

Abstract
Glioblastoma (GBM) is a type of intracranial brain tumor, for which there is no cure. In spite of advances in surgery, chemotherapy and radiotherapy, patients die within a year of diagnosis. Therefore, there is a critical need to develop novel therapeutic approaches for this disease. Gene therapy, which is the use of genes or other nucleic acids as drugs, is a powerful new treatment strategy which can be developed to treat GBM. Several treatment modalities are amenable for gene therapy implementation, e.g. conditional cytotoxic approaches, targeted delivery of toxins into the tumor mass, immune stimulatory strategies, and these will all be the focus of this review.
Both conditional cytotoxicity and targeted toxin mediated tumor death, are aimed at eliminating an established tumor mass and preventing further growth. Tumors employ several defensive strategies that suppress and inhibit anti-tumor immune responses. A better understanding of the mechanisms involved in eliciting anti-tumor immune responses has identified promising targets for immunotherapy. Immunotherapy is designed to aid the immune system to recognize and destroy tumor cells in order to eliminate the tumor burden. Also, immune-therapeutic strategies have the added advantage that an activated immune system has the capability of recognizing tumor cells at distant sites from the primary tumor, therefore targeting metastasis distant from the primary tumor locale. Pre-clinical models and clinical trials have demonstrated that in spite of their location within the central nervous system (CNS), a tissue described as ‘immune privileged’, brain tumors can be effectively targeted by the activated immune system following various immunotherapeutic strategies. This review will highlight recent advances in brain tumor immunotherapy, with particular emphasis on advances made using gene therapy strategies, as well as reviewing other novel therapies that can be used in combination with immunotherapy. Another important aspect of implementing gene therapy in the clinical arena is to be able to image the targeting of the therapeutics to the tumors, treatment effectiveness and progression of disease. We have therefore reviewed the most exciting non-invasive, in vivo imaging techniques which can be used in combination with gene therapy to monitor therapeutic efficacy over time.
INTRODUCTION
Brain cancers account for 1.4% of all cancers [1]. The most common primary brain tumor is the glioblastoma multiforme (GBM), that accounts for 40% of all brain tumors. While overall accounting for a small percentage of total cancer cases, GBM is highly aggressive with the average patient surviving no more than 12-18 months [2, 3]. GBM are highly proliferative, diffusely invasive, immunosuppressive, and highly vascularized tumors [4]. Current treatment of GBM involves surgery, chemo- and radiotherapy; however, these therapies are only marginally effective in altering the ultimate progression of this disease. After resection of the tumor mass, survival depends on whether chemo- and radiation therapies are able to eliminate the infiltrating cells [5] which invade healthy tissue. Genomic analysis of patients with GBM has led to the identified a subpopulation of GBM that has previously been unidentified. These patients respond well to current therapies and have a median survival time of more than 6 years [6]. However, the majority of patients with GBM respond very poorly to current treatments and the mean survival time following tumor resection is less than 18 months. Consequently, new effective therapies are urgently needed to improve survival and the quality of life in patients diagnosed with GBM.
Gene therapy, which is the use of nucleic acids as medicines, provides a novel alternative therapeutic approach which can be harnessed to develop more effective treatments for GBM. The most attractive therapeutic targets to develop gene therapies for GBM are conditional cytotoxic genes, targeted cytotoxins which selectively bind to and kill tumor cells, anti-angiogenic molecules which inhibit the formation of new blood vessels, pro-apoptotic genes, molecules that block signal trasduction pathways know to be activated in GBM, genes that express proteins that block growth factor receptors present in tumor cells, and also oncolytic viruses which will specifically replicate within cancer cells, leading to their death. Gene delivery vectors can also be used to deliver molecules which will enhance the immune system of the host with the aim of producing an effective systemic anti-tumor immune response. In this review, we will discuss the immune system within the CNS and how we can harness the power of gene therapy to mount an effective immune response against GBM. We will also discuss targeted therapeutic approaches which aim to deliver toxins selectively into tumor cells. In view of the importance of being able to monitor disease progression and persistence of gene therapies in vivo over time, we will also review current non-invasive imaging approaches which can be used in conjunction with gene therapy.
IMMUNE BASED GENE THERAPIES FOR GBM
Immunotherapy has become an exciting area of research for treating tumors where conventional treatment modalities have remained ineffective. The basic premise of a role of the immune system in rejection of tumors, specifically allografts, was demonstrated more than 50 years ago [7, 8]. This was an early example of how immune tolerance can lead to tumor growth. Tumors generally express different levels of proteins than normal surrounding tissue, and some of these proteins contain amino acid substitutions or deletions, or changes in phosphorylation or glycosylation. Any of these changes in proteins expressed by tumors can be sufficient for the immune system to recognize specifically proteins expressed by tumors as antigenic, and mount an immune response against these proteins. There is some anecdotal evidence in the literature of spontaneous regression of skin tumors, or even brain tumors following infection, and it is believed that the immune system can mediate tumor rejection in these cases [9-11]. However, this is not a common event and tumors secrete many anti-inflammatory mediators and other proteins that can stifle the development of an effective anti-tumor response. It has also emerged recently that regulatory T cells are commonly elevated in patients with tumors and they play an active role in suppressing the response of the immune system to the tumors [12]. With recent advances in our understanding of the components of the immune system that are involved in invoking or repressing immune responses it has become feasible to develop novel therapies that specifically target and eliminate tumors. A promising field of research may be to stimulate the immune system against the tumor by modulating the expression of cytokines using gene therapy. While immunotherapy is a promising approach to brain tumor treatment, both the anatomical structure and immune milieu of the brain in conjunction with the immunological features of brain tumors create a scenario in which effective immunotherapy faces many challenges. Below we will discuss some of the challenges that need to be surmounted before any of these therapies find their way to treat human GBM patients.
THE BRAIN IMMUNE SYSTEM
For the immune system to work effectively, it must be able to constantly monitor for the presence of foreign agents. This is known as the afferent arm of the immune system. Additionally, once a foreign agent is detected, a link between the innate and adaptive immune system must be made to activate the maturation and proliferation of cellular components of the adaptive immune response. This process is commonly referred to as the efferent arm of the immune system. In peripheral tissues and organs, innate immune cells such as macrophages and neutrophils constantly circulate and monitor for antigen expression and pathogen invasion. Macrophages and neutrophils phagocytose cell debris and other small particles, and macrophages display this processed antigen to T-cells. Neutrophils usually die shortly after phagocytosis and have not been considered very important as antigen presenting cells; however, recently it has been shown that cytokines produced at the site of inflammation can suppress apoptosis in neutrophils, and under these conditions neutrophils can become effective antigen presenting cells [13]. When appropriately stimulated, T-cells release cytokines and chemokines to induce activation of the adaptive immune response. Essential to activation of adaptive immunity is the presentation of antigen to T-cells by professional antigen presenting cells (APCs), the most efficient of whom are dendritic cells. Dendritic cells (DCs) migrate to the draining lymph nodes to present antigens to T-cells. Clonal expansion and activation CD4+ T-cells, CD8+ T-cells and B-cells that recognize this antigen then results in the eradication of the antigen by the immune system. This process can also lead to the induction of memory to prevent future infection from developing. A key requirement for DC activation and the initiation of both innate and adaptive immune resposnes is the stimulation of Toll-like receptors (TLR). TLRs are expressed on DCs and bind with conserved pathogen associated molecular patterns (PAMP) and increase the expression of co-stimulatory molecules on the surface of DCs and other APCs [14]. In the absence of co-stimulatory molecules, presentation of antigen to T-cells usually leads to T-cell tolerance.
The brain is an organ with specialized mechanisms to protect and maintain its functions. Among the specialized mechanisms is the interaction between the immune system and the brain parenchyma, which has an immune system that works in different ways when compared to peripheral sites, outside the central nervous system. For instance, if a pathogen is injected directly into the brain parenchyma, without reaching the brain ventricular system, a transient innate immune response occurs [15-24]; however, no adaptive immune response is mounted excusively from within the CNS. Immune responses in the brain are limited by several factors. Although immune cells are found within the cerebrospinal fluid (CSF), which drains into the central lymphatic system of the body [25], lymphatic surveillance, observed in the rest of the body, is not very efficient within the brain parenchyma. Foreign proteins and other antigens that exit the central nervous system (CNS) through the ventricular system can be phagocytosed and transported to peripheral lymphoid organs where they can elicit an adaptive immune response. Likewise, activated memory T-cells can detect antigen within the CNS, and target cells displaying antigenic epitopes. However, in the brain parenchyma of higher vertebrates there is a complete absence of lymphatic structures, and a noticeable absence of immune cells. Neutrophils, the largest population of circulating leukocytes involved in innate immunity, are rarely detected in either the CSF or brain parenchyma [25, 26]. DCs have not been detected in the normal brain [25, 27, 28] although they are observed in the meninges and choroid plexi [17, 20, 29-32]. Under inflammatory conditions both immature and mature DCs can infiltrate into the brain [28, 32-38]. While T-cells make up 80% of CSF cells [39], their presence within the brain parenchyma is limited unless an inflammatory condition exists. If a peripheral immune response is elicited against an antigen which is present within the brain parenchyma, then the activated immune cells can migrate into the CNS, and recognize the foreign antigen. This suggests that the afferent arm of adaptive immunity is deficient in the CNS and this in turn limits the response of the immune system to antigens which are exclusively located within the CNS.
CHALLENGES IN BRAIN TUMOR IMMUNOTHERAPY
Immunotherapy within the central nervous system has unique challenges based on the anatomical characteristics of the brain; however, the presence of a GBM adds another set of challenges (Fig. 1). GBM create an environment that is favorable to their continued growth, including evading detection by the immune system by creating an immunosuppressive environment, in part through the secretion of the powerful immunesuppressant TGF-β [40]. Glioma patients have been characterized as anergic to common bacterial antigens, unable to mount delayed type hypersensitivity responses, decreased circulating T-cells, depressed in vitro proliferation responses of T-cells to mitogens, decreased antibody response, and decreased antibody and T-cell cytotoxicity [41-47]. Interestingly, these peripheral dysfunctions can be partially reversed upon tumor resection, clearly implicating tumor involvement in global immunosuppression [48].
Fig. (1).
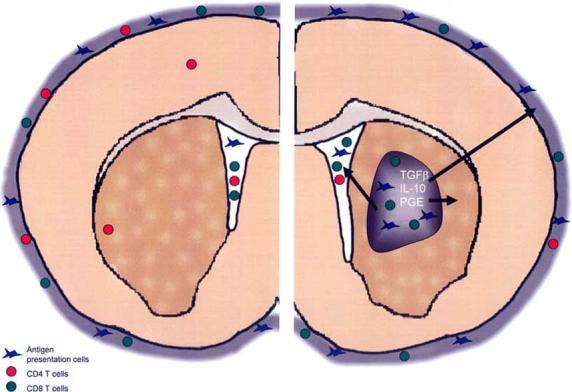
Immune cell surveillance of the normal and tumor bearing brain. In the normal brain, only memory CD4+ T-cells are found within the brain parenchyma while other immune cell types are restricted to the cerebrospinal fluid and meningeal layers. Tumors affect the local and systemic immune environment to evade immune detection by producing cytokines like TGFβ, IL-10 and PGE. CD4+ T-cell numbers are reduced systemically. Meanwhile tumors are infiltrated with macrophages and CD8+ T-cells whose normal immunological functions are blocked in the tumor environment.
While the glioma itself is infiltrated mainly with CD8+ T-cells [49-53], the number of peripheral T-cells in glioma patients are reduced [46]. Particularly affected are the number of CD4+ T-cells whose reduction disrupts the ratio of CD4+:CD8+ T-cells [54-56]. The T-cells isolated peripherally from glioma patients do not respond to mitogen in proliferation assays [57-59] nor do they serve as helper T-cells (TH) in vitro [56]. T-cells from glioma patients make and secrete less of the cytokine IL-2 essential for proper T-cell proliferation, and again, CD4+ TH cells are more profoundly affected [60, 61]. Human GBM are highly infiltrated with macrophages, which comprise up to 30% of the tumor mass [49]. Glioma patients’ monocytes have decreased MHC II expression on their cell surfaces as well as decreased IL-1β secretion [62], which limits their ability to properly present antigens to T-cells. In addition to immunosuppressive effects peripherally, tumor infiltrating monocytes may enhance glioma growth through the production of growth factors [49, 52, 63, 64], promotion of angiogenesis [65-67] and tumor invasion [68]. Together, this evidence indicates that the environment created by glioma cells actively affects the ability of the cellular and humoral adaptive immune response to function within the tumor or become activated in the periphery, limiting the ability of the immune system to eliminate the tumor (Fig. 1).
