

Abstract
The mammalian suprachiasmatic nucleus (SCN) is a master circadian pacemaker. It is not known which SCN neurons are autonomous pacemakers or how they synchronize their daily firing rhythms to coordinate circadian behavior. Vasoactive intestinal polypeptide (VIP) and the VIP receptor VPAC2 (encoded by the gene Vipr2) may mediate rhythms in individual SCN neurons, synchrony between neurons, or both. We found that Vip−/− and Vipr2−/− mice showed two daily bouts of activity in a skeleton photoperiod and multiple circadian periods in constant darkness. Loss of VIP or VPAC2 also abolished circadian firing rhythms in approximately half of all SCN neurons and disrupted synchrony between rhythmic neurons. Critically, daily application of a VPAC2 agonist restored rhythmicity and synchrony to VIP−/− SCN neurons, but not to Vipr2−/− neurons. We conclude that VIP coordinates daily rhythms in the SCN and behavior by synchronizing a small population of pacemaking neurons and maintaining rhythmicity in a larger subset of neurons.
The SCN of the mammalian hypothalamus coordinates diverse daily rhythms, including states of vigilance, locomotor activity and hormonal release, through rhythms in neuronal firing1. These rhythms ‘free-run’ with a circadian period in the absence of synchronizing (or entraining) cues such as environmental light cycles. When the SCN are electrically silenced or lesioned, behavioral and physiologic rhythms disappear2.
Rhythmic circadian firing within the SCN is dependent on cyclic expression of a family of ‘clock genes’. Mutations of period 1 (Per1) or Per2, cryptochrome 1 (Cry 1) or Cry2, casein kinase Iε (Csnk1e), RevErbα (Nr1d1), BMAL1 (MOP3, Arntl) or clock lead to altered or abolished circadian periodicity3. These results have led to a model in which circadian rhythms are generated and sustained by an intracellular transcription-translation negative feedback loop. In support of this model for cell-autonomous pacemaking, single SCN neurons dispersed at low density onto a multielectrode array (MEA) can express firing rate patterns with different circadian periods4, leading to the suggestion that all 20,000 SCN neurons are autonomous circadian pacemakers4-6. In the intact SCN, these neurons usually synchronize to one another with defined phase relationships7-10. How synchrony is maintained between SCN neurons is unclear, although GABA11 and gap-junction communication12-14 have been proposed as candidate mediators.
Recent evidence indicates that without intercellular communication, not only is synchrony between rhythmic SCN neurons lost, but at least some neurons lose rhythmicity. For example, when action potentials are blocked with tetrodotoxin (TTX), Period1 promoter–driven luciferase expression rhythms among SCN neurons desynchronize, and many neurons' rhythms damp out7. Further, whereas over 75% of neurons express circadian firing patterns in SCN explants15,when SCN neurons are dispersed and plated at lower densities, this percentage decreases and neuronal rhythms become less stable4,16. These data raise new questions about which SCN neurons are cell-autonomous circadian pacemakers and which require neurotransmission for rhythmicity.
VIP, a peptide neurotransmitter expressed by a subset (9–24%) of SCN neurons4,17, is well-positioned for a role in rhythmicity, synchrony or both. VIPergic neurons project densely throughout the SCN. A receptor for this peptide, VPAC2 (also known as Vipr2), is highly expressed in the SCN, with roughly 60% of SCN neurons responding to VIP with changes in firing rate18-20. Similar to environmental light, VIP or a VPAC2 agonist can phase-shift circadian rhythms of behavior and of firing rate, gene expression, and neuropeptide release across the SCN21-24. Many VIPergic neurons receive synapses containing VIP, suggesting reciprocal VIP signaling between neurons25,26. Mice over-expressing the VPAC2 receptor entrain more quickly to a shift in their light schedule and express a shortened period in constant darkness27, and mice lacking VPAC2 show markedly diminished or abolished behavioral rhythms in constant darkness28,29. Vipr2−/− SCN neurons show attenuated tissue-level rhythms in clock gene expression or ensemble firing rate18,29. Similarly, Vip−/− mice show diminished circadian behavior in constant darkness30 and lack normal SCN rhythms in inhibitory signaling31. VPAC2 antagonists block multiunit firing rhythmicity in wild-type SCN slices18. This has been interpreted to mean that a lack of VIP/VPAC2 signaling led to arrhythmicity of individual SCN neurons. However, the behavioral and SCN phenotypes of mutant mice could also result from desynchrony among rhythmic SCN neurons.
To determine the role of VIP within the SCN, we analyzed long-term recordings of behavior in constant darkness and recorded firing rates of individual neurons from Vip−/−, Vipr2−/− and wild-type SCN. Both mutations desynchronized most animals' behavioral rhythms into multiple low-amplitude circadian periods. Consistent with this, both mutations markedly reduced the percentage of rhythmic SCN neurons and eliminated synchrony between them. Notably, rhythmicity and synchrony were restored to Vip−/− neurons by daily application of a VPAC2 agonist. Our data show that many SCN neurons require VIP for rhythmicity, whereas others require it for synchrony. We conclude that a minority of SCN neurons are cell-autonomous circadian pacemakers, which coordinate rhythms in the majority through VIP.
RESULTS
Mice lacking VIP or VPAC2 show multiple circadian periods
Previous studies of locomotor activity in Vip−/− and Vipr2−/− mutant mice have shown either greatly reduced29 or multiple circadian periods in constant conditions28,30. This left unresolved whether VIP/VPAC2 signaling is responsible for rhythm generation or for synchronization among circadian pacemakers. To better clarify the behavioral phenotypes of these mice, we recorded running wheel activity from Vip−/−, Vipr2−/− and wild-type mice of the same genetic background (C57Bl/6). We designed a behavioral paradigm with three separate conditions to show deficits in the entrainment, generation or synchronization of circadian rhythms. In a 12 h/12 h light/dark cycle, activity of all wild-type and mutant mice was primarily nocturnal as previously reported (Fig. 1a,c). To test whether this rhythmic activity was entrained or merely suppressed by light, mice were then exposed to a skeleton photoperiod, consisting of two 11-h dark periods separated by 1-h light periods. Whereas wild-type mice consolidated an average of 80% of their total activity at the time corresponding to night in the 12 h/12 h light/dark cycle, Vip−/− and Vipr2−/− mice restricted only 40% of their activity to this period, showing roughly equal activity in both daily dark phases (Fig. 1; P < 0.0001, one-way ANOVA with post hoc Scheffé test). Consistent with the reported dissociation of activity in Vip−/− mice in a skeleton photoperiod30 and impaired responses to light pulses in Vipr2−/− mice28, these data indicate that loss of either VIP or VPAC2 similarly disrupts the normal entrainment and synchronization of circadian behavior.
