± 0.39 | 9.1 ± 0.81 | 7.5 ± 0.45 |
| NeuN | 22.8 ± 2.61 | 15.3 ± 1.67 | 6.6 ± 0.92 | 20.0 ± 1.16 | 9.4 ± 1.40 | 10.6 ± 0.78 | 15.3 ± 104 | 13.3 ± 1.22 | 5.5 ± 0.61 | |
| % | (34.2) | (63.3) | (75.8) | (30.0) | (86.7) | (70.6) | (41.2) | (68.2) | (100) | |
| II | RELN | 2.8 ± 0.31 | 2.5 ± 0.66 | 2.5 ± 0.66 | 2.8 ± 0.31 | 2.5 ± 0.47 | 1.9 ± 0.62 | 3.3 ± 0.29 | 3.9 ± 0.50 | 3.1 ± 0.48 |
| NeuN | 72.8 ± 2.81 | 54.1 ± 3.13 | 113.4 ± 3.98 | 56.8 ± 1.66 | 53.4 ± 2.3 | 145.6 ± 5.53 | 49.1 ± 1.85 | 64.5 ± 1.42 | 88.4 ± 3.95 | |
| % | (3.9) | (4.6) | (2.2) | (5.0) | (4.7) | (1.3) | (6.7) | (6.1) | (3.5) | |
| III | RELN | 0.62 ± 0.40 | 1.3 ± 0.66 | 0.3 ± 0.31 | 1.3 ± 0.47 | 1.6 ± 0.45 | 0.62 ± 0.40 | 1.71 ± 0.37 | 1.1 ± 0.39 | 0.93 ± 0.31 |
| NeuN | 55.6 ± 3.95 | 41.6 ± 1.57 | 101.3 ± 3.47 | 42.5 ± 0.95 | 37.2 ± 1.80 | 87.5 ± 4.91 | 32.0 ± 2.02 | 39.1 ± 1.42 | 84.1 ± 2.26 | |
| % | (1.1) | (3.0) | (0.3) | (2.9) | (4.2) | (0.7) | (5.4) | (2.8) | (1.1) | |
| IV | RELN | 0.31 ± 0.31 | 0 | 0 | 0.31 ± 0.31 | 0 | 0.31 ± 0.31 | 1.3 ± 0.51 | 0.15 ± 0.15 | 0.15 ± 0.15 |
| NeuN | 69.1 ± 4.41 | 24.7 ± 2.73 | 1.20 ± 5.41 | 63.8 ± 4.48 | 33.1 ± 2.98 | 114.4 ± 6.14 | 58.6 ± 2.04 | 35.5 ± 1.65 | 86.6 ± 2.40 | |
| % | (0.5) | (0) | (0) | (0.5) | (0) | (0.3) | (2.1) | (0.5) | (0.2) | |
| V | RELN | 0.31 ± 0.31 | 0 | 0 | 0 | 0 | 0 | 0.31 ± 0.21 | 0 | 0 |
| NeuN | 63.4 ± 5.30 | 42.2 ± 2.08 | 103.4 ± 5.87 | 46.6 ± 2.83 | 50.3 ± 3.58 | 81.9 ± 3.65 | 42.2 ± 1.98 | 64.7 ± 2.82 | 81.6 ± 3.78 | |
| % | (0.4) | (0) | (0) | (0) | (0) | (0) | (0.7) | (0) | (0) | |
| VI | RELN | 0 | 0 | 0 | 0 | 0 | 0 | 0.31 ± 0.21 | 0 | 0 |
| NeuN | 47.5 ± 1.77 | 42.8 ± 2.29 | 127.5 ± 4.23 | 42.2 ± 2.85 | 37.8 ± 2.19 | 101.2 ± 5.26 | 33.8 ± 1.72 | 38.1 ± 10.5 | 66.1 ± 2.48 | |
| % | (0) | (0) | (0) | (0) | (0) | (0) | (0.9) | (0) | (0) | |
Mean ± SEM number of Reln-expressing neurons (first row; bold), mean ± SEM number of NeuN-positive neurons (middle row), and percentage of Reln-expressing neurons (in parentheses) relative to the total number of neurons in cortical layers I–VI of prefrontal cortex (PFC), anterior cingulate cortex (CingC), and visual cortex (VC) of the patas monkey, macaque, and baboon. For definition of the area see Materials and Methods. RELN, Reln-expressing neurons; NeuN, NeuN-positive cells; %, Reln-expressing neurons/NeuN-positive cells.