IMMUNOSUPPRESSION BY BRAIN TUMORS
Effects on peripheral immune cells may be mediated by the immunosuppressive factors secreted intracranially by glioma cells [5, 40]. Originally called glioblastoma cell-derived T-cell suppressor factor [69], transforming growth factor-β (TGF-β) is the most well characterized of these factors [41, 63, 69-78]. TGF-β is not produced in the normal brain but is overexpressed by gliomas [73, 74] and can act either to enhance or suppress tumor growth depending on the stage of tumor development [4]. TGF-β directly inhibits B cell and T-cell proliferation, cytotoxic T-cell development, monocyte cytokine marker expression, cytokine secretion, and also promotes migration of tumor cells [40, 63, 69, 73, 75, 76, 79-85]. Within the glioma mass, TGF-β downregulates MHC class II expression, decreasing antigen presentation to CD4+ T-cells [86]. TGF-β suppresses the cytotoxic response triggered by tumor infiltrating T-cells, and has been linked to the promotion of angiogenesis [87], tumor stroma formation [73, 88] and decreasing the number infiltration of T-cells, Natural Killer (NK) cells and macrophages [89]. While many studies show links between TGF-β secretion and immunosuppression, others indicate that TGF-β alone is not sufficient to account for the immunosuppression observed in GBM patients [40, 90, 91].
Other potential cytokines involved in GBM-induced immunosuppression are IL-10 [90] and Prostaglandin E2 (PGE) [92, 93]. IL-10 was identified as a cytokine synthesis inhibitory factor affecting the production of a wide range of cytokines. The role of IL-10 is controversial and can both enhance and inhibit glioma progression. Although IL-10 secretion from glioma cells inhibits IFNγ and TNFα production, both of which are required for anti-tumor T-cell proliferation and maturation [90, 94]. Conversely, the use of IL-10 as a therapeutic agent enhanced anti tumor immune responses [95]. While IL-10’s role in glioma mediated immunosuppression is controversial, PGE directly decreases lymphokine activated killer cell activity, T-cell activation and proliferation, and causes down regulation of MHC class II molecules [96]. PGE may be important in the T-cell suppression observed in glioma, although in vitro levels of PGE required for such effects are not physiologically relevant thereby indicating it alone is not sufficient for glioma mediated immunosuppression [97].
Immunotherapy would overcome the difficulties currently encountered in brain cancer treatment. Researchers have been actively exploring the potential of gene therapy to deliver immune-target molecules to improve survival in pre-clinical models of glioma and also in clinical trials. Interleukins, interferons, co-stimulatory molecules, death ligands, transduced dendritic cells and immune cell growth factors have all been evaluated for anti-tumor effects and will be discussed in the following sections.
GENE THERAPY OF BRAIN TUMORS USING DEATH RECEPTOR LIGANDS
The TNF receptor superfamily is a large group of receptors that play important roles in cell survival, proliferation, differentiation and apoptosis. Many of these receptors appear to have evolved in parallel with novel components of the vertebrate immune system and are important mediators of many immune responses [98]. A subfamily of the TNF receptor superfamily are death receptors, characterized by the presence of a conserved intracellular motif called the death domain that can activate the apoptotic machinery of the cell. They regulate tissue homeostasis by inducing cell death after binding with the corresponding death ligand [99]. Glioma and other brain tumors have been shown to express death receptors such as the Fas receptor or TRAIL receptors (TRAILR1 and TRAILR2) [100, 101]. Expression of the corresponding death ligands, Fas ligand and TRAIL, in glioma using gene therapy has been studied for their efficacy to induce apoptosis in the tumor cells. Many of these studies have focused on the cytotoxicity in vitro, when glioma cell lines are infected with viral vectors expressing either Fas ligand or TRAIL [102-106]. Despite some promising results in pre-clinical models of glioma [107-109], Fas ligand and TRAIL have yet to demonstrate efficacy in treating human patients with brain tumors.
GENE THERAPY OF BRAIN TUMORS USING INTERFERONS
Interferons are secreted by cells in response to viral infections or other inflammatory signals and trigger potent anti-viral and anti-proliferative effects, including increasing the expression of death receptors and other components of the apoptotic program, arresting the cell cycle and stimulating the maturation of several effector immune cells [110, 111]. Two families of interferons have been described, based on their ability to bind with Interferon receptor I or Interferon receptor II; type I interferons include Interferon α (IFNα), Interferon β (IFNβ) and Interferon ω (IFNω), while the only type II interferon described to date is Interferon γ (IFNγ) [111]. Type I and type II interferon receptors are present on most somatic cell in the body, and are also expressed on many tumors. In part due to their potent anti-proliferative function and their ability to stimulate the immune system, interferons have been evaluated as therapeutic agents in a number of pre-clinical trials in brain tumors.
Type I Interferons
Type I interferons (IFNα, IFNβ, IFNω) are expressed by most somatic cells immediately following infection with a virus. They are also produced, in much greater quantities, by Interferon Producing Cells (IPCs) of the immune system. These cells have since been identified as plasmacytoid dendritic cells, a distinct lineage of dendritic cell found distributed throughout the body [112, 113]. Type I interferons possesses several tumorstatic or tumorcidal effects including cell cycle inhibition [114, 115] and induction of apoptosis [116, 117]. In addition, a cluster of type I interferon genes are commonly deleted from the genome of human gliomas [118, 119] indicating that this pathway may play a role in glioma progression. Consequently, several groups have assessed the potential of type I interferon gene delivery to improve survival in pre-clinical glioma models. In one study, it was found that direct intratumoral injection of replication deficient adenovirus expressing IFNα significantly improved survival in a mouse model of glioma. The authors found that survival was greatly enhanced when DCs were injected into the tumor mass, and induced a specific cytotoxic T-lymphocyte (CTL) response against the tumor cells [120]. Either activation of DCs by IFNα, or apoptosis induced in tumor cells by IFNα may have resulted in improved antigen uptake by DCs and induced a potent CTL immune response against the tumor [120]. IFNβ has also been successfully used as a gene therapy approach for treating brain tumors in pre-clinical models and also in clinical trials. In a mouse model of brain tumors using GL261 cells, it was found that intratumoral delivery of IFNβ gene in liposomes significantly reduced the tumor volume. Furthermore, the authors demonstrated that this effect was dependent on CD8+ but not CD4+ T-cells [121]. IFNβ loaded liposomes have been delivered directly into the surgical cavity of 5 patients with recurrent malignant glioma after tumor resection. Of these, 2 patients survived for more than 2 years before succumbing to the disease [122]. Another approach that is currently being tested in a Phase I dose escalation clinical trial is the use of adenoviral vectors to deliver IFNβ directly into the surgical cavity following resection of glioma [123].
Type II Interferons
Interferon γ (IFNγ) is the only interferon known to bind to the type II IFN receptor. Intracranial delivery of IFNα using gene therapy reduced tumor growth in pre-clinical models of glioma by activation of the immune system [124] and inhibition of angiogenesis [125]. Delivery of IFNα and TNFα in combination into established mouse glioma using recombinant adenoviral vectors led to a significant increase in survival [126]. However, these results were not as striking as those obtained for IFNα and IFNβ, suggesting that IFNγ may not be the most efficacious choice for translation into clinical trials.
GENE THERAPY OF BRAIN TUMORS USING INTERLEUKINS
Interleukins (IL) are a family of small cytokines produced by leukocytes that play a key role in coordinating the response of other immune cells during an immune response. Many interleukins are important for proliferation or activation of immune cell subtypes, including IL-2, IL-3 and IL-4. However, as we described above, some interleukins such as IL-10 actually inhibit immune responses. The efficacy of Interleukin-2 (IL-2), IL-4, and IL-12 have been most thoroughly studied in pre-clinical brain tumor models and will be discussed here.
IL-2
IL-2 is an interleukin produced primarily by T-cells. It promotes the proliferation of a variety of T-cells in vitro, including TH and cytotoxic (TC) T-cells [127-129]. In vitro studies have demonstrated that IL-2 is required for T-cell proliferation, activation and effector responses. Consequently, it has been extensively studied as a potential therapeutic agent for treating glioma. It was reasoned that by increasing T-cell proliferation and activation at the site of the tumor, the anti-tumor immune response could be increased. When syngeneic 9L tumor cells engineered to express IL-2 were introduced into the CNS of rats, it was found that tumors developed in the CNS. These grew more slowly than wild-type tumors, but all of the animals eventually succumbed to the disease irrespective of IL-2 expression [130]. It was later found by the same group that IL-2 producing cells transplanted into the CNS could effectively eliminate established tumors by inducing immunity in peripheral tissues. This was accompanied by an increase in cytolytic T-cell activity against 9L cells when analyzed by Chromium release assay [131]. Combined administration of IL-2 with IFNγ, or IL-2 with IL-12 and P53 were also found to delay tumor growth in rodent models of glioma [132, 133]. Unfortunately, none of these combined therapies were very effective at eliminating tumor cells. With the recent development of IL-2 deficient mice and IL-2 receptor deficient mice, it has become apparent that T-cell proliferation is redundant in vivo and IL-2 deficiency does not decrease the numbers of T-cells at the site of infection. Instead of indiscriminately inducing T-cell proliferation, it is now believed that the principle function of IL-2 is to stimulate the proliferation of regulatory T-cells (TReg). CD4+/CD25+ TReg cells are absent in mice lacking either IL-2 or IL-2 receptor, and autoimmune disorders invariably develop in these mice, underscoring the importance of IL-2 in maintaining T-cell tolerance [134-136]. Consequently, IL-2 may not be a useful transgene for inducing robust immune responses against brain tumors.
IL-4
IL-4 is commonly associated with the induction of a type II TH response. TH2 responses are most important when defending against larger pathogens such as parasites, and also plays an important role in allergy [137]. IL-4 is produced by T-cells and regulates the maturation and proliferation of B-cells, mast cells and T-cells [138-142]. IL-4 expressing neuronal progenitor cells were injected into established tumors in both rats and mice. This was found to increase the survival of glioma bearing animals compared with controls [143]. In a related publication, 9L tumor cells were transfected with IL-4, IL-12, GM-CSF or IFNα. Although subcutaneous injection of 9L cells expressing IL-4, GM-CSF and IFNα was found to promote immunity to intracranial administration of parental 9L cells and prevented the establishment of intracranial tumors, only IL-4 was found to improve survival in animals with pre-existing tumors. One drawback of this approach was that rat 9L cells were transfected with mouse cytokines [144], which may have resulted in the incorrect assessment on the efficacy of GM-CSF, IL-12 and INFα when compared with IL-4. In fact, an earlier study by Sampson et al., found that GM-CSF and not IL-4 was effective in eliminating tumors in a mouse model of glioma [145]. Notwithstanding, a pilot study designed to study the efficacy of tumor cells expressing IL-4 and HSV-TK in combination to bolster the anti-glioma immune response has been undertaken in patients with malignant glioma [146].
IL-12
IL-12 is a cytokine that is associated with polarizing T-cells towards a type I TH response (TH1) by mediating the differentiation of naïve T-cells. Systemic administration of recombinant IL-12 has been demonstrated to improve survival in a variety of rodent tumor models [147, 148]. C57BL/6 mice bearing intracranial GL26 gliomas were treated by intratumoral injection of adenovirus expressing IL-12 (AdmIL-12), or control virus. IL-12 was found to significantly improve survival in these animals, indicating that IL-12 was able to delay tumor growth. Although enhanced infiltration of CD8+ and CD4+ T-cells was detected in the tumor tissue of animals injected with AdmIL-12, no significant increase in target cell lysis was noted compared animals treated with control virus [149]. This data suggests that while IL-12 is capable of inducing increased T-cell proliferation and infiltration into the tumor, antigen presentation is not markedly increased and the majority of these T-cells are as inefficient at tumor lysis as in control animals. To circumvent this problem, Yamanaka et al., designed a DC that constitutively produces Semliki Forest Virus carrying the IL-12 transgene (SFV IL-12) [150]. C57BL/6 mice bearing B16 tumors in the CNS were treated after 7 days with dendritic cells producing either SFV-IL-12 or control viruses. It was found that immunization with dendritic cells pulsed with SFV-IL-12 resulted in long-term survival in 40% of animals. This was associated with an increase in IFNγ production by murine splenocytes [150].
DENDRITIC CELLS
A limitation with using either interferons or interleukins is that although they stimulate various effector immune cells, they do not improve antigen presentation to DCs. Thus, although there is an increase in tumor infiltrating T-cells at the tumor, there is no increase in the percentage of T-cells that are stimulated by tumor antigens. To address this problem both co-injection of DCs and gene therapy utilizing co-stimulatory molecules or DC growth factors have been attempted.