Figure 1.

Mice with disrupted VIP/VPAC2 signaling express multiple circadian periods. (a,c) Double-plotted actograms showing representative wheel-running activity of wild-type (C57Bl/6), Vip−/− and Vipr2−/− mice in a light/dark schedule (days 1–10), a skeleton photoperiod (days 11–20), and constant darkness (days 20–90). Gray shading indicates the time when lights were on. VIP and VPAC2 mutant mice appeared normal while on a 12 h/12 h light/dark schedule but were active during both dark phases each day in a skeleton photoperiod, whereas wild-type mice were strictly nocturnal. (b,d) Peaks in the corresponding χ2 periodogram for each animal show the dominant and secondary period(s) during days 21–90 in constant darkness. Peaks above the diagonal line (indicating the 99.9% confidence level) between 16 and 34 h were considered significant circadian periods. Note that mutants in a and b (representing two-thirds of mutants of both genotypes) expressed multiple circadian periods, whereas those in c and d (representing one-third of mutants) showed more coherent circadian behavior, with a single, shorter, free-running period. (e) Mice of all three genotypes expressed significant circadian periodicity. The percentage of mice expressing a single circadian period (gray) was higher in wild-type mice than in the two mutant genotypes, whereas a large percentage of mutant mice expressed multiple circadian periods (black). (f) The circadian amplitude of rhythms was diminished in both mutants relative to wild-type. Bars show circadian amplitudes (mean ± s.e.m.) at the dominant period as determined by χ2 periodogram analysis. Asterisk (*) indicates P < 0.00001, one-way ANOVA with Scheffé post hoc test.
We next assessed locomotor activity over 70 d in constant darkness to more clearly define the circadian defect of mutants in free-running conditions. All wild-type mice ran with a single, stable circadian period (Fig. 1e). In contrast, two-thirds of Vip−/− (9 of 14) and Vipr2−/− mice (9 of 15) simultaneously expressed two or more statistically significant circadian periods. Free-running rhythms were also of lower circadian amplitude in mutants than in wild-type mice (Fig. 1f; P < 0.00001, one-way ANOVA with Scheffé test). The average amplitude of rhythmicity at the dominant period, assessed by χ2 periodogram, was 939 ± 159 for Vip−/− mice, 1,162 ± 311 for Vipr2−/− mice and 4,868 ± 403 for wild-type mice (all measurements mean ± s.e.m. unless otherwise noted; see Methods for details). The multiple free-running periods expressed by individual mice with disrupted VIP/ VPAC2 signaling demonstrate that circadian pacemaking persists, but synchronization among pacemakers is severely impaired.
The dominant period of mutant mice (Vip−/−, 22.9 ± 0.2 h; Vipr2−/−, 22.2 ± 0.2 h) was also shorter than that of wild-type mice (23.4 ± 0.05 h; P < 0.05, one-way ANOVA with Scheffé test), consistent with published reports30. When all statistically significant periods expressed by mutant mice were taken into account, however, their distributions were significantly broader than wild-type (P < 0.005, Brown-Forsythe's and Levene's tests for equal variance), but with no difference between their mean periods (P > 0.7, Kruskal-Wallis one-way ANOVA with Scheffé post hoc test). We conclude that without VIP/VPAC2 signaling, mice are capable of expressing circadian rhythms, but that these rhythms are both weaker and less synchronized than wild-type rhythms.
Most SCN neurons require VIP signaling for rhythmicity
To assess the role of VIP/VPAC2 signaling in SCN rhythmicity, we recorded for 5–14 d the spontaneous firing patterns of individual Vip−/−, Vipr2−/−, and wild-type neurons dispersed onto multielectrode arrays. We found that nearly 70% of wild-type mouse SCN neurons (54 of 80 neurons, from five cultures) fired at rates that fluctuated in a circadian pattern (Fig. 2). In contrast, approximately 30% of Vip−/− neurons (67 of 213, from eight cultures) and Vipr2−/− neurons (73 of 267, from eight cultures) expressed circadian firing rate rhythms. A similar proportion of rhythmic neurons was seen in recordings from organotypic slices of Vipr2−/− SCN (29%, 5 of 17, from three slices; data not shown), indicating that rhythmicity in this subset of neurons was not an artifact of the dispersal process. The peak-to-trough amplitudes of firing rate rhythms did not differ between mutant and wild-type SCN neurons (2.5 ± 0.2 Hz for Vip−/−, 2.3 ± 0.2 Hz for Vipr2−/−, and 2.3 ± 0.2 Hz for wild-type; P > 0.7, one-way ANOVA with Scheffé test). However, the circadian amplitudes of firing rhythms as assessed by χ2 periodogram were significantly reduced in Vip−/− and Vipr2−/− SCN neurons (97 ± 14 and 102 ± 13, respectively) relative to wild-type neurons (182 ± 22; P < 0.0005, one-way ANOVA with Scheffé test). The decreased circadian amplitude of mutant neurons is likely to reflect decreased cycle-to-cycle stability of circadian period, which is normally regulated through intercellular signaling in the SCN16.
Figure 2.

A reduced proportion of Vip−/− and Vipr2−/− SCN neurons fires rhythmically in vitro. (a) Representative firing rate traces showing examples of circadian and arrhythmic neurons from each genotype. The χ2 periodogram to the left of each plot shows the period estimation of each neuron as in Figure 1. (b) The percentage of SCN neurons with a circadian firing pattern was reduced for both mutants relative to wild-type. (c) The circadian amplitudes (mean ± s.e.m.) of mutant rhythms were lower than in the wild type. Asterisk (*) indicates P < 0.0005, one-way ANOVA with Scheffé post hoc test.