Figure 1.

Photomicrographs of 20-μm sections showing the distribution of NeuN- and Reln-expressing cells in the PFC, CingC, and VC of the baboon. Note that immunostaining for Reln strongly labels a small population of cells predominantly located in layer I. In addition to these Reln-expressing cells, there appears to be a faint immunolabeling of Reln on many pyramidal cells in deeper cortical layers. (Calibration bar = 200 μm.)
Figure 3.

Photomicrographs showing immunolabeling for GAD67 (A), Dab1 (B), Reln (C), and integrin α3 subunit in the patas PFC. (A) Immunostaining for GAD67 reveals a GAD67-positive GABAergic cell (solid arrow) as well as numerous punctate GAD67-positive terminal endings in the neuropil and surrounding excitatory pyramidal cells (open arrows) of layer V. (B) Immunostaining for Dab1 in layer V shows that it is expressed in excitatory pyramidal cells. (C) Immunostaining for Reln reveals cells that appear to contain Reln, such as the GABAergic bitufted cell indicated with a solid arrow in layer III, as well as many pyramidal cells that appear to have a light dusting of Reln (open arrows). (D) Photomicrograph of layer V showing that the integrin α3 subunit appears to be well expressed on pyramidal cells and their apical dendrites. (Calibration bar = 20 μm.)
Coexistence of Reln and GAD67.
Using confocal microscopy and different fluorescent labels for Reln and GAD67 in layers I and II of baboon PFC, we found that cells that express Reln also express GAD67 (Fig. 2). The colocalization of GAD67 and Reln is demonstrated in the overlay of Fig. 2. Very likely these cells are bitufted (double bouquet) or multipolar GABAergic interneurons. This colocalization was verified in layers I and II of the three cortical areas of the three species of the nonhuman primates. Because axon terminals from GABAergic interneurons impinge upon the somata and the initial segments of pyramidal neuron apical dendrites, this input may be responsible for the light Reln immunostaining observed in the pyramidal neurons shown in Fig. 3 A and C. Thus, the weaker immunostaining in the second population of cells is possibly because of the innervation of pyramidal neurons by GABAergic cell axons expressing high-density Reln immunoreactivity. Hence, despite the appearance, one can argue that there is only one population of cortical cells that store Reln, and these are the GABAergic cells. In fact, we could never find pyramidal neuron expressing either GAD67 or Reln mRNA (8, 9).
Figure 2.

Confocal microscope images showing the double labeling for Reln and GAD67 in layers I and II in 20-μm sections from the PFC of baboon brain. (Left) The expression of Reln (fluorescein; green) in the somata and proximal dendrite of three cells. (Middle) The expression of GAD67 (Texas Red; red) in the same section. The colocalization frequency between Reln and GAD67 (Right, Overlay) illustrates that Reln appears to be expressed in GABAergic cells in the baboon PFC. (Calibration bar = 20 μm.)
Immunostaining for Dab1 and Integrin α3 Subunits.
Fig. 3B shows the immunostaining for Dab1 in a pyramidal neuron of layer V from the PFC of a patas monkey, whereas Fig. 3D shows two pyramidal neurons immunopositive for the α3 integrin subunit. In both cases, the immunostaining is evident in the soma and proximal apical dendrites. Similarly, pyramidal neurons in various layers of the different cortical areas also expressed both the adapter protein Dab1 and the integrin α3 subunit. On the basis of these findings, we proceeded to investigate whether we could find evidence of a possible interaction between Reln and integrins at the dendritic spine level.
Reln in ECM and Its Synaptic Colocalization with Integrin α3 Subunits.