Presentation of tumor antigen to T-cells and subsequent activation of the antigen-specific T-cell can occur directly, via MHC expression on the surface of the tumor cells to activated antigen specific T cells during the effector phase of the immune response, or indirectly, utilizing specialized APCs called DCs, during the activation of naïve T lymphocytes within the lymph nodes. Direct presentation of antigen is required for cytotoxic T-cell mediated killing of tumor cells and occurs when activated tumor-antigen specific CD8+ T-cells bind via the T-cell receptor (TCR) to MHC class I molecules, present on the surface of tumor cells, that display tumor antigen. Co-stimulatory molecules such as B7.1 (CD80) are expressed on the cell surface of DCs and other APCs and are required to first activate naïve T-cells following binding of the TCR with MHC molecules displaying the tumor antigen. In fact, antigen presentation to naïve T-cells in the absence of co-stimulatory molecules leads to T-cell tolerance rather than effector T-cell function. Usually, specialized APCs such as DCs are first required to present antigen to naïve, antigen specific T-cells in order to activate these T-cells. However, the possibility of using a gene therapy approach to express co-stimulatory molecules on MHC-expressing tumor cells and activate tumor specific T-cells. This was thought would circumvent the necessity of dendritic cells to first activate naïve T-cells. In a mouse model of glioma, it was found that B7.1 expression on tumor cells within the CNS was sufficient to improve survival in 60% animals for longer than 120 days [151]. Survival was significantly greater than animals injected with wild-type tumor cells, which survived for less than 20 days. However, the tumor model used in this study was injected into the subarachnoid space, a region of the brain with a large number of infiltrating immune cells, including T-cells and dendritic cells [151]. In two separate studies, one using transfected glioma cells and the other using adenovirus expressing the B7.1 transgene to infect glioma cells in vitro before intracranial implantation, a very weak anti-tumor response was observed This suggests that B7.1 expression alone is not sufficient to induce strong anti-tumor immunity against brain tumors [152, 153], most likely because priming of naïve T lymphocytes normally occurs within the microenvironment of the lymph nodes.
Other groups have used purified dendritic cells alone, or in combination with other therapies, to increase the levels of APCs at the site of the tumor. In effect, this approach is designed to develop a dendritic cell vaccine against the tumor by stimulating various components of the immune system. This has proved to be a successful approach in pre-clinical trials and there are currently at least 20 Phase II and one Phase III clinical trials ongoing in the US that involve injection of dendritic cells either directly into the tumor, or in the periphery [154-159].
Our group has developed a novel approach for increasing DC infiltration into the tumor. First generation, replication-defective adenoviral vectors (RAd) were used to deliver human soluble Fems-like tyrosine 3 ligand (Flt3L) directly into the tumor. Flt3L is a potent inducer of dendritic cell differentiation, proliferation, and activation [160-162]. Considering that administration of purified recombinant Flt3L induces infiltration of dendritic cells into tumors and tumor regression [163], our hypothesis was that expression of high levels of this transgene within the brain tumor using RAd vectors would increase intratumoral DCs infiltration and enhance the immune response against the tumor. Intracranial injection of RAdFlt3L significantly improved survival in a rat model of glioma. A large increase in macrophages and CD8+ T-cells infiltrating into the tumor was observed when compared with animals treated with controls [164]. This suggests that overexpression of Flt3L in the tumor stimulates an anti-tumor immune response that can eliminate tumor from the CNS.
HSV1-Thymidine Kinase (TK) has become the most popular and widely tested approach using gene therapy for killing glioma cells in pre-clinical models [165-167]. TK converts a non-toxic compound called ganciclovir into a toxic product that interferes with DNA synthesis and induces cell death in glioma cells [165]. Patients with GBM were analyzed in a randomized clinical trial that either received standard therapy including surgical resection and radiation therapy or intracranial administration of TK in a retroviral vector in addition to standard therapy. Although there was no difference in the levels of peripheral blood mononuclear cells in either group, the authors demonstrated that patients that received TK in addition to regular therapy had elevated IFNγ producing cells when stimulated with autologous tumor cells. Furthermore, serum concentrations of IL-12 and soluble Fas ligand were also elevated in these patients when measured by ELISA [168]. This was evidence that TK improved the ability of the immune system to recognize the tumor, albeit weakly. A growing body of evidence shows that tumoricidal therapies could induce some degree of tumor immunity [168]. Apoptotic tumor cells are phagocytosed and processed efficiently by dendritic cells and mediate much stronger anti-tumor immune responses. We hypothesized that TK expression in the CNS, in combination with immune stimulation using Flt3L, would greatly enhance the immune response against the tumor by providing apoptotic tumor cells to a large population of activated dendritic cells. This recapitulates the strategies used by researchers to pulse DCs in vitro with tumor antigen, i.e. coincubation of DCs with apoptotic glioma cells is known to dramatically improve antigen presentation by facilitating the uptake and processing of tumor cells by DCs.
To test whether Flt3L, or adenoviral vectors expressing other cytokines, could improve survival when combined with the cytotoxic RAdTK vector, viral vectors were injected into a large tumor model in rats. Animals injected with RAdFlt3L alone were all dead by day 20, while RAdTK treatment alone only improved survival in 20% of animals. However, when animals were treated with RAdFlt3L and RAdTK together, 70% survived for more than 175 days (Fig. 2). The use of first generation adenoviruses expressing IL-12, which promotes a TH1 immune response, CD40L, a co-stimulatory molecule had no effect on survival even when combined with RAdTK, suggesting that these immunostimulatory molecules were unable to promote a strong immune response against the tumor [169]. Thus, increasing dendritic cells within the tumor was a better approach to stimulating an immune response against the tumor than either increasing T-cell function, or increasing the expression of co-stimulatory molecules on tumor cells. Depletion of immune cells from glioma bearing animals before administration of RAdFlt3L and RadTK showed that CD4+ T-cells and macrophages were required for tumor regression [169]. In contrast, depletion of either CD8+ T-cells or NK cells did not affect the survival of animals treated with RAdFlt3L and RAdTK, suggesting that these cells did not play an important role in tumor regression [169].
Fig. (2).
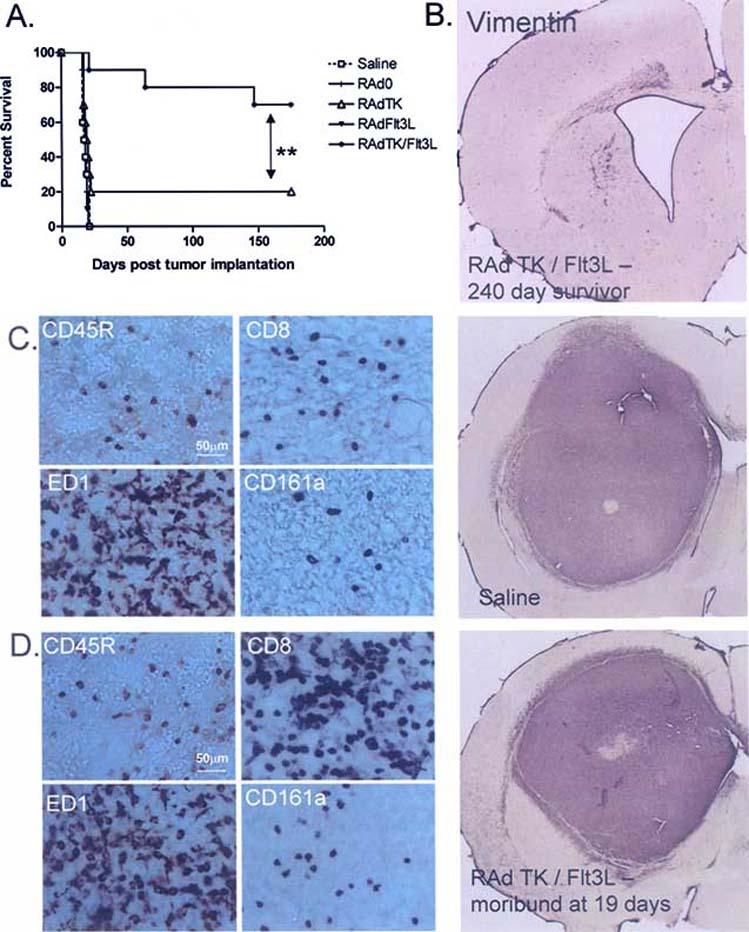
Immunotherapy of brain tumors using RAdFlt3L and RAdTK. A. Survival curve from rats treated ten days after tumor implantation with replication deficient adenoviruses. RAdTK+Flt3L results in tumor regression in 80% of animals (modified from Cancer Research [169]). B. Brain section from a glioma survivor 240 days after tumor implantation followed by RAdTK and RAdFlt3L treatment. Brain sections were stained with vimentin to detect any reminant of tumor cells. C and D. Brain sections from animals treated with either saline or RAd TK+Flt3L 10 days after tumor implantation. While 80% of animals treated with RAd TK+Flt3L survive, neuropathological analysis of those who secumb shows immune cell infiltration that is distinct from saline treated controls. As with human tumors, rat CNS-1 gliomas are heavily infiltrated with macrophages (ED1 staining) regardless of treatment modality. B cells (CD45R staining), cytotoxic T cells (CD8 staining), and Natural Killer cells (CD161a staining) are all increased in treated animals compared to controls.
A major advantage of stimulating DCs with tumor antigen in vivo instead of ex vivo is that DCs encounter the tumor antigen in their natural environment. Other co-factors involved in antigen processing and presentation are already available, simplifying the manipulation required to maximize anti-tumor immune responses. In addition, these gene therapies can be administered to patients at the same time as tumor resection by surgery, while ex vivo manipulation of DCs requires a week or more before the patient can receive the immunotherapy. This means that the tumor is rapidly hit with the anti-tumor immune response, is not given any chance to recover from surgical resection and the therapy can take advantage of any inflammation naturally associated with the tumor resection.
CHEMOTHERAPY AND IMMUNOTHERAPY
Many immunotherapy trials are conducted in patients that have received or are currently receiving chemotherapy. The majority of contemporary chemotherapeutic agents are often designed to preferentially induce toxic chemical insults on tumor cells, leading to cell death by apoptosis, necrosis, or autophagy. Molecular targets are identified that make tumor cells more susceptible to chemical agents, for example, many target the rapid proliferation of tumor cells compared with normal somatic cells of the body. It was quickly realized that the immune response against the tumor is augmented in patients receiving chemotherapy at the same time. Low levels of cell death, when detected by immune cells, leads to the suppression of inflammation by stimulating the production of IL-10, TGF-β and prostanoids [170]. Apoptosis occurs frequently even in healthy tissues and this anti-inflammatory response is believed to prevent unnecessary inflammation and tissue damage in otherwise healthy tissues. Massive levels of cell death leads also to the release of heat shock proteins (HSPs) and uric acid, both known to be powerful inflammatory signals [171, 172]. Studies using pre-clinical models have indicated that immunotherapy and chemotherapy can be synergistic in the treatment of brain tumors. For example, intracranial delivery of IL-2 alone marginally improves survival in an intracranial model of metastatic melanoma in the brain of mice. However, combined therapy with systemic administration of the chemotherapeutic drugs carmustine or carboplatin and IL-2-transduced tumor cells dramatically enhanced long-term survival with 70% of animals surviving for 70 days in a model where all controls were dead by day 20 [173]. It is important to note that radiotherapy and many chemotherapeutic agents currently used against GBM are also toxic to cells of the immune system. In particular, they tend to be toxic to rapidly dividing cells and this may serve to disrupt or delay the development of immunity against the tumor when combined with immunotherapy. A pre-clinical study noted that administration of dexamethasone, a glucocorticosteroid given to reduce pressure within the CNS casued by chemotherapy, in combination with IL-4 gene therapy completely abrogated the positive effects of IL-4 immunotherapy on the tumor bearing animals [174]. The dexamethasone dose found to cause complete immune suppression (250 μg/Kg/day) is equivalent to the dose given to human patients together with chemotherapy (4mg qid). Even doses 5 times lower than this still significantly inhibited the therapeutic benefits of IL-4 [174]. As a consequence, in clinical trials that use immunotherapy in combination with conventional treatment modalities, the immunotherapy is given first, and chemotherapy is given later after immunity against the tumor is allowed to develop. In this scenario, chemotherapy given later tends to augment the immune response that develops. For example, in a retrospective study of glioblastoma patients that received DC vaccination and conventional chemotherapy after surgery, it was reported that chemotherapy can improve the anti-tumor immune response in patients receiving immunotherapy. Vaccinated patients with glioblastoma that received subsequent chemotherapy were found to be tumor free for significantly longer than patients that received either vaccination alone, or chemotherapy alone and had elevated anti-tumor responses and CD8+ T-cell receptor excision circle content was predictive of response to chemotherapy [175]. These studies in preclinical animal models and clinical trials suggest that by enhancing the host immune response and reducing tumor burden, a synergistic effect leads to a more efficacious therapy. A very attractive approach to specifically reduce tumor burden and release tumor antigens which could be harnessed to stimulate a specific immune response against the tumor, is the use of targeted toxins. Although targeted toxins are already in clinical trials for treating GBM [176, 177], below we will review the potential of combining targeted toxins and gene therapy to develop safer and long lived therapies for GBM.