It has previously been noted that the ensemble firing rate of SCN neurons in Vipr2−/− mutants is reduced at all times of the day, leading to the suggestion that circadian defects may be due to membrane hyperpolarization in mutant SCN neurons18. Long-term recordings from individual SCN neurons, however, showed that the average firing rates of Vip−/− (n = 213) and Vipr2−/− neurons (n = 267) did not differ significantly from the average firing rate of wild-type neurons in pairwise comparisons (n = 80, 1.9 ± 0.2 Hz for Vip−/−, 1.6 ± 0.1 Hz for Vipr2−/−, and 1.5 ± 0.2 Hz for wild-type; P > 0.05, Scheffé post hoc test). We found a weakly significant effect among the three genotypes by ANOVA; mutant neurons' firing rates tended to be higher, not lower, than wild-type (P < 0.04). Within each genotype, rhythmic and arrhythmic neurons had similar respective average firing rates (± s.e.m.) of 2.0 ± 0.3 Hz and 1.8 ± 0.1 Hz for Vip−/−, 1.5 ± 0.1 Hz and 1.7 ± 0.1 Hz for Vipr2−/−, and 1.4 ± 0.3 Hz and 1.5 ± 0.3 Hz for wild-type SCN. The number of neurons recorded per electrode array also did not differ between genotypes (data not shown), suggesting that there is no increase in the proportion of silent neurons in mutant SCN cultures. These data indicate that the decreased proportion of rhythmic neurons in mutant SCN is the result of neurons losing rhythmicity, not a secondary effect of chronically depressed firing.
Rhythmic neurons desynchronize without VIP signaling
We used high-density dispersals (> 10,000 cells mm−2) from mutant and wild-type SCN to assess the role of VIP/VPAC2 signaling in synchronization of firing rate rhythms. Rhythmic wild-type neurons within a given high-density culture showed similar periods and statistically significant clustering of circadian phases (Fig. 3a,b; Rayleigh test, P < 0.05). The distribution of phases seen in these cultures was similar to that reported previously for SCN neurons in organotypic slices9,10. Moreover, the distribution of periods for rhythmic wild-type neurons was relatively narrow, both within and between SCN cultures (Figs. 3c and 4, respectively). Thus, whereas firing rate rhythms of dispersed SCN neurons in low-density cultures do not synchronize4,32, those in higher-density dispersals are capable of maintaining synchrony. We conclude that intercellular distance is critical for period synchronization in the SCN.
Figure 3.
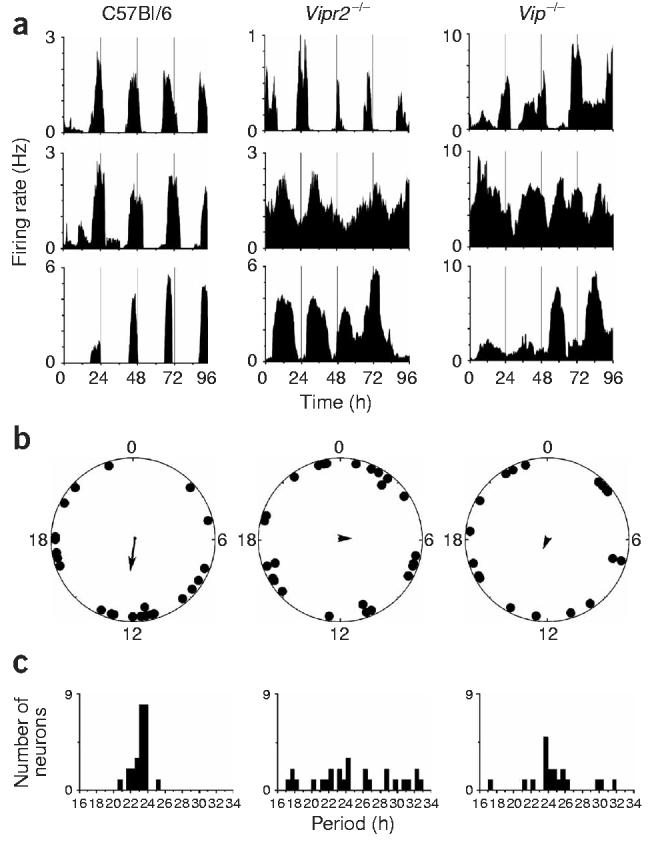
Genetic knockout of Vip or Vipr2 abolishes synchrony among rhythmic SCN neurons in the same culture (a) Representative rhythmic firing rate records from wild-type, Vipr2−/− or Vip−/− SCN neurons in the same high density culture. Rhythmic neurons in the wild-type culture expressed robust rhythms with similar periods and phases, whereas those in the mutant cultures had variable periods and phases. (b) Relative circadian phases of all rhythmic neurons (n = 24 wild-type, 19 Vip−/− and 26 Vipr2−/−) from the same three cultures as in a. Data are plotted within circles that represent the fourth day of recording. Arrows indicate the average phase of each distribution. Arrow length reflects the r-value of each phase distribution. Wild-type neurons showed statistically significant synchronization (Rayleigh test, P < 0.05, r = 0.36), but Vipr2−/− and Vip−/− neurons did not (P > 0.7 and P > 0.9, r = 0.09 and 0.05, respectively). (c) Distribution of periods for the same neurons as in b. Vipr2−/− and Vip−/− SCN neurons showed a significantly broader distribution than did wild-type neurons in the same culture (P < 0.05, Brown-Forsythe's and Levene's tests for equal variance).
Figure 4.
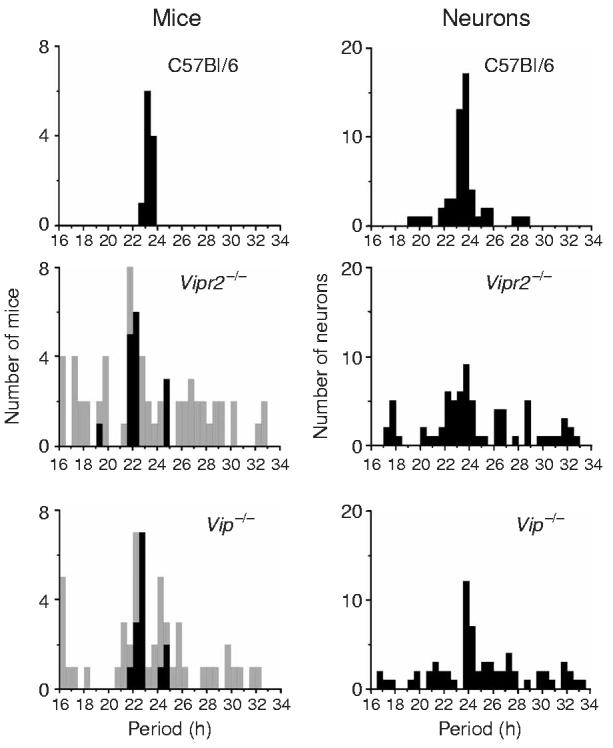
The distributions of circadian periods of locomotor activity in mice (left) are similar to those of firing rate rhythms in SCN neurons (right) for the three genotypes. Although the average dominant periods (black bars) of Vip−/− and Vipr2−/− mutant mice were shorter than those of wild-type mice (P < 0.05, one-way ANOVA with Scheffé post hoc test), the average of all significant circadian periods (gray bars) was similar between genotypes (P > 0.7, Kruskal-Wallis one-way ANOVA with Scheffé post hoc test). Similarly, the average periods of SCN neuronal rhythms did not change with genotype (P > 0.1, Kruskal-Wallis one-way ANOVA). The distributions of all behavioral and firing rate periods were significantly broader in the mutant mice and neurons (P < 0.005 for behavior and P < 0.00005 for neurons, Brown-Forsythe's and Levene's tests for equal variance), indicating a loss of circadian synchrony.