In the frontal cortex of rats, we described the presence of Reln immunoreactive halos in proximity of apical dendritic spines (6, 8, 9). Here, we show that, in rat, Reln immunoreactivity of the cortical neuropil is found in proximity of postsynaptic domains of dendritic spines facing unidentified axon terminals in layers I and II (Fig. 4A). Also, in the histological preparations shown in Fig. 1, there are Reln-immunoreactive halos in layers I and II of the three cortical areas of three species of nonhuman primates. The electronmicrographs of ultrathin sections of patas PFC show that Reln-immunolabeling is present in the postsynaptic dendritic spines of nonhuman primates, which appear to be innervated by a presynaptic button containing an array of synaptic vesicles (Figs. 4B and 5A). It was not possible to definitely identify such terminals as GABAergic, but because Reln is mostly located in GABAergic neurons (Fig. 2), we suggest that these presynaptic buttons are very likely GABAergic. The Reln immunoreactivity shown in Figs. 4A and 5A is concentrated in the postsynaptic density and surrounds the dendritic spine plasma membrane. Fig. 5B shows the double-immunolabeling of Reln and the integrin α3 subunit in a dendrite and a dendritic postsynaptic spine. Fig. 5 B and C clearly demonstrates the contiguity of Reln immunoreactivity with the gold immunolabeling of the α3 subunits of integrin receptors. Hence, these results support the hypothesis that Reln diffusing into the ECM of the cortical neuropil once released from the Reln-containing GABAergic interneurons reaches the dendritic spines of pyramidal neurons where integrin receptors are located.
Figure 4.

Electron micrographs of ultrathin sections taken through the superficial layers of patas (A) and rat (B) PFC, immunostained for Reln. In both patas and rat, the immunoperoxidase reaction product in dendritic spines is concentrated in the area of the postsynaptic densities. (Magnification, ×50,000.)
Figure 5.
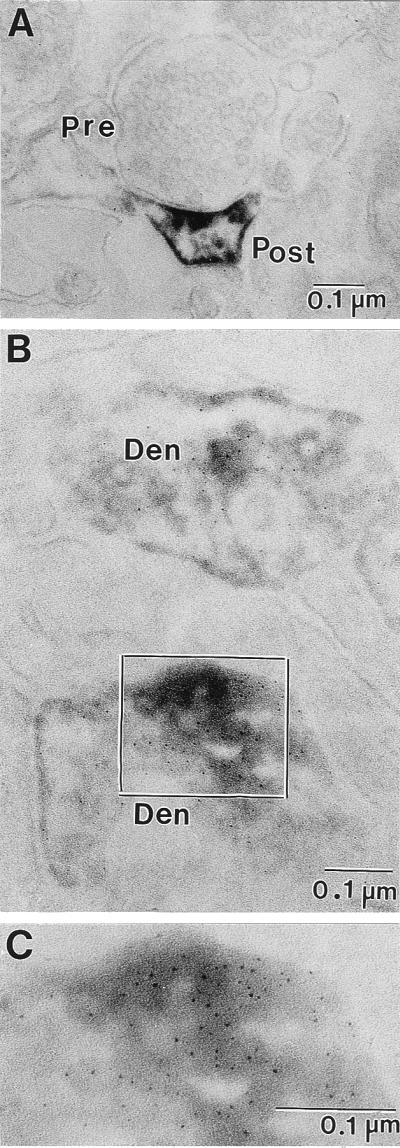
Electron micrographs of ultrathin sections of Reln immunolabeling in the patas PFC layers I and II. (A) Immunoperoxidase reaction product in a dendritic spine is concentrated in the area of the postsynaptic density as well as the surrounding dendritic plasma membrane. (B) Double-immunolabeling showing the colocalization of Reln (immunoperoxidase-diaminobenzidine) and integrin α3-subunit (silver-enhanced 1-nm ImmunoGold) in ultrathin sections from patas PFC layers I and II. Both dendritic processes present in this electron micrograph are immunopositive for Reln and integrin α3-subunit. (C) Higher magnification of the area outlined in B. Pre, presynaptic bouton with synaptic vesicles; Post, postsynaptic spine; Den, dendritic process. (Magnification: A = ×75,000; B = ×125,000; C = ×212,500.)
Because most pyramidal neurons were found to express Dab1 (Fig. 3B), one can suggest that a Reln transduction pathway including Dab1 protein may be operative in adult brain cortex of nonhuman primates.
Discussion
In the cortex of adult rat, Reln is very likely secreted from GABAergic interneurons (bitufted, horizontal, and multipolar cells) (8, 9, 25) into the ECM by a constitutive mechanism requiring a specific signal peptide expressed at the carboxy-terminal domain of Reln. It is important to stress that Reln is not contained in synaptic vesicles and that such secretion occurs in absence of transmitter regulation and does not require Ca2+ (10).