TARGETED TOXINS FOR GLIOMA THERAPY
Novel chemotherapeutic agents that can induce widespread tumor cytotoxicity without adverse toxicity to components of the immune system or healthy surrounding tissue are highly desirable, as these would improve the efficacy of immunotherapies currently being developed [178]. Receptors for the urokinase-type plasminogen activator (uPA) [179], transferrin [180-182], pleiotropic immunoregulatory cytokines [183-188] and growth factors [189, 190] are overexpressed by human brain tumors but are virtually absent in the normal brain. Such specificity makes these receptors very attractive targets for targeted therapeutic approaches in glioma, minimizing any putative adverse side effects to normal brain tissue. Thus, ligands of these receptors, such as IL-13, uPA, EGF and transforming growth factor α (TGF-α) have been fused to the catalytic and translocation domains of highly cytotoxic bacterial products, including Pseudomona [185, 190, 191] and Diphteria toxins [179, 187, 189, 192], in order to selectively kill glioma cells (Fig. 3).
Fig. (3).

Targeted toxins for the treatment of glioma. Human brain tumors have been shown to overexpress several receptors, including urokinase-type plasminogen activator receptor, transferring receptor, pleiotropic immunoregulatory cytokine receptors and growth factor receptors. The expression of these receptors seems to be more abundant in malignant tumors, than in benign, slow growing tumors. Since these receptors are virtually absent in the normal brain, they have been targeted in several therapeutic approaches in the treatment of glioma, to avoid toxicity to normal brain tissue. Ligands of these receptors, such as IL-13, IL-4, uPA, transferrin, EGF and TGF have been fused to the catalytic and translocation domains of highly cytotoxic bacterial products, including Pseudomonas and Diphteria toxins (T), in order to kill selectively malignant glioma cells, but preserving surrounding normal brain tissue.
IL-13
Established glioma cell lines, primary glioblastoma cell cultures and surgical glioma biopsies, express a variant of the IL-13 receptor, IL13Rα2, different from its physiological counterpart, i.e., IL13/IL4R [183-185, 192, 193]. The chimeric toxin composed of IL-13 and truncated Pseudomonas exotoxin, also termed IL-13 toxin, exerts a potent cytotoxic effect in most human glioblastoma cells tested in culture [185, 187, 191] and in vivo, in human xenografts consisting of glioma cells implanted in the flank of nude mice [194]. Moreover, the intratumoral administration of IL13-PE toxin into intracranial human glioma xenografts in immunodeficient mice showed highly cytotoxic effects without undesirable side effects [195]. To optimize the targeting of GMB-associated IL-13α2 receptor, a mutated human IL-13 which exhibits 50 fold higher affinity for the IL-13α2 receptor present in human glioma cells when compared to the wild-type IL-13 was engineered [196, 197]. Fusion of this muIL-13 to PE resulted in an even more active cytotoxin on glioma tumors both in vitro and in vivo [196]. Importantly, the muIL13 no longer interacts with the principal chain of IL4R, thus becoming ineffective in its binding to this receptor and signaling through the physiological IL13/IL4R of normal cells. This in turn decreases the already low toxicity of the chimeric toxin to normal cells [196]. Thus, although this mutant has negligible affinity by IL-13 receptor of normal cells, it exerts an enhanced cytotoxic effect towards glioma cells. The fact that IL13Rα2 is over-expressed not only in glioma cells, but also in other malignancies, including renal cell carcinoma [198], ovarian carcinoma [199], colon adenocarcinoma [185], epidermoid carcinoma [191], AIDS-associated Kaposi’s sarcoma [200], prostate carcinoma [201] and pancreatic cancer [202], makes IL13Rα2 a unique target for anti-cancer therapy. In a PhaseI/II clinical trial, patients with glioblastoma multiforme were intratumorally injected with IL-13 toxin 8 days before surgical resection [176]. Necrotic areas were found in the tumors from half the patients, suggesting that the toxin successfully induced tumoral cell death.
IL-4
Human malignant glioma cell lines, primary cell cultures, and tumor specimens derived from surgical samples also express high affinity IL-4 receptors [186, 188]. A circular permuted IL-4 fused to a mutated form of Pseudomonas exotoxin, showed highly cytotoxic effect to cancer cells, but was not toxic to normal cells that express detectable IL-4 receptors, such as, B-cells, T-cells, and monocytes [203]. In vivo models of human glioblastoma in immunodeficient animals demonstrated that this immunotoxin also exhibits remarkable anti-tumor activity [204, 205]. Phase I and Phase I/II clinical trial were developed for the treatment of recurrent glioblastoma multiforme, showing that IL-4 cytotoxin intratumoral administration to patients has an acceptable safety profile, being well tolerated at low doses [177]. These studies suggest that this cytotoxin has anti-tumor activity, inducing necrosis in the tumor parenchyma, without histological evidence of toxicity to normal brain tissues [206]. Although local toxicity, such as intracranial edema, was reported, it seems to be due to tumor necrosis or occasionally to the volume of infusion.
Transferrin
The cytotoxic activity of targeted toxins constructed with human transferrin fused to Pseudomonas toxin or with Diphteria toxin was detected in human brain tumor cell lines [180, 182]. In ex vivo experiments, pediatric brain tumor tissues were shown to be sensitive to transferrin-diphteria toxin [182], being that the toxin efficacy correlated with tumor grade. Transferrin receptor expression was high in the more aggressive and malignant tumors, such as glioblastoma multiforme and medulloblastoma, which were extremely sensitive to transferrin-diphteria toxin, while slow-growing and benign tumors expressed lower levels of receptor and were not as greatly affected by the toxin. Nude rats inoculated intracranially with human glioblastoma biopsy specimens, which develop highly infiltrative brain tumors, received direct interstitial infusion of transferrin-diphtheria toxin, showing strong anti-tumor efficacy [207]. In clinical trials, patients with recurrent malignant brain tumors, which were refractory to conventional therapy, were locally treated with the transferrin-diphteria toxin administered by high-flow interstitial microinfusion [208]. Although episodes of local toxicity in some of the patients were reported, direct interstitial infusion was shown to successfully distribute the toxin in the tumor and infiltrated brain areas, achieving anti-tumor responses without severe neurologic or systemic toxicity [209].
EGFR
Overexpresssion of EGFR was found in more than 50% of high grade gliomas by several authors [187, 210-212]. EGFR ligands, such as EGF and TGF, have been fused to Pseudomonas and Diphteria toxins. A chimeric toxin consisting of TGF and Pseudomonas toxin was systemically administered to nude mice bearing glioblastoma xenografts in the flank, inducing tumor regression [190]. However, mice bearing intracranial tumors required intratumoral administration of the toxin to increase survival time. Overexpresssion of EGFR was found in more than 50% of high grade gliomas by several authors [181, 210-212]. EGFR ligands, such as EGF and TGF, have been fused to Pseudomonas and Diphteria toxins. Diphtheria toxin was fused to EGF and systemically administered to nude mice bearing subcutaneous human glioma. The toxicity of the fusion protein at high doses included loss of activity, reduced oral intake, and dehydration, elevated blood urea nitrogen, creatinine, aspartate transaminase, and alanine transaminase and renal tubular necrosis. However, tumor regression was seen in all animals, while relapses occurred 25% of the animals [189]. A chimeric toxin consisting of TGF and Pseudomonas toxin was systemically administered to nude mice bearing glioblastoma xenografts in the flank, inducing tumor regression [190]. However, mice bearing intracranial tumors required intratumoral administration of the toxin to increase survival time. In a Phase I trial the dose limiting toxicity of this chimeric toxin was determined after convection-enhanced delivery in patients with recurrent malignant brain tumors. In this study, which included 20 patients, the maximal tolerated dose could not be established, being the overall median survival 23 weeks after intracranial administration of the toxin [213]. Considering that EGFR is overexpressed not only in malignant brain tumors, but also in other neoplasias, including squamous cell carcinomas, adenocarcinomas, sarcomas, brain and germ line tumours [214], this receptor constitutes a promising target for anticancer therapy.
Although the expression of one or more of these receptors is wide spread between malignant brain tumors, which of these is overexpressed in each particular patient needs to be determined prior the treatment for the therapy to be successful or the administration of a cocktail of chimeric toxins could address the individual sensitivity to this novel and promising therapy. A drawback to the use of these toxins in clinical setting is that the half-life after intracranial administration is short requiring long periods of constant intracranial infusion. An approach that combines gene therapy with the targeted toxin technology may circumvent this problem by continually expressing chimeric toxins within the CNS.
CONSIDERATIONS WHEN USING IMMUNOTHERAPY AND GENE THERAPY TO TREAT GBM
Auto-Immune Disorders Induced Through Immunotherapy
Immunotherapy used to treat melanoma has been associated with autoimmune response to melanocytes, resulting in autoimmune vitiligo [215, 216]. A major consideration with using immunotherapy to treat brain tumors, or any other brain disease, is that the CNS contains a number of antigens not normally visible to the immune system. Tolerance of the immune system to these antigens does not develop and later exposure of the immune system to these antigens may potentially induce a harmful autoimmune response. Recent research has illuminated many of the mechanisms leading to autoimmune disorders against CNS antigen, and in particular, in models of multiple sclerosis. It has been postulated that APCs may be able to take up CNS antigen process it and present it to naïve, antigen specific T-cells without the need to migrate to proximal lymph nodes. Furthermore, persistent presentation of dominant proteolipid protein (PLP) antigen is necessary to stimulate auto-immune diseases [217, 218]. We have analyzed the motor coordination and immune cell infiltrates into the CNS of long-term survivors of glioma treated with RAdTK and RAdFlt3L. Lewis rats were used, as these have previously been shown to be susceptible to experimental autoimmune encephalomyelitis (EAE) [219, 220]. None of the animals were observed to display any overt symptoms of EAE. However, elevated CD8+ T-cells infiltrating in the corticospinal tract and meninges of all survivors treated with RAdFlt3L, RAdIL-12 and RAdCD40L, suggests that this may be a side effect of immunotherapy against CNS tumors [169] although none of the long term surviving animals developed clinical auto-immune disease.
IMAGING GENE THERAPY AND DISEASE PROGRESSION IN VIVO
Accurate Assessment of Tumor Regression and Gene Therapy Distribution
By inducing anti-tumor immunity, it is important to note that tumor regression will occur more slowly than with conventional chemotherapeutic agents. This is due to the nature of an immune response, which, after identifying antigen for destruction, undergoes a phase of clonal expansion where effector cells are produced in large numbers. This process can take a week or more, before these cells mature and enter the tumor, where they target tumor cells and induce cell death. In order to correctly monitor the response of the patient to the tumor, imaging systems currently available in the clinic and novel imaging technologies will be required. A number of imaging techniques are available and are applied in the field of neuro-oncology, including MRI scans, PET scans, PEBBLEs, 3T1HMR Spectroscopy and Infrared (IR) Imaging Spectroscopy [221]. Several useful applications exist for each imaging technique, especially for gliomas within the brain. MRI and PET scans provide an informed diagnosis of the tumor, determining the size, vascularity, severity and type (primary vs. metastic). They also track the progression of the tumor much more efficiently than biopsies, which can give an incorrect assessment of tumor size, due to the heterogeneous nature of gliomas. MRI and PET scans are non-invasive methods which can be used for checking the effectiveness of radiation treatment and gene therapy and provide a means to track the tumor pre- and post-operatively, denoting the area that needs to be excised (or that requires surgical treatment) and identifying remaining regions containing tumor infiltrates after resection. They also serve to monitor and assess the rate and extent of tumor regression following therapy, and in particular, will allow the physician to monitor any regions of the brain that may contain tumor infiltrates.