We found a lack of synchronization among mutant SCN neurons in the same high-density culture. Rhythmic Vip−/− and Vipr2−/− SCN neurons assumed random phase relationships to one another (Figs. 3a,b, Rayleigh test, P > 0.9 and P > 0.7, respectively). Mutant neurons within the same culture also showed significantly broader distributions of periods than wild-type neurons (Fig. 3c; P < 0.05, Brown-Forsythe's and Levene's tests).
The multiple behaviorally expressed periods correlate with the desynchronized rhythms among Vip−/− and Vipr2−/− SCN neurons. Combining results from all cultures, we found that, similarly to locomotor rhythms, the mean periodicity of firing rate rhythms did not depend on genotype (Fig. 4; 25.0 ± 4.0 h for Vip−/−, 24.4 ± 4.0 h for Vipr2−/−, and 23.6 ± 1.7 h for wild-type; Kruskal-Wallis one-way ANOVA, P > 0.1). The distribution of periods, however, was significantly broader for Vip−/− and Vipr2−/− SCN neurons than for wild-type neurons (P < 0.00005, Brown-Forsythe's and Levene's tests). Thus, the broad range of periods and the loss of neuronal synchrony correlate with the broadly distributed, multiple behavioral periods expressed by Vip−/− and Vipr2−/− mice. These results indicate that VIP/VPAC2 signaling normally restricts the range of periods in SCN neurons, synchronizing locomotor rhythms to a single circadian period.
VIP is not required for normal complement of SCN neurons
Because VIP signaling has been implicated in neural development33, it is possible that disruption of VIP signaling could result in aberrant cell numbers or connectivity within the SCN. Here we found that the average percentage of neurons expressing arginine vasopressin (AVP) was similar in dispersed cultures of wild-type (14.4 ± 1.6% of 3,588 neurons from nine cultures), Vip−/− (11.7 ± 0.6% of 1,311 neurons from three cultures) and Vipr2−/− SCN (15.4 ± 0.6% of 3,516 neurons from eight cultures; P > 0.1, one-way ANOVA with Scheffé test). The mean percentage of neurons expressing VIP was also similar for wild-type and Vipr2−/− SCN cultures (wild-type, 8.3 ± 0.7%; Vipr2−/−, 9.9 ± 0.6%). VIPergic and AVPergic neurons grew extensive processes and showed similar morphology in both wild-type and mutant cultures. Thus, based on two of the primary neurochemical markers for SCN neurons17, we did not find evidence for major cell loss or disrupted development of SCN neurons from Vip−/− or Vipr2−/− mice.
Circadian VPAC2 signaling restores SCN rhythms and synchrony
To determine whether VPAC2 signaling is sufficient to maintain firing rate rhythms in individual SCN neurons, we treated Vip−/− mutant SCN cultures with a VPAC2-specific agonist (Ro 25-1553 (ref. 18), 150 nM) every 24 h for 6 d. To minimize disturbance to cultures during these treatments, we applied a small volume (7.5 μl) of concentrated agonist to 1 ml of culture medium on the array each day. In four separate recordings of Vip−/− SCN cultures, circadian rhythmicity was restored to 71% of previously arrhythmic neurons by daily Ro 25-1553 treatment (Fig. 5a; 65 of 92 neurons). These rhythms anticipated daily treatments (Fig. 5b, n = 60 of 65 neurons). In addition, firing rhythms of initially rhythmic neurons entrained to drug application (Fig. 5a). Both previously rhythmic and arrhythmic neurons within a treated Vip−/− culture showed statistically significant clustering of circadian phases, with the minimum of firing occurring approximately 10–12 h after the final agonist treatment (Fig. 5c, Rayleigh test, P < 0.05). The restored synchrony in these cultures was similar to that of wild-type cultures (Fig. 3b). Finally, the periods of the neurons that were rhythmic both before and during treatment were significantly more restricted during daily agonist application (Fig. 5d; P < 0.005, Brown-Forsythe's and Levene's tests). We conclude that firing rate rhythms of Vip−/− neurons entrained to the daily application of a VPAC2 agonist.
Figure 5.

Daily application of VPAC2 agonist Ro 25-1553 restores rhythmicity to Vip−/− SCN neurons. (a) Firing rate records of three initially arrhythmic Vip−/− neurons and a rhythmic neuron treated with 150 nM Ro 25-1553 every 24 h for 6 d (arrows). The top two traces are from representative neurons in which rhythmicity was restored, and the bottom trace is from a neuron that remained arrhythmic. The third trace is from a representative rhythmic neuron entrained by agonist treatment. Rhythmicity persisted for at least 48 h after the last treatment, indicating that cyclic firing was entrained by drug application. (b) Firing patterns of rhythmic neurons, averaged over the last 4 d of treatment, increased in anticipation of the drug treatment, indicating that the daily change in firing rate was not simply an acute response to the agonist. (c) Relative phases of initially rhythmic (n = 16, left) and arrhythmic neurons with restored rhythms (n = 22, right) from a representative Vip−/− culture on the sixth day of Ro 25-1553 treatment. Arrowheads at hour 6 denote the time of agonist application. Both subsets of neurons showed statistically significant synchronization (Rayleigh test, P < 0.05, r = 0.43 and 0.39 for rhythmic and arrhythmic neurons, respectively) with the mean bathyphase of firing (arrows) 10–12 h after agonist application. (d) The broad distribution of periods for rhythmic Vip−/− neurons before treatment (left) was significantly narrowed during agonist treatment (right; P < 0.005, Brown-Forsythe's and Levene's tests). (e) The relative proportion of rhythmic (black) to arrhythmic (white) neurons from Vip−/− cultures was restored to wild-type levels by daily VPAC2 agonist treatment, but there was little change in the proportion of rhythmic Vipr2−/− neurons with the same treatment.