To extrapolate from rat to human, it would be necessary to show that Reln also colocalizes with GAD67 in bitufted or horizontal GABAergic neurons in the human brain cortex. Unfortunately, such studies could not be carried out in human postmortem brain because the poor preservation of postmortem tissue prevents electron microscopic analyses. For such study, the brain must be fixed in situ by means of perfusion, immediately after death; thus, we turned our attention to perfused brains of three species of nonhuman primates. The particular proximity of these species to humans in the evolutionary tree allows us to predict whether a colocalization of Reln and GAD67, similar to that described in rats, can be expected in humans. The results obtained replicate in nonhuman primates brain cortex our previous findings in rats (8, 9), showing GAD67 and Reln colocalization in GABAergic cells. Because in postmortem brain of psychotic patients and nonpsychiatric subjects, Reln-immunopositive neurons are located more abundantly in layer I (6), it is now plausible to extrapolate that, in human brain cortex, Reln also may localize in GABAergic neurons and can be secreted into the ECM by a constitutive mechanism operative in the proximity of apical dendritic spines of pyramidal neurons.
The Reln-immunoreactive cell counts listed in Table 1 clearly show that strongly Reln-immunoreactive cells are located more abundantly in layer I, but are also present in layers II and III of the PFC, CingC, and VC of the three species under study. In contrast, in all three species, the neuronal density labeled with NeuN antibody is significantly lower in layer I than in the other layers. In agreement with previous studies (26, 27), done without computer-assisted technologies and NeuN labeling, the VC, in all three species, has the highest density of NeuN-positive neurons.
In Fig. 1, one can notice that, in addition to strongly stained immunoreactive Reln cells, faintly Reln-immunopositive cells are abundant in layers III and IV. These are presumably somata of pyramidal neurons that are innervated by GAD67-immunoreactive terminal endings (Fig. 3A) containing Reln (Fig. 3C), which is expressed in GABAergic neurons (Fig. 2). In rats, we have shown that cortical pyramidal neurons are also weakly immunopositive for Reln, but are devoid of Reln mRNA (8, 9). Thus, it can be inferred that the axon terminals of GABAergic cells, impinging upon the somata of pyramidal neurons are responsible for the weak Reln immunostaining observed in these cells.
The findings of the present work pertinent to the function of spines of pyramidal cell apical dendrites are: (i) the rat and primate brain cortices express Reln immunoreactivity in conjunction with postsynaptic densities located in apical dendritic spines; (ii) the α3 subunit of integrin receptors and the Dab1 adapter protein are expressed in primate pyramidal neurons of layers II–VI; (iii) the colocalization of Reln and clusters of α3 integrin receptor subunits in postsynaptic densities can be interpreted as an evidence that Reln is a putative endogenous ligand for integrin receptors (Fig. 5); and (iv) so far, these synapses are found located in the neuropil (layers I and II).
Taken together, this evidence supports the model of a Reln-integrin pathway activating the phosphorylation of Dab1 protein in dendritic spines and the modulation of the expression of cytoskeleton proteins. This finding adds significance to the reports that the knockout of α3 integrin receptor subunit is associated with the appearance of anatomical and behavioral phenotypes similar to those found in reeler−/− mice (12).
The innervation by GABAergic neurons of the postsynaptic densities expressed in the spines of cortical layers I and II is presently under investigation for a putative role in learning. In fact, we now know that the presence of dendritic protein synthesis is important to establish a long-term increase of synaptic strength associated with learning (28, 29). The dendritic spines are, in fact, the site where duplication of synaptic spines associated with long-term potentiation (LTP) and learning may occur (30, 31) and are also the site where the stimulation of protein synthesis associated with LTP and learning may take place (28, 29). Studying the function of the gene product (FMRP) lacking in the fragile X mental retardation, Greenough and colleagues (28, 29) have shown that this protein is synthesized at synapses in response to glutamate receptor activation, a process that facilitates learning and regulates LTP induction. FMRP, in turn, catalyzes the postsynaptic assembly of polyribosomes, targeting newly synthesized mRNA to synapses, in a process that may be essential to normal maturation and pruning of synaptic spines. Is this process in part also regulated by Reln-integrin interaction?