Magnetic Resonance Imaging (MRI)
MRI uses a powerful magnetic field to determine the nuclear magnetic spin and resonance properties of a small volume of tissue. Different tissues have different nuclear magnetic spin and resonance properties, and by collating this information, a scan of the tissue or body can be produced. The use of MRI to detect tumors is based on the observation that tumors and normal tissue differ in the time required for nuclear magnetic relaxation [222]. Before surgery, imaging allows the surgeon to know if the glioma is embedded in an inaccessible area within the brain. If surgery is reasonable, imaging enables the opportunity to determine whether or not the tumor has been completely or partially removed. MRI diffusion imaging provides fibre-tract mapping, thus enabling the surgeon to plan the surgical strategy more efficiently. Once the tumor is removed, intraoperative MRIs can detect remaining tumor infiltrates within the brain. To aid surgical accuracy, 3D imaging techniques are being developed to reveal all of the heterogeneous aspects of GBM [223]. These systems will improve the oncologist’s understanding and knowledge of the tumor; size, location, and infiltration into surrounding CNS tissue and allowing more successful tumor resection.
Many imaging techniques are able to track the progression and grade of the tumor. Often imaging relates directly to the histopathological characteristics and the clinical behavior of the tumor. For example, a biopsy may indicate a low-grade tumor; however, while that particular section of the tumor expresses traits of a low-grade tumor, other portions of the tumor may express high-grade tumor characteristics such as invasion or metastasis. By comparing old MRIs to more recent ones, oncologists can establish a treatment appropriate for that specific tumor progression pattern. Successive imaging surveillance allows early detection of tumor progression, for progression is variable depending on age, etc. MRI’s may also elucidate clinical behavior unexplainable by biopsy sections. By comparing changes in vascular volume to changes of tumor size after gene therapy treatment, one can be able to verify whether or not the treatment is effective.
Positron Emission Tomography (PET)
Usually, brain tumors are visualized with different, complimentary imaging techniques. While MRI detects changes in tissue density, and is useful for studying the anatomy surrounding the brain tumor, PET scanning is often a more sensitive technique for detecting small tumors. Unlike MRI, PET scanning detects the decay of a radioactive molecules. Chemical tracers such as amino acids labeled with radioactive atoms such as Carbon-11, Nitrogen-13 Oxygen-15, Fluorine-18, or Iodine-124 can be injected into patients with brain tumors. These can detect early lesions of the tumor using PET scans, whereas CT and MRI scans are often unable to identify the presence of small or narrow regions of tumor. Because certain amino acids accumulate intensely at an early stage of the tumor, the tumor mass can be readily distinguished from normal brain tissue using radiolabelled amino acid tracers [224, 225]. This is of particular use when trying to identify tumor metastases, or in the case of glioma to detect regions of the brain with small numbers of infiltrating tumor cells.
The resolution of PET is currently less than 1 mm, sufficient to allow researchers that use rodent models of brain tumors to utilize micro-PET imaging. This allows researchers to accurately monitor the growth kinetics of the tumor in response to numerous therapies. Numerous tracking methods exist, each one depending on the expression of the reporter gene. A truncated form of the dopamine receptor (DR2R80A) and TK can both be used as reporter genes in tumor cells implanted into rodents and have been tracked by PET scan imaging [226-229]. Modified, radiolabelled dopamine analogues and TK substrates can be injected systemically into the animal where they diffuse across the blood brain barrier and subsequently accumulate in the CNS in regions with elevated expression of the reporter genes DR2R80A or TK (Fig. 4A). TK is a viral protein, and is not normally expressed in the CNS. Consequently, TK allows the identification of this reporter gene expression to a very high degree of sensitivity. By expressing the reporter gene in glioma cells, researchers have monitored the progression of the tumor, as well as size and growth after therapy and/or resection. Therapeutic efficacy of the treatments to be tested is determined based on levels of reporter gene expression in live rodents, thus eliminating the need to euthanize the subjects. This reduces the number of animals required for each study and improves the ability of researchers to assess the efficacy of their therapy [224]. In contrast, Dopamine receptors are present in the CNS. However, DR2R80A can be used to image in the brain striatum as this region is completely devoid of Dopamine receptors. A major advantage with PET over other imaging systems is that several markers can be used simultaneously, thus enabling the researcher multiple tracing options.
Fig. (4).
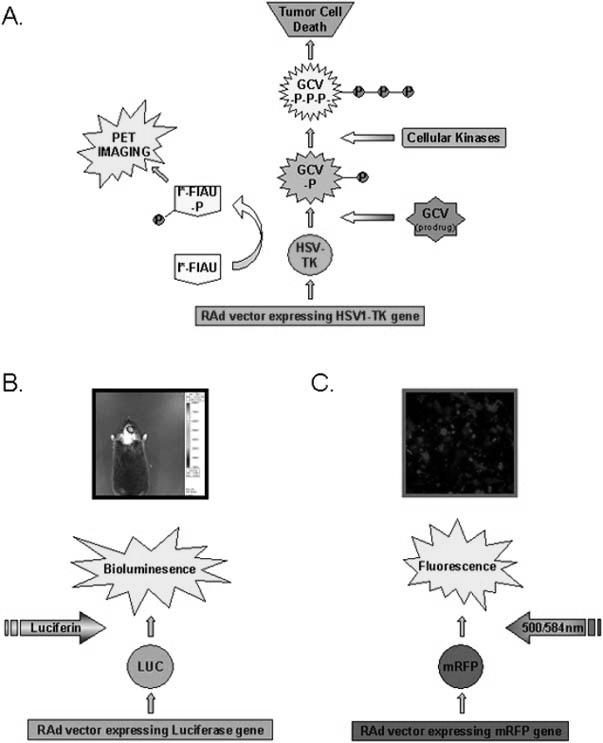
Stratagies for live animal imaging A. HSV1-Thymidine kinase (TK) is expressed by a first generation adenoviral vector (RAd) and catalyzes the phosphorylation of the prodrug ganciclovir (GCV) to generate GCV-monophosphate (GCV-P). Cellular kinases further phosphorylate to form a nucleotide analogue GCV-triphosphate (GCV-P-P-P) which interferes with DNA synthesis and induces apoptosis in dividing cells. In addition, the HSV1-TK protein interacts with the imaging substrate (124I)-labelled-2’-fluoro-2′-deoxy-1-ß-D-arabinofuranosyl-5-iodouracil (I*-FIAU) to form a phosphorylated version of the molecule (I*-FIAU-P) which can be used for PET imaging. I*-FIAU is membrane permeable and can diffuse freely through the plasma membrane of cells. However, following phosphorylation by TK, I*-FIAU-P becomes hydrophylic and remains trapped in the cell expressing TK. Consequently, the concentration of radioactive 124I increases only in cells expressing TK and is rapidly cleared from the rest of the body. B. A RAd expressing Firefly Luciferase was injected into the striatum of a mouse. Seven days later the mouse was injected with the substrate D-Luciferin and the animal was imaged using a CCD-based in vivo imaging system (IVIS, Xenogen). The intensity of light production was evaluated and is represented using a color scale with red being regions with most intensity and blue being regions with lowest intensity, as indicated on the scale to the right of the image. C. A RAd expressing monomeric red fluorescent protein (mRFP) was infected onto a monolayer of 293 cells. The monolayer was imaged using a inverted fluorescent microscope 24 hours after infection (Axiovert 200, ZEISS).
Bioluminescence Imaging
PET detects gamma rays, emitted following the radioactive decay of short-lived isotopes to build a 3 dimensional image within the CNS. In pre-clinical rodent models, light rays emitted following the conversion of substrate into product can also be detected through layers of tissue using a bioluminescence imaging system with a cooled charge couple device (CCD) camera (Fig. 4b). Firefly and Renilla luciferase catalyzes the emission of light during the conversion of substrate into product. It has been shown that D-luciferin is an exclusive substrate for firefly luciferase, while coelenterazine is only metabolized by renilla luciferase in vitro and in vivo, and this has opened up the possibility of imaging two separate targets in vivo, one with Firefly luciferase and the other with Renilla luciferase [230, 231]. Unlike PET, bioluminescence imaging does not give high resolution, 3 dimensional images of the tumor. However, the size of the tumor is proportional to the amount of light emitted, and this can be used to estimate the tumor volume. Furthermore, equipment for bioluminescent imaging is more affordable than microPET and safer as it does not involve the use of radiolabelled dyes.
Expression of Luciferase has been used by researchers to monitor the growth kinetics of intracranial tumors, and to track the migration of neural precursor cells (NPCs) in vivo. Tumor cells were generated that express Renilla luciferase and these were implanted into nude mice. Immortalized NPCs were generated that express Firefly luciferase and the therapeutic transgene soluble TRAIL (sTRAIL). Migration of these NPCs into brain regions containing tumors was tracked using the Firefly luciferase substrate D-Luciferin. Tumor volume was estimated based on total light produced when mice were injected with the Renilla luciferase substrate ceolenterazine. Mice that received NPCs expressing sTRAIL displayed significantly slower growth kinetics compared with control NPC injected animals [108]. Luciferase substrates can also be engineered to require an activation step by a cellular enzyme before being catalyzed by luciferase enzyme. For example, D-Luciferin has been conjugated with DEVD and is only converted to product with the liberation of light after it has been processed by active Caspase 3, a hallmark of apoptotic cells. This has allowed researchers to monitor the activation of Caspase 3, and consequently the extent of apoptosis, in vivo in a mouse model of glioma following treatment with sTRAIL [109].
Future Directions of Imaging in Gene Therapy
An exciting future direction for imaging with PET scanning and bioluminescence imaging is in the field of gene therapy. Assays exist for the majority of chemotherapeutic agents, to determine the pharmacokinetic and pharmacodynamic properties of the compounds in the patient. This is often found to vary substantially and the dose of a substance must be increased or decreased accordingly to obtain maximum therapeutic benefit. No reliable, relatively non-invasive method has yet been developed that can accurately assess the distribution and expression of therapeutic transgenes within the brain following injection of viral vectors. This is a major drawback in gene therapy and severely limits the ability to modify the dose if required. It is hoped that incorporation of genes into these viral vectors that can be detected using PET or bioluminescence imaging techniques will allow for a more detailed assessment of transgene expression and distribution in the patient. Different reporter genes can be incorporated into the same vector. In one application, a lentivirus was constructed to express both TK and firefly luciferase. When cells stably infected with lentivirus were injected into mice, both luciferase and TK could be imaged using Bioluminescent imaging and microPET, respectively [232]. Likewise, three reporter genes were detected independently in live mice bearing 293T cells transfected with TK, Renilla luciferase and monomeric red fluorescence protein (mRFP1). Excitation of mRFP1 occurs at wavelengths of 500nm and 584nm while the maximum emission occurs around 607nm (Fig. 4c). The activity of each reporter gene was shown to be preserved, suggesting that multiple reporter genes can be used in combination both in a clinical setting and also for pre-clinical research [233]. A second useful application will be to monitor the infiltration of specific immune cells into the tumor following immunotherapy, allowing more accurate quantification of the extent of the immune response against the tumor.
CONCLUSIONS AND FUTURE PROSPECTS
Novel treatments for brain cancer (GBM) require the development of approaches which encompass several mechanisms of action, including different modes of cell death and immune stimulation. These novel therapies would work synergistically with the best treatment options currently available; i.e., surgery, radiotherapy and chemotherapy. Gene-based therapies are actively pursued to treat GBM, include oncolytic viruses to selectively replicate in cancer cells, causing their destruction; anti-angiogenic targets that aim at depriving the growing tumor of new blood vessels needed for it to spread; targeted toxins, which after delivery into tumor cells will cause their death; immune-stimulatory targets aimed at eliciting an anti-tumor immune response to inhibit tumor growth and prevent the spread of metastatic disease. Other exciting approaches are targeted at inhibiting signal transduction pathways, activated in GBM and shown to mediate tumor progression.
Although many of these approaches have shown excellent efficacy with low or no toxicity in preclinical animal models, their success has not been reproduced in human clinical trials. There are several reasons for this, such as the need to enhance the specificity and efficacy of the gene transfer vectors and the therapies. This can be achieved by the use of targeted vectors, which have been engineered so that they can only infect tumor cells; or the use of the cancer cells’ specific promoters which will drive the expression of therapeutic molecules exclusively in the tumor. The ability to generate a systemic long lived anti-tumor immune response is also a critical advancement which would prove very powerful for the treatment of GBM, which inevitably recurs. Immune stimulatory approaches mediated through the delivery of genes which induce immune cell recruitment and/or activation is an approach which is being actively pursued for the treatment of GBM. For the clinical implementation of these therapies, it is imperative to be able to monitor disease progression and persistence of the gene therapy in vivo, using non-invasive imaging techniques. This is an exciting technology which will enable the monitoring of the persistence of the therapeutic vectors and also their putative bio-distribution.
Perhaps the biggest challenge before these gene-based therapies can be successfully and safely implemented to treat GBM in human patients, is the issue of the toxicity and bio-distribution of the gene transfer vectors used. Also, the pre-existing immune response to the vectors can have very serious deleterious effects, not only by decreasing the effects of the therapy by inhibiting therapeutic gene expression, but also by causing severe, systemic, immune-related side effects. Again, this is an area which is the focus of many investigations and has led to the development of better and safer vectors.