We also made four separate recordings of Vipr2−/− SCN neurons under identical conditions. Of 155 arrhythmic Vipr2−/− neurons treated with Ro 25-1553, only 39 (25%) fired rhythmically during agonist treatment, whereas a similar number of rhythmic neurons lost rhythmicity (n = 36). In addition, the distribution of periods of initially rhythmic Vipr2−/− neurons did not change during daily agonist treatment (P > 0.7, Brown-Forsythe's and Levene's tests), indicating that Ro 25-1553 specifically acts through VPAC2 receptors to coordinate rhythms in the SCN.
Restoration of circadian function was also evident in the percentage of recorded neurons that were rhythmic before and during Ro 25-1553 treatment. In Vip−/− cultures, 41 of 133 total recorded neurons (23%) were initially rhythmic, whereas during agonist treatment, 93 of 133 (70%) showed significant rhythmicity (Fig. 5e), similar to the 68% of all wild-type SCN neurons with spontaneous circadian rhythms (Fig. 2b). In contrast, 49 of 204 (24%) Vipr2−/− SCN neurons were initially rhythmic, whereas only 52 (25%) were rhythmic during drug treatment (Fig. 5e). Together, these data indicate that circadian VPAC2 signaling is sufficient to maintain rhythmicity and synchrony in separate subsets of SCN neurons.
DISCUSSION
Our data provide the first evidence that VIP signaling through its receptor, VPAC2, has two distinct roles in the SCN—maintaining circadian rhythmicity in a subset of neurons, and maintaining synchrony between intrinsically rhythmic neurons. The loss of VIP or VPAC2 results in functionally similar deficits in both circadian behavior and SCN firing rhythms. Both mutations approximately halve the number of rhythmic neurons in the SCN, so that only about 30% remain rhythmic. This is the first demonstration of a role for an extracellular signal in the generation of firing rate rhythms in individual SCN neurons. The loss of rhythmicity in VIP-deficient SCN could indicate that VIPergic pacemakers normally promote rhythmicity in other cell types or that VIP is a part of the pacemaking loop in a subset of SCN neurons. That arrhythmic Vip−/− neurons can anticipate daily application of a VPAC2 agonist with a circadian increase in firing rate provides notable evidence that VIP may not only maintain rhythms in these neurons, but also entrain them. Which cells require VIP input for rhythmicity is not known, but the broad distribution of VPAC2 receptors in the SCN34 suggests that they are likely to be a heterogeneous class of neurons. The 30% of SCN neurons that do not require VIP for sustained rhythmicity may include VIPergic neurons themselves (roughly 10–20% of SCN neurons4,17) and at least one other neuronal class. Thus, the SCN is comprised of functionally heterogeneous populations: neurons that require VIP signaling to sustain rhythmicity and pacemaking neurons that do not.
VIP signaling is also necessary for synchronization of firing rate rhythms between neurons in the SCN. Consistent with this, a recent report found that separating the dorsal and ventral SCN, which eliminates VIP input to the dorsal SCN, causes rhythmic neurons in the dorsal SCN to drift out of phase with one another while rhythms in the ventral SCN remain synchronized7. The lack of synchronization we found among rhythmic mutant neurons in vitro mirrors the multiple periods and diminished strength of rhythmicity seen behaviorally in mutant mice. Although we cannot rule out the contribution of VIP within other brain areas such as cortical or thalamic neurons, it is likely that the behavioral phenotype arises initially at the level of SCN neurons. We found no evidence of multiple periodicities of firing rhythms in individual neurons, indicating that the behavioral desynchronization of mutant mice is due to desynchrony among pacemaking neurons. This, combined with the decreased number of rhythmic SCN neurons in mutant mice, is likely to cause the weakening of behavioral rhythms recorded in constant darkness.
The SCN and behavioral phenotypes of VIP- and VPAC2-mutants seem remarkably similar to neural and behavioral phenotypes of Drosophila lacking pigment dispersing factor (PDF). In these mutant flies, populations of clock neurons in the dorsal brain lose rhythmicity in constant conditions35, whereas pacemaking ventral lateral neurons lose synchrony36. How approximately one-third of mutant mice maintain short-period rhythmicity in constant darkness is still unclear. Notably, the same phenotype is seen in roughly one-third of Drosophila lacking PDF36. It may be that feedback onto the pacemakers from behaviors such as locomotion and sleep37,38 suffices to coordinate their activity.
How VIP/VPAC2 signaling mediates its effects on firing rhythmicity and rhythm synchronization is not yet known. VIP is rhythmically transcribed and released in neonatal SCN15,39,40, and circadian transcription has recently been described in adult SCN as well41. VIP could act indirectly, through its known effects on other neurotransmitter pathways, such as GABA31. Rhythmic GABA application has been shown to synchronize the firing of SCN neurons in vitro, making this signal a possible intermediary, although it is unknown whether GABA in the SCN is required for synchronization11. Alternatively, VIP signaling could directly activate clock gene transcription in neurons through its effects on intracellular calcium and cAMP signaling42-44. Future experiments should resolve whether VIP acts in a circadian manner or constitutively and whether it directly establishes and entrains rhythms in SCN neurons.
Taken together, the present data show that approximately 70% of all SCN neurons fire rhythmically, but roughly half of these require VIP/VPAC2 signaling for circadian rhythmicity. The neurons that remain rhythmic in the absence of this signaling pathway lose their ability to synchronize to each other. We conclude that VIPergic signaling serves two functions in the SCN: promoting rhythmicity in a subset of non-pacemaking SCN neurons, and synchronizing pacemaking neurons. Thus, it is likely that VIP-expressing neurons themselves are circadian pacemakers in the SCN, critical for establishing and synchronizing rhythms in other SCN neurons and in behavior.
METHODS
Behavioral recording
Mice were maintained in the Hilltop animal facility at Washington University. Mutant mice were backcrossed onto a C57Bl/6 background (wild-type mice originally purchased from Charles River Laboratories) for at least eight generations before these experiments. Adult Vipr2−/− (n = 15), Vip−/− (n = 14), and C57Bl/6 mice (n = 11) were housed individually in cages equipped with running wheels and maintained in light-tight chambers illuminated with fluorescent bulbs (2.4 ± 0.5 × 1018 photons s−1 m−2; General Electric). Running wheel activity was recorded in 6-min bins (Clocklab, Actimetrics) during 10 d in 12 h/12 h light/dark, then during 10 d in a skeleton photoperiod consisting of two 11-h dark periods per day separated by 1-h light pulses. Running wheel activity was then recorded in constant darkness for 50–90 d. All procedures were approved by the Animal Care and Use Committee of Washington University and conformed to US National Institutes of Health guidelines.