The function of dendrite spines, which may be postsynaptic to inhibitory inputs, in the mammalian central nervous system, is still not well understood. Although changes in spine morphology may mediate synaptic plasticity, the extent of basal spine motility and its regulation and function remains controversial in its interpretation. However, the spine dynamic is neurodevelopmentally regulated in both cortex and cerebellum (Purkinje cells). In this regard, we point out that Reln is synthesized in glutamatergic cerebellar granule cells and acts on Purkinje cell dendritic spines during development. It is possible that both in cerebellum and cortex the spine motility has a function that is regulated by a Reln interaction with integrin receptors that may modulate changes in cytoskeleton function. In fact, blockade or induction of neuronal activity fails to change spine motility (32), but dysfunction of actin polymerization abates spine motility (32). It would seem likely that if spine motility is an intrinsic feature of the central nervous system plasticity, this neuronal property also may be regulated by dynamic aspects of cytoskeleton function. This spine motility may turn out to be important in regulating brain function as an adjunct element that changes the value of excitability independently from afferent stimulation.
Recently, a decrease of dendritic spine density has been reported to exist in the neocortex of schizophrenia patients (33, 34). This observation allows us to give a functional perspective of the Reln and GAD67 deficit we detected in the cortex, hippocampus, and cerebellum of schizophrenia and bipolar disorder postmortem brains.
Is psychosis expressed in schizophrenia and bipolar disorder patients related to a defect of dendritic spine dynamics? This is a question that may be addressed by studying the role of Reln in apical dendritic spine motility and protein synthesis.
Acknowledgments
We thank Dr. Floyd E. Bloom, Department of Neuropharmacology, The Scripps Research Institute, La Jolla, CA, for constructive criticisms and suggestions in the preparation of the manuscript. This work was supported by National Institutes of Health Grant 56500 (to E.C.), National Institutes of Health Grant 49-486 (to A.G.), and a University of Illinois, Chicago, Campus Research Board grant (to C.P.).
Abbreviations
- Reln
reelin
- GAD67, glutamic acid decarboxylase 67
ECM, extracellular matrix
- GABA
γ-aminobutyric acid
- PFC
prefrontal cortex
- CingC
anterior cingulate cortex
- VC
primary visual cortex
- NeuN
neuronal nuclei
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.050589797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.050589797
References
- 1.D'Arcangelo G, Curran T. Bioessays. 1998;20:235–244. doi: 10.1002/(SICI)1521-1878(199803)20:3<235::AID-BIES7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 2.Curran T, D'Arcangelo G. Brain Res Rev. 1998;26:285–294. doi: 10.1016/s0165-0173(97)00035-0. [DOI] [PubMed] [Google Scholar]
- 3.DeSilva U, D'Arcangelo G, Braden V V, Chen J, Miao G G, Curran T, Green E D. Genome Res. 1997;7:157–164. doi: 10.1101/gr.7.2.157. [DOI] [PubMed] [Google Scholar]
- 4.Lambert de Rouvroit C, Goffinet A M. Adv Anat Embryol Cell Biol. 1998;150:1–106. [PubMed] [Google Scholar]
- 5.D'Arcangelo G, Miao G G, Chen S C, Soares H D, Morgan J L, Curran T. Nature (London) 1995;347:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 6.Impagnatiello F, Guidotti A, Pesold C, Dwivedi Y, Caruncho H, Pisu M G, Uzunov D P, Smalheiser N, Davis J, Pandey G, et al. Proc Natl Acad Sci USA. 1998;95:15718–15723. doi: 10.1073/pnas.95.26.15718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akbarian S, Kim J J, Potkin S G, Hagman J O, Tafuzzoli A, Bunney W E, Jr, Jones E G. Arch Gen Psychiatry. 1995;52:258–266. doi: 10.1001/archpsyc.1995.03950160008002. [DOI] [PubMed] [Google Scholar]