ACKNOWLEDGEMENTS
Grant support: NIH/National Institute of Neurological Disorders and Stroke grant 1R01 NS44556.01; National Institute of Diabetes and Digestive and Kidney Diseases grant 1 RO3 TW006273-01 (M.G. Castro); NIH/National Institute of Neurological Disorders and Stroke grants 1 RO1 NS 42893.01, U54 NS045309-01, and 1R21 NS047298-01, and Bram and Elaine Goldsmith Chair in Gene Therapeutics (P.R. Lowenstein); The Linda Tallen and David Paul Kane Annual Fellowship (M.G. Castro and P.R. Lowenstein); and the Board of Governors at Cedars Sinai Medical Center.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. We thank S. Melmed, R. Katzman, and D. Meyer for their academic leadership and superb administrative and organizational support.
REFERENCES
- [1].Ali-Osman F. Contemporary Cancer Research. Humana Press; Totowa: 2005. Brain Tumors. [Google Scholar]
- [2].Legler JM, Ries LA, Smith MA, Warren JL, Heineman EF, Kaplan RS, Linet MS. Cancer surveillance series [corrected]: brain and other central nervous system cancers: recent trends in incidence and mortality. J. Natl. Cancer Inst. 1999;91(16):1382–90. doi: 10.1093/jnci/91.16.1382. [DOI] [PubMed] [Google Scholar]
- [3].Surawicz TS, Davis F, Freels S, Laws ER, Jr., Menck HR. Brain tumor survival: results from the National Cancer Data Base. J. Neurooncol. 1998;40(2):151–60. doi: 10.1023/a:1006091608586. [DOI] [PubMed] [Google Scholar]
- [4].Platten M, Wick W, Weller M. Malignant glioma biology: role for TGF-beta in growth, motility, angiogenesis, and immune escape. Microsc. Res. Tech. 2001;52(4):401–10. doi: 10.1002/1097-0029(20010215)52:4<401::AID-JEMT1025>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- [5].Yang L, Ng KY, Lillehei KO. Cell-mediated immunotherapy: a new approach to the treatment of malignant glioma. Cancer Control. 2003;10(2):138–47. doi: 10.1177/107327480301000205. [DOI] [PubMed] [Google Scholar]
- [6].Freije WA, Castro-Vargas FE, Fang Z, Horvath S, Cloughesy T, Liau LM, Mischel PS, Nelson SF. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004;64(18):6503–10. doi: 10.1158/0008-5472.CAN-04-0452. [DOI] [PubMed] [Google Scholar]
- [7].Kaliss N. Regression or survival of tumor homoiografts in mice pretreated with injections of lyophilized tissues. Cancer Res. 1952;12(5):379–82. [PubMed] [Google Scholar]
- [8].Kaliss N, Molomut N. The effect of prior injections of tissue antiserums on the survival of cancer homoiografts in mice. Cancer Res. 1952;12(2):110–2. [PubMed] [Google Scholar]
- [9].Barnetson RS, Halliday GM. Regression in skin tumours: a common phenomenon. Australas J. Dermatol. 1997;38(Suppl 1):S63–5. doi: 10.1111/j.1440-0960.1997.tb01013.x. [DOI] [PubMed] [Google Scholar]
- [10].Sarkar RR, Banerjee S. Cancer self remission and tumor stability - a stochastic approach. Math. Biosci. 2005;196(1):65–81. doi: 10.1016/j.mbs.2005.04.001. [DOI] [PubMed] [Google Scholar]
- [11].Bowles AP, Jr., Perkins E. Long-term remission of malignant brain tumors after intracranial infection: a report of four cases. Neurosurgery. 1999;44(3):636–42. doi: 10.1097/00006123-199903000-00110. discussion 642-3. [DOI] [PubMed] [Google Scholar]
- [12].Chattopadhyay S, Chakraborty NG, Mukherji B. Regulatory T cells and tumor immunity. Cancer Immunol. Immunother. 2005 doi: 10.1007/s00262-005-0699-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ishikawa F, Miyazaki S. New biodefense strategies by neutrophils. Arch. Immunol. Ther. Exp. (Warsz) 2005;53(3):226–33. [PubMed] [Google Scholar]
- [14].Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004;5(10):987–95. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- [15].Andersson PB, Perry VH, Gordon S. The acute inflammatory response to lipopolysaccharide in CNS parenchyma differs from that in other body tissues. Neuroscience. 1992;48(1):169–86. doi: 10.1016/0306-4522(92)90347-5. [DOI] [PubMed] [Google Scholar]
- [16].Cartmell T, Southgate T, Rees GS, Castro MG, Lowenstein PR, Luheshi GN. Interleukin-1 mediates a rapid inflammatory response after injection of adenoviral vectors into the brain. J. Neurosci. 1999;19(4):1517–23. doi: 10.1523/JNEUROSCI.19-04-01517.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Harling-Berg CJ, Park TJ, Knopf PM. Role of the cervical lymphatics in the Th2-type hierarchy of CNS immune regulation. J. Neuroimmunol. 1999;101(2):111–27. doi: 10.1016/s0165-5728(99)00130-7. [DOI] [PubMed] [Google Scholar]
- [18].Matyszak MK, Perry VH. A comparison of leucocyte responses to heat-killed bacillus Calmette-Guerin in different CNS compartments. Neuropathol. Appl. Neurobiol. 1996;22(1):44–53. [PubMed] [Google Scholar]
- [19].McMenamin MM, Byrnes AP, Charlton HM, Coffin RS, Latchman DS, Wood MJ. A gamma34.5 mutant of herpes simplex 1 causes severe inflammation in the brain. Neuroscience. 1998;83(4):1225–37. doi: 10.1016/s0306-4522(97)00513-7. [DOI] [PubMed] [Google Scholar]
- [20].Perry VH. A revised view of the central nervous system microenvironment and major histocompatibility complex class II antigen presentation. J. Neuroimmunol. 1998;90(2):113–21. doi: 10.1016/s0165-5728(98)00145-3. [DOI] [PubMed] [Google Scholar]
- [21].Stevenson PG, Freeman S, Bangham CR, Hawke S. Virus dissemination through the brain parenchyma without immunologic control. J. Immunol. 1997;159(4):1876–84. [PubMed] [Google Scholar]
- [22].Stevenson PG, Hawke S, Sloan DJ, Bangham CR. The immunogenicity of intracerebral virus infection depends on anatomical site. J. Virol. 1997;71(1):145–51. doi: 10.1128/jvi.71.1.145-151.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Thomas CE, Schiedner G, Kochanek S, Castro MG, Lowenstein PR. Peripheral infection with adenovirus causes unexpected long-term brain inflammation in animals injected intracranially with first-generation, but not with high-capacity, adenovirus vectors: toward realistic long-term neurological gene therapy for chronic diseases. Proc. Natl. Acad. Sci. USA. 2000;97(13):7482–7. doi: 10.1073/pnas.120474397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wood MJ, Charlton HM, Wood KJ, Kajiwara K, Byrnes AP. Immune responses to adenovirus vectors in the nervous system. Trends Neurosci. 1996;19(11):497–501. doi: 10.1016/S0166-2236(96)10060-6. [DOI] [PubMed] [Google Scholar]
- [25].Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol. 2003;3(7):569–81. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- [26].Perry VH, Andersson PB. The inflammatory response in the CNS. Neuropathol. Appl. Neurobiol. 1992;18(5):454–9. doi: 10.1111/j.1365-2990.1992.tb00811.x. [DOI] [PubMed] [Google Scholar]
- [27].Lowenstein PR. Immunology of viral-vector-mediated gene transfer into the brain: an evolutionary and developmental perspective. Trends Immunol. 2002;23(1):23–30. doi: 10.1016/s1471-4906(01)02063-4. [DOI] [PubMed] [Google Scholar]
- [28].McMenamin PG. Distribution and phenotype of dendritic cells and resident tissue macrophages in the dura mater, leptomeninges, and choroid plexus of the rat brain as demonstrated in wholemount preparations. J. Comp. Neurol. 1999;405(4):553–62. [PubMed] [Google Scholar]
- [29].Aloisi F, Ria F, Adorini L. Regulation of T-cell responses by CNS antigen-presenting cells: different roles for microglia and astrocytes. Immunol. Today. 2000;21(3):141–7. doi: 10.1016/s0167-5699(99)01512-1. [DOI] [PubMed] [Google Scholar]
- [30].Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- [31].Fischer HG, Reichmann G. Brain dendritic cells and macrophages/microglia in central nervous system inflammation. J. Immunol. 2001;166(4):2717–26. doi: 10.4049/jimmunol.166.4.2717. [DOI] [PubMed] [Google Scholar]
- [32].Serafini B, Columba-Cabezas S, Di Rosa F, Aloisi F. Intracerebral recruitment and maturation of dendritic cells in the onset and progression of experimental autoimmune encephalomyelitis. Am. J. Pathol. 2000;157(6):1991–2002. doi: 10.1016/S0002-9440(10)64838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fischer HG, Bonifas U, Reichmann G. Phenotype and functions of brain dendritic cells emerging during chronic infection of mice with Toxoplasma gondii. J. Immunol. 2000;164(9):4826–34. doi: 10.4049/jimmunol.164.9.4826. [DOI] [PubMed] [Google Scholar]
- [34].Matyszak MK, Perry VH. The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience. 1996;74(2):599–608. doi: 10.1016/0306-4522(96)00160-1. [DOI] [PubMed] [Google Scholar]
- [35].Pashenkov M, Huang YM, Kostulas V, Haglund M, Soderstrom M, Link H. Two subsets of dendritic cells are present in human cerebrospinal fluid. Brain. 2001;124(Pt 3):480–92. doi: 10.1093/brain/124.3.480. [DOI] [PubMed] [Google Scholar]
- [36].Pashenkov M, Teleshova N, Kouwenhoven M, Smirnova T, Jin YP, Kostulas V, Huang YM, Pinegin B, Boiko A, Link H. Recruitment of dendritic cells to the cerebrospinal fluid in bacterial neuroinfections. J. Neuroimmunol. 2002;122(12):106–16. doi: 10.1016/s0165-5728(01)00451-9. [DOI] [PubMed] [Google Scholar]
- [37].Reichmann G, Schroeter M, Jander S, Fischer HG. Dendritic cells and dendritic-like microglia in focal cortical ischemia of the mouse brain. J. Neuroimmunol. 2002;129(12):125–32. doi: 10.1016/s0165-5728(02)00184-4. [DOI] [PubMed] [Google Scholar]
- [38].Sallusto F, Schaerli P, Loetscher P, Schaniel C, Lenig D, Mackay CR, Qin S, Lanzavecchia A. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur. J. Immunol. 1998;28(9):2760–9. doi: 10.1002/(SICI)1521-4141(199809)28:09<2760::AID-IMMU2760>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- [39].Svenningsson A, Andersen O, Edsbagge M, Stemme S. Lymphocyte phenotype and subset distribution in normal cerebrospinal fluid. J. Neuroimmunol. 1995;63(1):39–46. doi: 10.1016/0165-5728(95)00126-3. [DOI] [PubMed] [Google Scholar]
- [40].Dix AR, Brooks WH, Roszman TL, Morford LA. Immune defects observed in patients with primary malignant brain tumors. J. Neuroimmunol. 1999;100(12):216–32. doi: 10.1016/s0165-5728(99)00203-9. [DOI] [PubMed] [Google Scholar]
- [41].Bodmer S, Strommer K, Frei K, Siepl C, de Tribolet N, Heid I, Fontana A. Immunosuppression and transforming growth factor-beta in glioblastoma. Preferential production of transforming growth factor-beta. 1989;143(10):3222–9. [PubMed] [Google Scholar]
- [42].Brooks WH, Caldwell HD, Mortara RH. Immune responses in patients with gliomas. Surg. Neurol. 1974;2(6):419–23. [PubMed] [Google Scholar]
- [43].Brooks WH, Netsky MG, Normansell DE, Horwitz DA. Depressed cell-mediated immunity in patients with primary intracranial tumors. Characterization of a humoral immunosuppressive factor. J. Exp. Med. 1972;136(6):1631–47. doi: 10.1084/jem.136.6.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Brooks WH, Roszman TL, Mahaley MS, Woosley RE. Immunobiology of primary intracranial tumours. II. Analysis of lymphocyte subpopulations in patients with primary brain tumours. Clin. Exp. Immunol. 1977;29(1):61–6. [PMC free article] [PubMed] [Google Scholar]