Cell culture
SCN were obtained from 1- to 7-d-old Vipr2−/−, Vip−/− or C57Bl/6 mice housed in a 12 h/12 h light/dark cycle. Genotypes for all animals used in these studies were confirmed using PCR techniques described elsewhere18,30. For slice cultures, 150-μm–thick coronal slices of bilateral SCN were cultured on Millicell-CM membranes (Millipore) for 2–5 d and then inverted onto collagen-coated multielectrode arrays (MEAs, Multichannel Systems) as previously described45. Slices were maintained on arrays for another 3–5 d in 150 μl of CO2-buffered medium46 supplemented with 10% newborn calf serum (Invitrogen) before recording. For dispersed cultures, SCNs were punched from 300-μm-thick slices, and cells were dissociated using papain5. Viable cells (from 4–8 SCN slices) were plated at a density of at least 10,000 cells mm−2 on each MEA. Cylindrical gaskets of Sylgard polymer (Dow Corning) of 2 mm internal diameter were placed around arrays to maintain high cell densities during plating and recording. Dispersed cultures were maintained in 1 ml of CO2-buffered medium with serum for 1–2 weeks before recording. After 7 d in vitro, cultures were treated with 20 μM cytosine arabinoside (Ara-C, Sigma) to control glial proliferation.
Immunostaining
SCN dispersals on glass coverslips were treated with colchicine (Sigma; 50 μg/ml) overnight before staining. Cells were fixed using 4% paraformaldehyde, 4% sucrose and immunolabeled for neuropeptides using standard methods47. Cultures were labeled with rabbit anti-VIP (Diasorin; 1:2,000) and mouse anti-AVP (monoclonal, PS41, ATCC; 1:50). Secondary antibodies were donkey anti–rabbit IgG conjugated to Cy2 (Jackson Immuno Research; 1:50) and donkey anti–mouse IgG conjugated to Texas Red (Jackson; 1:200). Cultures were subjected to phase-contrast imaging to discriminate neuronal cell bodies, and phase-bright cells were counted as neurons as previously described4. Some cultures were stained with rabbit anti–glial fibrillary acidic protein (anti-GFAP, Sigma; 1:100) and mouse anti-MAP2 (Calbiochem; 1:100) to confirm this discrimination of neurons from glia. Immunopositive cell counts were made by two independent observers blind to experimental conditions.
Multielectrode array recording
Long-term recordings of firing rate from SCN neurons were carried out using arrays of sixty 10-μm–diameter electrodes (100-μm spacing; Multichannel Systems). Culture chambers fixed to arrays were covered with a fluorinated ethylene-polypropylene membrane48 before transfer to a recording incubator. The recording incubator was maintained at 37 °C with 5% CO2 throughout all experiments. Extracellular voltage signals were recorded for a minimum of 5 d from either 60 or 120 electrodes (one or two arrays) simultaneously. Action potentials exceeding a defined voltage threshold were digitized into 2-ms time stamped cutouts (MC-Rack software, Multichannel Systems). Spikes from individual neurons were discriminated offline using a principal component analysis–based system (Offline Sorter, Plexon), and firing rates were calculated over 10-min recording epochs (NeuroExplorer, Plexon). Correct discrimination of single-neuron activity was ensured by the presence of a clear refractory period in autocorrelograms of firing records (NeuroExplorer).
The VPAC2 agonist Ro 25-1553 (ref. 18) was diluted in deionized water and stored at −20° C. A volume of 7.5 μl of concentrated stock (for a final concentration of 150 nM) was applied every 24 h for 6 d to Vip−/− and Vipr2−/− SCN cultures on MEAs. To minimize disturbance to cultures during these treatments, MEAs were removed from the incubator for a maximum of 30 s.
Data analysis
Continuous single-neuron firing records lasting at least 4 d were used for analysis of rhythmicity. Circadian amplitude and period of behavioral and firing rate rhythmicity were determined using χ2 periodogram analysis49 from continuous recordings of at least 4 d from neurons or 70 d in constant darkness from mice (Clocklab). Rhythmicity was considered statistically significant if the χ2 periodogram value exceeded the 99.9% confidence interval (Qp value), and circadian amplitude was measured as the amplitude above the Qp value at the dominant period. Mean periods, circadian amplitudes and peak-to-trough amplitudes of rhythmicity and mean firing rates were compared for mice and neurons of the three genotypes using one-way ANOVA and Scheffé post hoc tests. Distributions of periods were compared between the three genotypes using both Levene's and Brown-Forsythe's tests for equal variance. Phase analysis was carried out using relative bathyphases of individual neurons' rhythms (determined with Clocklab software) over a given day16. Rayleigh tests50 were used to determine whether distributions of phases around the circadian day were significantly different from random (in indicator of synchronization). For comparison of neurons' rhythmicity or periodicity with and without Ro 25-1553, we analyzed firing rate records of equal duration before and during treatment by χ2 periodogram.
ACKNOWLEDGMENTS
We thank P. Taghert, R. Van Gelder, D. Granados-Fuentes, and U. Abraham for helpful discussions; D. Piatchek and J. Diani of the Washington University Hilltop animal facility; H. Dave, T. Fadelu, and L. Prolo for expert technical assistance and animal care; and P. Robberecht (University of Brussels) for providing VPAC2 agonist Ro 25-1553. This work was supported by a US National Science Foundation graduate research fellowship (S.J.A.) and by the National Institutes of Health (grants MH63104, MH62517 and MH073302).