- 8.Pesold C, Impagnatiello F, Pisu M G, Uzunov D P, Costa E, Guidotti A, Caruncho H J. Proc Natl Acad Sci USA. 1998;95:3221–3226. doi: 10.1073/pnas.95.6.3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pesold C, Liu W S, Guidotti A, Costa E, Caruncho H J. Proc Natl Acad Sci USA. 1999;96:3217–3222. doi: 10.1073/pnas.96.6.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lacor, P. N., Grayson, D. R., Auta, J., Sugaya, I., Costa, E. & Guidotti, A. (March 21, 2000) Proc. Natl. Acad. Sci. USA, 10.1073/pnas.050589597. http://www.pnas.org/cgi/doi/10.1073/pnas.050589597 [DOI] [PMC free article] [PubMed]
- 11.Giancotti F G, Rouslahti E. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 12.Anton E S, Kreidberg J A, Rakic P. Neuron. 1999;22:277–289. doi: 10.1016/s0896-6273(00)81089-2. [DOI] [PubMed] [Google Scholar]
- 13.Rice D S, Curran T. Genes Dev. 1999;13:2758–2773. doi: 10.1101/gad.13.21.2758. [DOI] [PubMed] [Google Scholar]
- 14.Howell B W, Hawkes R, Soriano P, Cooper J A. Nature (London) 1997;389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- 15.Gilmore E C, Ohshima T, Goffinet A M, Kulkarni A B, Herrup K. J Neurosci. 1998;18:6370–6377. doi: 10.1523/JNEUROSCI.18-16-06370.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwon Y T, Tsai L H. J Comp Neurol. 1998;395:510–522. doi: 10.1002/(sici)1096-9861(19980615)395:4<510::aid-cne7>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 17.Howell B W, Lanier L M, Frank R, Gertler F B, Cooper J A. Mol Cell Biol. 1999;19:5179–5188. doi: 10.1128/mcb.19.7.5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Senzaki K, Ogawa M, Yagi T. Cell. 1999;99:635–647. doi: 10.1016/s0092-8674(00)81552-4. [DOI] [PubMed] [Google Scholar]
- 19.Hiesberger T, Trommsdorff M, Howell B W, Goffinet A, Mumby M C, Cooper J A, Herz J. Neuron. 1999;24:481–489. doi: 10.1016/s0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- 20.D'Arcangelo G, Homayouni R, Keshvara L, Rice D S, Sheldon M, Curran T. Neuron. 1999;24:471–479. doi: 10.1016/s0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- 21.Paxinos G, Huang X F, Toga A W. The Rhesus Monkey Brain in Stereotaxic Coordinates. San Diego: Academic; 2000. [Google Scholar]
- 22.Pesold C, Caruncho H J, Impagnatiello F, Berg M J, Fritschy J M, Guidotti A, Costa E. Neuroscience. 1997;79:477–487. doi: 10.1016/s0306-4522(96)00609-4. [DOI] [PubMed] [Google Scholar]
- 23.Williams R W, Rakic P. J Comp Neurol. 1998;278:344–352. doi: 10.1002/cne.902780305. [DOI] [PubMed] [Google Scholar]
- 24.Somogyi P, Tamas G, Lujan R, Buhl E H. Brain Res Rev. 1998;26:113–135. doi: 10.1016/s0165-0173(97)00061-1. [DOI] [PubMed] [Google Scholar]
- 25.Alcantara S, Ruiz M, D'Arcangelo G, Ezan E, de Lecea L, Curran T, Sotelo C, Soriano E. J Neurosci. 1998;18:7779–7799. doi: 10.1523/JNEUROSCI.18-19-07779.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rockel A J, Hiorns R W, Powell T P. Brain. 1980;103:221–244. doi: 10.1093/brain/103.2.221. [DOI] [PubMed] [Google Scholar]
- 27.O'Kusky J, Colonier M. J Comp Neurol. 1982;210:278–290. doi: 10.1002/cne.902100307. [DOI] [PubMed] [Google Scholar]
- 28.Weiler I J, Irwin S A, Klintsova A Y, Spencer C M, Brazetlton A D, Miyashiro K, Comery T A, Patel B, Eberwine J, Greenough T. Proc Natl Acad Sci USA. 1997;94:5395–5400. doi: 10.1073/pnas.94.10.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klintsova A Y, Greenough W T. Curr Opin Neurobiol. 1999;9:203–208. doi: 10.1016/s0959-4388(99)80028-2. [DOI] [PubMed] [Google Scholar]
- 30.Toni N, Buchs P A, Nikonenko I, Bron C R, Muller D. Nature (London) 1999;402:421–425. doi: 10.1038/46574. [DOI] [PubMed] [Google Scholar]
- 31.Maletic-Savatic M, Malinow R, Svoboda K. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- 32.Dunaevsky A, Tashiro A, Majewska A, Mason C, Yuste R. Proc Natl Acad Sci USA. 1999;96:13438–13443. doi: 10.1073/pnas.96.23.13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garey L J, Ong W Y, Patel T S, Kanani M, Davis A, Mortimer A M, Barnes T R E, Hirsch S R. J Neurol Neurosurg Psychiatry. 1998;65:446–453. doi: 10.1136/jnnp.65.4.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glantz L A, Lewis D A. Arch Gen Psych. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]