- [45].Brooks WH, Roszman TL, Rogers AS. Impairment of rosette-forming T lymphoctyes in patients with primary intracranial tumors. Cancer. 1976;37(4):1869–73. doi: 10.1002/1097-0142(197604)37:4<1869::aid-cncr2820370435>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- [46].Mahaley MS, Jr., Brooks WH, Roszman TL, Bigner DD, Dudka L, Richardson S. Immunobiology of primary intracranial tumors. Part 1: studies of the cellular and humoral general immune competence of brain-tumor patients. J. Neurosurg. 1977;46(4):467–76. doi: 10.3171/jns.1977.46.4.0467. [DOI] [PubMed] [Google Scholar]
- [47].Young HF, Kaplan AM. Cellular immune deficiency in patients with glioblastoma. Surg. Forum. 1976;27(62):476–8. [PubMed] [Google Scholar]
- [48].Brooks WH, Latta RB, Mahaley MS, Roszman TL, Dudka L, Skaggs C. Immunobiology of primary intracranial tumors. Part 5: Correlation of a lymphocyte index and clinical status. J. Neurosurg. 1981;54(3):331–7. doi: 10.3171/jns.1981.54.3.0331. [DOI] [PubMed] [Google Scholar]
- [49].Giometto B, Bozza F, Faresin F, Alessio L, Mingrino S, Tavolato B. Immune infiltrates and cytokines in gliomas. Acta Neurochir. (Wien) 1996;138(1):50–6. doi: 10.1007/BF01411724. [DOI] [PubMed] [Google Scholar]
- [50].Rossi ML, Cruz-Sanchez F, Hughes JT, Esiri MM, Coakham HB, Moss TH. Mononuclear cell infiltrate and HLA-DR expression in low grade astrocytomas. An immunohistological study of 23 cases. Acta Neuropathol. (Berl) 1988;76(3):281–6. doi: 10.1007/BF00687776. [DOI] [PubMed] [Google Scholar]
- [51].Rossi ML, Hughes JT, Esiri MM, Coakham HB, Brownell DB. Immunohistological study of mononuclear cell infiltrate in malignant gliomas. Acta Neuropathol. (Berl) 1987;74(3):269–77. doi: 10.1007/BF00688191. [DOI] [PubMed] [Google Scholar]
- [52].Schneider J, Hofman FM, Apuzzo ML, Hinton DR. Cytokines and immunoregulatory molecules in malignant glial neoplasms. J. Neurosurg. 1992;77(2):265–73. doi: 10.3171/jns.1992.77.2.0265. [DOI] [PubMed] [Google Scholar]
- [53].Stevens A, Kloter I, Roggendorf W. Inflammatory infiltrates and natural killer cell presence in human brain tumors. Cancer. 1988;61(4):738–43. doi: 10.1002/1097-0142(19880215)61:4<738::aid-cncr2820610417>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- [54].Roszman T, Elliott L, Brooks W. Modulation of T-cell function by gliomas. Immunol. Today. 1991;12(10):370–4. doi: 10.1016/0167-5699(91)90068-5. [DOI] [PubMed] [Google Scholar]
- [55].Kruse CA, Mitchell DH, Lillehei KO, Johnson SD, McCleary EL, Moore GE, Waldrop S, Mierau GW. Interleukin-2-activated lymphocytes from brain tumor patients. A comparison of two preparations generated in vitro. Cancer. 1989;64(8):1629–37. doi: 10.1002/1097-0142(19891015)64:8<1629::aid-cncr2820640813>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- [56].Roszman TL, Brooks WH, Steele C, Elliott LH. Pokeweed mitogen-induced immunoglobulin secretion by peripheral blood lymphocytes from patients with primary intracranial tumors. Characterization of T helper and B cell function. J. Immunol. 1985;134(3):1545–50. [PubMed] [Google Scholar]
- [57].Ashkenazi E, Deutsch M, Tirosh R, Weinreb A, Tsukerman A, Brodie C. A selective impairment of the IL-2 system in lymphocytes of patients with glioblastomas: increased level of soluble IL-2R and reduced protein tyrosine phosphorylation. Neuroimmunomodulation. 1997;4(1):49–56. doi: 10.1159/000097315. [DOI] [PubMed] [Google Scholar]
- [58].Elliott LH, Brooks WH, Roszman TL. Activation of immunoregulatory lymphocytes obtained from patients with malignant gliomas. J. Neurosurg. 1987;67(2):231–6. doi: 10.3171/jns.1987.67.2.0231. [DOI] [PubMed] [Google Scholar]
- [59].McVicar DW, Davis DF, Merchant RE. In vitro analysis of the proliferative potential of T cells from patients with brain tumor: glioma-associated immunosuppression unrelated to intrinsic cellular defect. J. Neurosurg. 1992;76(2):251–60. doi: 10.3171/jns.1992.76.2.0251. [DOI] [PubMed] [Google Scholar]
- [60].Ausiello CM, Palma C, Maleci A, Spagnoli GC, Amici C, Antonelli G, Casciani CU, Cassone A. Cell mediated cytotoxicity and cytokine production in peripheral blood mononuclear cells of glioma patients. Eur. J. Cancer . 1991;27(5):646–50. doi: 10.1016/0277-5379(91)90235-6. [DOI] [PubMed] [Google Scholar]
- [61].Elliott LH, Brooks WH, Roszman TL. Cytokinetic basis for the impaired activation of lymphocytes from patients with primary intracranial tumors. J. Immunol. 1984;132(3):1208–15. [PubMed] [Google Scholar]
- [62].Woiciechowsky C, Asadullah K, Nestler D, Schoning B, Glockner F, Docke WD, Volk HD. Diminished monocytic HLA-DR expression and ex vivo cytokine secretion capacity in patients with glioblastoma: effect of tumor extirpation. J. Neuroimmunol. 1998;84(2):164–71. doi: 10.1016/s0165-5728(97)00236-1. [DOI] [PubMed] [Google Scholar]
- [63].Black KL, Chen K, Becker DP, Merrill JE. Inflammatory leukocytes associated with increased immunosuppression by glioblastoma. J. Neurosurg. 1992;77(1):120–6. doi: 10.3171/jns.1992.77.1.0120. [DOI] [PubMed] [Google Scholar]
- [64].Fries G, Perneczky A, Kempski O. Glioblastoma-associated circulating monocytes and the release of epidermal growth factor. J. Neurosurg. 1996;85(4):642–7. doi: 10.3171/jns.1996.85.4.0642. [DOI] [PubMed] [Google Scholar]
- [65].Lewis CE, Leek R, Harris A, McGee JO. Cytokine regulation of angiogenesis in breast cancer: the role of tumor-associated macrophages. J. Leukoc. Biol. 1995;57(5):747–51. doi: 10.1002/jlb.57.5.747. [DOI] [PubMed] [Google Scholar]
- [66].Sunderkotter C, Goebeler M, Schulze-Osthoff K, Bhardwaj R, Sorg C. Macrophage-derived angiogenesis factors. Pharmacol Ther. 1991;51(2):195–216. doi: 10.1016/0163-7258(91)90077-y. [DOI] [PubMed] [Google Scholar]
- [67].Sunderkotter C, Steinbrink K, Goebeler M, Bhardwaj R, Sorg C. Macrophages and angiogenesis. J. Leukoc. Biol. . 1994;55(3):410–22. doi: 10.1002/jlb.55.3.410. [DOI] [PubMed] [Google Scholar]
- [68].Hildenbrand R, Dilger I, Horlin A, Stutte HJ. Urokinase and macrophages in tumour angiogenesis. Br. J. Cancer. 1995;72(4):818–23. doi: 10.1038/bjc.1995.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Fontana A, Hengartner H, de Tribolet N, Weber E. Glioblastoma cells release interleukin 1 and factors inhibiting interleukin 2-mediated effects. J. Immunol. 1984;132(4):1837–44. [PubMed] [Google Scholar]
- [70].Bodmer S, Huber D, Heid I, Fontana A. Human glioblastoma cell derived transforming growth factor-beta 2: evidence for secretion of both high and low molecular weight biologically active forms. J. Neuroimmunol. 1991;34(1):33–42. doi: 10.1016/0165-5728(91)90096-p. [DOI] [PubMed] [Google Scholar]
- [71].Constam DB, Philipp J, Malipiero UV, ten Dijke P, Schachner M, Fontana A. Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J. Immunol. 1992;148(5):1404–10. [PubMed] [Google Scholar]
- [72].de Martin R, Haendler B, Hofer-Warbinek R, Gaugitsch H, Wrann M, Schlusener H, Seifert JM, Bodmer S, Fontana A, Hofer E. Complementary DNA for human glioblastoma-derived T cell suppressor factor, a novel member of the transforming growth factor-beta gene family. EMBO J. 1987;6(12):3673–7. doi: 10.1002/j.1460-2075.1987.tb02700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kuppner MC, Hamou MF, Sawamura Y, Bodmer S, de Tribolet N. Inhibition of lymphocyte function by glioblastoma-derived transforming growth factor beta 2. J. Neurosurg. 1989;71(2):211–7. doi: 10.3171/jns.1989.71.2.0211. [DOI] [PubMed] [Google Scholar]
- [74].Olofsson A, Miyazono K, Kanzaki T, Colosetti P, Engstrom U, Heldin CH. Transforming growth factor-beta 1, -beta 2, and -beta 3 secreted by a human glioblastoma cell line. Identification of small and different forms of large latent complexes. J. Biol. Chem. 1992;267(27):19482–8. [PubMed] [Google Scholar]
- [75].Sasaki A, Naganuma H, Satoh E, Nagasaka M, Isoe S, Nakano S, Nukui H. Secretion of transforming growth factor-beta 1 and -beta 2 by malignant glioma cells. Neurol. Med. Chir. (Tokyo) 1995;35(7):423–30. doi: 10.2176/nmc.35.423. [DOI] [PubMed] [Google Scholar]
- [76].Siepl C, Bodmer S, Frei K, MacDonald HR, De Martin R, Hofer E, Fontana A. The glioblastoma-derived T cell suppressor factor/transforming growth factor-beta 2 inhibits T cell growth without affecting the interaction of interleukin 2 with its receptor. Eur. J. Immunol. 1988;18(4):593–600. doi: 10.1002/eji.1830180416. [DOI] [PubMed] [Google Scholar]
- [77].Weller M, Constam DB, Malipiero U, Fontana A. Transforming growth factor-beta 2 induces apoptosis of murine T cell clones without down-regulating bcl-2 mRNA expression. Eur. J. Immunol. 1994;24(6):1293–300. doi: 10.1002/eji.1830240608. [DOI] [PubMed] [Google Scholar]
- [78].Wrann M, Bodmer S, de Martin R, Siepl C, Hofer-Warbinek R, Frei K, Hofer E, Fontana A. T cell suppressor factor from human glioblastoma cells is a 12.5-kd protein closely related to transforming growth factor-beta. EMBO J. 1987;6(6):1633–6. doi: 10.1002/j.1460-2075.1987.tb02411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Platten M, Wick W, Wild-Bode C, Aulwurm S, Dichgans J, Weller M. Transforming growth factors beta(1) (TGF-beta(1)) and TGF-beta(2) promote glioma cell migration via Up-regulation of alpha(V)beta(3) integrin expression. Biochem. Biophys. Res. Commun. 2000;268(2):607–11. doi: 10.1006/bbrc.2000.2176. [DOI] [PubMed] [Google Scholar]
- [80].Huber D, Philipp J, Fontana A. Protease inhibitors interfere with the transforming growth factor-beta-dependent but not the transforming growth factor-beta-independent pathway of tumor cell-mediated immunosuppression. J. Immunol. 1992;148(1):277–84. [PubMed] [Google Scholar]
- [81].Kehrl JH, Roberts AB, Wakefield LM, Jakowlew S, Sporn MB, Fauci AS. Transforming growth factor beta is an important immunomodulatory protein for human B lymphocytes. J. Immunol. 1986;137(12):3855–60. [PubMed] [Google Scholar]
- [82].Kehrl JH, Wakefield LM, Roberts AB, Jakowlew S, Alvarez-Mon M, Derynck R, Sporn MB, Fauci AS. Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J. Exp. Med. 1986;163(5):1037–50. doi: 10.1084/jem.163.5.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kuppner MC, Hamou MF, Bodmer S, Fontana A, de Tribolet N. The glioblastoma-derived T-cell suppressor factor/transforming growth factor beta 2 inhibits the generation of lymphokine-activated killer (LAK) cells. Int. J. Cancer. 1988;42(4):562–7. doi: 10.1002/ijc.2910420416. [DOI] [PubMed] [Google Scholar]