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
References
- 1.Kalsbeek A, Buijs RM. Output pathways of the mammalian suprachiasmatic nucleus: coding circadian time by transmitter selection and specific targeting. Cell Tissue Res. 2002;309:109–118. doi: 10.1007/s00441-002-0577-0. [DOI] [PubMed] [Google Scholar]
- 2.Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- 3.Van Gelder RN, Herzog ED, Schwartz WJ, Taghert PH. Circadian rhythms: in the loop at last. Science. 2003;300:1534–1535. doi: 10.1126/science.1085446. [DOI] [PubMed] [Google Scholar]
- 4.Welsh DK, Logothetis DE, Meister M, Reppert SM. Individual neurons dissociated from rat suprachiasmatic nucleus express independently phased circadian firing rhythms. Neuron. 1995;14:697–706. doi: 10.1016/0896-6273(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 5.Herzog ED, Takahashi JS, Block GD. Clock controls circadian period in isolated suprachiasmatic nucleus neurons. Nat. Neurosci. 1998;1:708–713. doi: 10.1038/3708. [DOI] [PubMed] [Google Scholar]
- 6.Honma S, Shirakawa T, Katsuno Y, Namihira M, Honma K-I. Circadian periods of single suprachiasmatic neurons in rats. Neurosci. Lett. 1998;250:157–160. doi: 10.1016/s0304-3940(98)00464-9. [DOI] [PubMed] [Google Scholar]
- 7.Yamaguchi S, et al. Synchronization of cellular clocks in the suprachiasmatic nucleus. Science. 2003;302:1408–1412. doi: 10.1126/science.1089287. [DOI] [PubMed] [Google Scholar]
- 8.Quintero JE, Kuhlman SJ, McMahon DG. The biological clock nucleus: a multiphasic oscillator network regulated by light. J. Neurosci. 2003;23:8070–8076. doi: 10.1523/JNEUROSCI.23-22-08070.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaap J, et al. Heterogeneity of rhythmic suprachiasmatic nucleus neurons: Implications for circadian waveform and photoperiodic encoding. Proc. Natl. Acad. Sci. USA. 2003;100:15994–15999. doi: 10.1073/pnas.2436298100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herzog ED, Geusz ME, Khalsa SBS, Straume M, Block GD. Circadian rhythms in mouse suprachiasmatic nucleus explants on multimicroelectrode plates. Brain Res. 1997;757:285–290. doi: 10.1016/s0006-8993(97)00337-5. [DOI] [PubMed] [Google Scholar]
- 11.Liu C, Reppert SM. GABA synchronizes clock cells within the suprachiasmatic circadian clock. Neuron. 2000;25:123–128. doi: 10.1016/s0896-6273(00)80876-4. [DOI] [PubMed] [Google Scholar]
- 12.Colwell CS. Rhythmic coupling among cells in the suprachiasmatic nucleus. J. Neurobiol. 2000;43:379–388. doi: 10.1002/1097-4695(20000615)43:4<379::aid-neu6>3.0.co;2-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Long MA, Jutras MJ, Connors BW, Burwell RD. Electrical synapses coordinate activity in the suprachiasmatic nucleus. Nat. Neurosci. 2005;8:61–66. doi: 10.1038/nn1361. [DOI] [PubMed] [Google Scholar]
- 14.Jiang ZG, Yang YQ, Allen CN. Tracer and electrical coupling of rat suprachiasmatic nucleus neurons. Neuroscience. 1997;77:1059–1066. doi: 10.1016/s0306-4522(96)00539-8. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura W, Honma S, Shirakawa T, Honma K-I. Regional pacemakers composed of multiple oscillator neurons in the rat suprachiasmatic nucleus. Eur. J. Neurosci. 2001;14:666–674. doi: 10.1046/j.0953-816x.2001.01684.x. [DOI] [PubMed] [Google Scholar]
- 16.Herzog ED, Aton SJ, Numano R, Sakaki Y, Tei H. Temporal precision in the mammalian circadian system: a reliable clock from less reliable neurons. J. Biol. Rhythms. 2004;19:35–46. doi: 10.1177/0748730403260776. [DOI] [PubMed] [Google Scholar]
- 17.Abrahamson EE, Moore RY. Suprachiasmatic nucleus in the mouse: retinal innervation, intrinsic organization and efferent projections. Brain Res. 2001;916:172–191. doi: 10.1016/s0006-8993(01)02890-6. [DOI] [PubMed] [Google Scholar]
- 18.Cutler DJ, et al. The mouse VPAC2 receptor confers suprachiasmatic nuclei cellular rhythmicity and responsiveness to vasoactive intestinal polypeptide in vitro. Eur. J. Neurosci. 2003;17:197–204. doi: 10.1046/j.1460-9568.2003.02425.x. [DOI] [PubMed] [Google Scholar]
- 19.Reed HE, et al. Effects of vasoactive intestinal polypeptide on neurones of the rat suprachiasmatic nuclei in vitro. J. Neuroendocrinol. 2002;14:639–646. doi: 10.1046/j.1365-2826.2002.00826.x. [DOI] [PubMed] [Google Scholar]
- 20.Cagampang FR, Sheward WJ, Harmar AJ, Piggins HD, Coen CW. Circadian changes in the expression of vasoactive intestinal peptide 2 receptor mRNA in the rat suprachiasmatic nuclei. Brain Res. Mol. Brain Res. 1998;54:108–112. doi: 10.1016/s0169-328x(97)00327-6. [DOI] [PubMed] [Google Scholar]
- 21.Piggins HD, Antle MC, Rusak B. Neuropeptides phase shift the mammalian circadian pacemaker. J. Neurosci. 1995;15:5612–5622. doi: 10.1523/JNEUROSCI.15-08-05612.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watanabe K, Vanecek J, Yamaoka S. In vitro entrainment of the circadian rhythm of vasopressin-releasing cells in suprachiasmatic nucleus by vasoactive intestinal polypeptide. Brain Res. 2000;877:361–366. doi: 10.1016/s0006-8993(00)02724-4. [DOI] [PubMed] [Google Scholar]
- 23.Reed HE, Meyer-Spasche A, Cutler DJ, Coen CW, Piggins HD. Vasoactive intestinal polypeptide (VIP) phase-shifts the rat suprachiasmatic nucleus clock in vitro. Eur. J. Neurosci. 2001;13:839–843. doi: 10.1046/j.0953-816x.2000.01437.x. [DOI] [PubMed] [Google Scholar]
- 24.Nielsen HS, Hannibal J, Fahrenkrug J. Vasoactive intestinal polypeptide induces per1 and per2 gene expression in the rat suprachiasmatic nucleus late at night. Eur. J. Neurosci. 2002;15:570–574. doi: 10.1046/j.0953-816x.2001.01882.x. [DOI] [PubMed] [Google Scholar]
- 25.van den Pol AN, Gorcs T. Synaptic relationships between neurons containing vasopressin, gastrin-releasing peptide, vasoactive intestinal polypeptide, and glutamate decarboxylase immunoreactivity in the suprachiasmatic nucleus: dual ultrastructural immunocytochemistry with gold- substituted silver peroxidase. J. Comp. Neurol. 1986;252:507–521. doi: 10.1002/cne.902520407. [DOI] [PubMed] [Google Scholar]
- 26.Daikoku S, Hisano S, Kagotani Y. Neuronal associations in the rat suprachiasmatic nucleus demonstrated by immunoelectron microscopy. J. Comp. Neurol. 1992;325:559–571. doi: 10.1002/cne.903250408. [DOI] [PubMed] [Google Scholar]
- 27.Shen S, et al. Overexpression of the human VPAC2 receptor in the suprachiasmatic nucleus alters the circadian phenotype of mice. Proc. Natl. Acad. Sci. USA. 2000;97:11575–11580. doi: 10.1073/pnas.97.21.11575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hughes AT, Fahey B, Cutler DJ, Coogan AN, Piggins HD. Aberrant gating of photic input to the suprachiasmatic circadian pacemaker of mice lacking the VPAC2 receptor. J. Neurosci. 2004;24:3522–3526. doi: 10.1523/JNEUROSCI.5345-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harmar AJ, et al. The VPAC(2) receptor is essential for circadian function in the mouse suprachiasmatic nuclei. Cell. 2002;109:497–508. doi: 10.1016/s0092-8674(02)00736-5. [DOI] [PubMed] [Google Scholar]
- 30.Colwell CS, et al. Disrupted circadian rhythms in VIP and PHI deficient mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003;285:R939–R949. doi: 10.1152/ajpregu.00200.2003. [DOI] [PubMed] [Google Scholar]
- 31.Itri J, Michel S, Waschek JA, Colwell CS. Circadian rhythm in inhibitory synaptic transmission in the mouse suprachiasmatic nucleus. J. Neurophysiol. 2004;92:311–319. doi: 10.1152/jn.01078.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Honma S, Nakamura W, Shirakawa T, Honma K. Diversity in the circadian periods of single neurons of the rat suprachiasmatic nucleus depends on nuclear structure and intrinsic period. Neurosci. Lett. 2004;358:173–176. doi: 10.1016/j.neulet.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 33.Moody TW, Hill JM, Jensen RT. VIP as a trophic factor in the CNS and cancer cells. Peptides. 2003;24:163–177. doi: 10.1016/s0196-9781(02)00290-5. [DOI] [PubMed] [Google Scholar]
- 34.Kallo I, et al. Transgenic approach reveals expression of the VPAC2 receptor in phenotypically defined neurons in the mouse suprachiasmatic nucleus and in its efferent target sites. Eur. J Neurosci. 2004;19:2201–2211. doi: 10.1111/j.0953-816X.2004.03335.x. [DOI] [PubMed] [Google Scholar]
- 35.Peng Y, Stoleru D, Levine JD, Hall JC, Rosbash M. Drosophila free-running rhythms require intercellular communication. PLoS Biol. 2003;1:E13. doi: 10.1371/journal.pbio.0000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin Y, Stormo GD, Taghert PH. The neuropeptide pigment-dispersing factor coordinates pacemaker interactions in the Drosophila circadian system. J. Neurosci. 2004;24:7951–7957. doi: 10.1523/JNEUROSCI.2370-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deboer T, Vansteensel MJ, Detari L, Meijer JH. Sleep states alter activity of suprachiasmatic nucleus neurons. Nat. Neurosci. 2003;10:1086–1090. doi: 10.1038/nn1122. [DOI] [PubMed] [Google Scholar]
- 38.Mistlberger RE. Effects of daily schedules of forced activity on free-running rhythms in the rat. J. Biol. Rhythms. 1991;6:71–80. doi: 10.1177/074873049100600108. [DOI] [PubMed] [Google Scholar]
- 39.Shinohara K, Honma S, Katsuno Y, Abe H, Honma K-I. Two distinct oscillators in the rat suprachiasmatic nucleus in vitro. Proc. Natl. Acad. Sci. USA. 1995;92:7396–7400. doi: 10.1073/pnas.92.16.7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ban Y, Shigeyoshi Y, Okamura H. Development of vasoactive intestinal peptide mRNA rhythm in the rat suprachiasmatic nucleus. J. Neurosci. 1997;17:3920–3931. doi: 10.1523/JNEUROSCI.17-10-03920.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dardente H, et al. Daily and circadian expression of neuropeptides in the suprachiasmatic nuclei of nocturnal and diurnal rodents. Brain Res. Mol. Brain Res. 2004;124:143–151. doi: 10.1016/j.molbrainres.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 42.Travnickova-Bendova Z, Cermakian N, Reppert SM, Sassone-Corsi P. Bimodal regulation of mPeriod promoters by CREB-dependent signaling and CLOCK/BMAL1 activity. Proc. Natl. Acad. Sci. USA. 2002;99:7728–7733. doi: 10.1073/pnas.102075599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akiyama M, Minami Y, Nakajima T, Moriya T, Shibata S. Calcium and pituitary adenylate cyclase-activating polypeptide induced expression of circadian clock gene mPer1 in the mouse cerebellar granule cell culture. J. Neurochem. 2001;78:499–508. doi: 10.1046/j.1471-4159.2001.00452.x. [DOI] [PubMed] [Google Scholar]
- 44.Itri J, Colwell CS. Regulation of inhibitory synaptic transmission by vasoactive intestinal peptide (VIP) in the mouse suprachiasmatic nucleus. J. Neurophysiol. 2003;90:1589–1597. doi: 10.1152/jn.00332.2003. [DOI] [PubMed] [Google Scholar]
- 45.Ikeda M, et al. Circadian dynamics of cytosolic and nuclear Ca2+ in single suprachiasmatic nucleus neurons. Neuron. 2003;38:253–263. doi: 10.1016/s0896-6273(03)00164-8. [DOI] [PubMed] [Google Scholar]
- 46.Granados-Fuentes D, Saxena MT, Prolo LM, Aton SJ, Herzog ED. Olfactory bulb neurons express functional, entrainable circadian rhythms. Eur. J. Neurosci. 2004;19:898–906. doi: 10.1111/j.0953-816x.2004.03117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Banker G, Goslin K. In: Culturing Nerve Cells. Banker G, Goslin K, editors. MIT Press; Cambridge, Massachusetts, USA: 1991. pp. 75–118. [Google Scholar]
- 48.Potter SM, DeMarse TB. A new approach to neural cell culture for long-term studies. J. Neurosci. Methods. 2001;110:17–24. doi: 10.1016/s0165-0270(01)00412-5. [DOI] [PubMed] [Google Scholar]
- 49.Sokolove PG, Bushell WN. The chi square periodogram: its utility for analysis of circadian rhythms. J. Theor. Biol. 1978;72:131–160. doi: 10.1016/0022-5193(78)90022-x. [DOI] [PubMed] [Google Scholar]
- 50.Batschelet E. In: Mathematics in Biology. Sibson R, Cohen JE, editors. Academic; New York: 1981. pp. 31–54. [Google Scholar]