- [84].Ranges GE, Figari IS, Espevik T, Palladino MA., Jr. Inhibition of cytotoxic T cell development by transforming growth factor beta and reversal by recombinant tumor necrosis factor alpha. J. Exp. Med. 1987;166(4):991–8. doi: 10.1084/jem.166.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Rook AH, Kehrl JH, Wakefield LM, Roberts AB, Sporn MB, Burlington DB, Lane HC, Fauci AS. Effects of transforming growth factor beta on the functions of natural killer cells: depressed cytolytic activity and blunting of interferon responsiveness. J. Immunol. 1986;136(10):3916–20. [PubMed] [Google Scholar]
- [86].Zuber P, Kuppner MC, De Tribolet N. Transforming growth factor-beta 2 down-regulates HLA-DR antigen expression on human malignant glioma cells. Eur. J. Immunol. . 1988;18(10):1623–6. doi: 10.1002/eji.1830181023. [DOI] [PubMed] [Google Scholar]
- [87].Koochekpour S, Merzak A, Pilkington GJ. Vascular endothelial growth factor production is stimulated by gangliosides and TGF-beta isoforms in human glioma cells in vitro. Cancer Lett. 1996;102(12):209–15. doi: 10.1016/0304-3835(96)04161-4. [DOI] [PubMed] [Google Scholar]
- [88].Jensen RL. Growth factor-mediated angiogenesis in the malignant progression of glial tumors: a review. Surg. Neurol. 1998;49(2):189–95. doi: 10.1016/s0090-3019(97)00218-8. discussion 196. [DOI] [PubMed] [Google Scholar]
- [89].Uhl M, Aulwurm S, Wischhusen J, Weiler M, Ma JY, Almirez R, Mangadu R, Liu YW, Platten M, Herrlinger U, Murphy A, Wong DH, Wick W, Higgins LS, Weller M. SD-208, a novel transforming growth factor beta receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res. 2004;64(21):7954–61. doi: 10.1158/0008-5472.CAN-04-1013. [DOI] [PubMed] [Google Scholar]
- [90].Hishii M, Nitta T, Ishida H, Ebato M, Kurosu A, Yagita H, Sato K, Okumura K. Human glioma-derived interleukin-10 inhibits antitumor immune responses in vitro. Neurosurgery. 1995;37(6):1160–6. doi: 10.1227/00006123-199512000-00016. [DOI] [PubMed] [Google Scholar]
- [91].Munz C, Naumann U, Grimmel C, Rammensee HG, Weller M. TGF-beta-independent induction of immunogenicity by decorin gene transfer in human malignant glioma cells. Eur. J. Immunol. 1999;29(3):1032–40. doi: 10.1002/(SICI)1521-4141(199903)29:03<1032::AID-IMMU1032>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- [92].Castelli MG, Chiabrando C, Fanelli R, Martelli L, Butti G, Gaetani P, Paoletti P. Prostaglandin and thromboxane synthesis by human intracranial tumors. Cancer Res. 1989;49(6):1505–8. [PubMed] [Google Scholar]
- [93].Kuppner MC, Sawamura Y, Hamou MF, de Tribolet N. Influence of PGE2- and cAMP-modulating agents on human glioblastoma cell killing by interleukin-2-activated lymphocytes. J. Neurosurg. 1990;72(4):619–25. doi: 10.3171/jns.1990.72.4.0619. [DOI] [PubMed] [Google Scholar]
- [94].Huettner C, Czub S, Kerkau S, Roggendorf W, Tonn JC. Interleukin 10 is expressed in human gliomas in vivo and increases glioma cell proliferation and motility in vitro. Anticancer Res. 1997;17(5A):3217–24. [PubMed] [Google Scholar]
- [95].Berman RM, Suzuki T, Tahara H, Robbins PD, Narula SK, Lotze MT. Systemic administration of cellular IL-10 induces an effective, specific, and long-lived immune response against established tumors in mice. J. Immunol. 1996;157(1):231–8. [PubMed] [Google Scholar]
- [96].Wojtowicz-Praga S. Reversal of tumor-induced immunosuppression: a new approach to cancer therapy. J. Immunother. 1997;20(3):165–77. [PubMed] [Google Scholar]
- [97].Couldwell WT, Dore-Duffy P, Apuzzo ML, Antel JP. Malignant glioma modulation of immune function: relative contribution of different soluble factors. J. Neuroimmunol. 1991;33(2):89–96. doi: 10.1016/0165-5728(91)90052-9. [DOI] [PubMed] [Google Scholar]
- [98].Collette Y, Gilles A, Pontarotti P, Olive D. A co-evolution perspective of the TNFSF and TNFRSF families in the immune system. Trends Immunol. 2003;24(7):387–94. doi: 10.1016/s1471-4906(03)00166-2. [DOI] [PubMed] [Google Scholar]
- [99].Curtin JF, Cotter TG. Live and let die: regulatory mechanisms in Fas-mediated apoptosis. Cell Signal. 2003;15(11):983–92. doi: 10.1016/s0898-6568(03)00093-7. [DOI] [PubMed] [Google Scholar]
- [100].Roth W, Weller M. Chemotherapy and immunotherapy of malignant glioma: molecular mechanisms and clinical perspectives. Cell. Mol. Life Sci. 1999;56(56):481–506. doi: 10.1007/s000180050447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Weller M, Kleihues P, Dichgans J, Ohgaki H. CD95 ligand: lethal weapon against malignant glioma? Brain Pathol. 1998;8(2):285–93. doi: 10.1111/j.1750-3639.1998.tb00154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Xia S, Rosen EM, Laterra J. Sensitization of glioma cells to Fas-dependent apoptosis by chemotherapy-induced oxidative stress. Cancer Res. 2005;65(12):5248–55. doi: 10.1158/0008-5472.CAN-04-4332. [DOI] [PubMed] [Google Scholar]
- [103].Choi C, Jeong E, Benveniste EN. Caspase-1 mediates Fas-induced apoptosis and is up-regulated by interferon-gamma in human astrocytoma cells. J. Neurooncol. 2004;67(12):167–76. doi: 10.1023/b:neon.0000021896.52664.9e. [DOI] [PubMed] [Google Scholar]
- [104].Naumann U, Waltereit R, Schulz JB, Weller M. Adenoviral (full-length) Apo2L/TRAIL gene transfer is an ineffective treatment strategy for malignant glioma. J. Neurooncol. 2003;61(1):7–15. doi: 10.1023/a:1021248329980. [DOI] [PubMed] [Google Scholar]
- [105].Rubinchik S, Yu H, Woraratanadharm J, Voelkel-Johnson C, Norris JS, Dong JY. Enhanced apoptosis of glioma cell lines is achieved by co-delivering FasL-GFP and TRAIL with a complex Ad5 vector. Cancer Gene Ther. 2003;10(11):814–22. doi: 10.1038/sj.cgt.7700651. [DOI] [PubMed] [Google Scholar]
- [106].Maleniak TC, Darling JL, Lowenstein PR, Castro MG. Adenovirus-mediated expression of HSV1-TK or Fas ligand induces cell death in primary human glioma-derived cell cultures that are resistant to the chemotherapeutic agent CCNU. Cancer Gene Ther. 2001;8(8):589–98. doi: 10.1038/sj.cgt.7700348. [DOI] [PubMed] [Google Scholar]
- [107].Ambar BB, Frei K, Malipiero U, Morelli AE, Castro MG, Lowenstein PR, Fontana A. Treatment of experimental glioma by administration of adenoviral vectors expressing Fas ligand. Hum. Gene Ther. 1999;10(10):1641–8. doi: 10.1089/10430349950017644. [DOI] [PubMed] [Google Scholar]
- [108].Shah K, Bureau E, Kim DE, Yang K, Tang Y, Weissleder R, Breakefield XO. Glioma therapy and real-time imaging of neural precursor cell migration and tumor regression. Ann. Neurol. 2005;57(1):34–41. doi: 10.1002/ana.20306. [DOI] [PubMed] [Google Scholar]
- [109].Shah K, Tung CH, Breakefield XO, Weissleder R. In vivo imaging of S-TRAIL-mediated tumor regression and apoptosis. Mol. Ther. 2005;11(6):926–31. doi: 10.1016/j.ymthe.2005.01.017. [DOI] [PubMed] [Google Scholar]
- [110].Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, Borden EC. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. 2003;8(3):237–49. doi: 10.1023/a:1023668705040. [DOI] [PubMed] [Google Scholar]
- [111].Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005;5(5):375–86. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- [112].Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S, Liu YJ. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284(5421):1835–7. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- [113].Kadowaki N, Antonenko S, Liu YJ. Distinct CpG DNA and polyinosinic-polycytidylic acid double-stranded RNA, respectively, stimulate CD11c-type 2 dendritic cell precursors and CD11c+ dendritic cells to produce type I IFN. J. Immunol. . 2001;166(4):2291–5. doi: 10.4049/jimmunol.166.4.2291. [DOI] [PubMed] [Google Scholar]
- [114].Tanabe T, Kominsky SL, Subramaniam PS, Johnson HM, Torres BA. Inhibition of the glioblastoma cell cycle by type I IFNs occurs at both the G1 and S phases and correlates with the upregulation of p21(WAF1/CIP1) J. Neurooncol. 2000;48(3):225–32. doi: 10.1023/a:1006476408190. [DOI] [PubMed] [Google Scholar]
- [115].Garrison JI, Berens ME, Shapiro JR, Treasurywala S, Floyd-Smith G. Interferon-beta inhibits proliferation and progression through S phase of the cell cycle in five glioma cell lines. J. Neurooncol. 1996;30(3):213–23. doi: 10.1007/BF00177272. [DOI] [PubMed] [Google Scholar]
- [116].Porta C, Hadj-Slimane R, Nejmeddine M, Pampin M, Tovey MG, Espert L, Alvarez S, Chelbi-Alix MK. Interferons alpha and gamma induce p53-dependent and p53-independent apoptosis, respectively. Oncogene. 2005;24(4):605–15. doi: 10.1038/sj.onc.1208204. [DOI] [PubMed] [Google Scholar]
- [117].Sandoval R, Xue J, Pilkinton M, Salvi D, Kiyokawa H, Colamonici OR. Different requirements for the cytostatic and apoptotic effects of type I interferons. Induction of apoptosis requires ARF but not p53 in osteosarcoma cell lines. J. Biol. Chem. 2004;279(31):32275–80. doi: 10.1074/jbc.M313830200. [DOI] [PubMed] [Google Scholar]
- [118].Miyakoshi J, Dobler KD, Allalunis-Turner J, McKean JD, Petruk K, Allen PB, Aronyk KN, Weir B, Huyser-Wierenga D, Fulton D, et al. Absence of IFNA and IFNB genes from human malignant glioma cell lines and lack of correlation with cellular sensitivity to interferons. Cancer Res. 1990;50(2):278–83. [PubMed] [Google Scholar]
- [119].James CD, He J, Carlbom E, Nordenskjold M, Cavenee WK, Collins VP. Chromosome 9 deletion mapping reveals interferon alpha and interferon beta-1 gene deletions in human glial tumors. Cancer Res. 1991;51(6):1684–8. [PubMed] [Google Scholar]
- [120].Tsugawa T, Kuwashima N, Sato H, Fellows-Mayle WK, Dusak JE, Okada K, Papworth GD, Watkins SC, Gambotto A, Yoshida J, Pollack IF, Okada H. Sequential delivery of interferon-alpha gene and DCs to intracranial gliomas promotes an effective antitumor response. Gene Ther. 2004;11(21):1551–8. doi: 10.1038/sj.gt.3302300. [DOI] [PubMed] [Google Scholar]
- [121].Natsume A, Tsujimura K, Mizuno M, Takahashi T, Yoshida J. IFN-beta gene therapy induces systemic antitumor immunity against malignant glioma. J. Neurooncol. 2000;47(2):117–24. doi: 10.1023/a:1006441030976. [DOI] [PubMed] [Google Scholar]
- [122].Yoshida J, Mizuno M, Fujii M, Kajita Y, Nakahara N, Hatano M, Saito R, Nobayashi M, Wakabayashi T. Human gene therapy for malignant gliomas (glioblastoma multiforme and anaplastic astrocytoma) by in vivo transduction with human interferon beta gene using cationic liposomes. Hum. Gene Ther. 2004;15(1):77–86. doi: 10.1089/10430340460732472. [DOI] [PubMed] [Google Scholar]
- [123].Eck SL, Alavi JB, Judy K, Phillips P, Alavi A, Hackney D, Cross P, Hughes J, Gao G, Wilson JM, Propert K. Treatment of recurrent or progressive malignant glioma with a recombinant adenovirus expressing human interferon-beta (H5.010CMVhIFN-beta): a phase I trial. Hum. Gene Ther. 2001;12(1):97–113. doi: 10.1089/104303401451013. [DOI] [PubMed] [Google Scholar]
- [124].Saleh M, Jonas NK, Wiegmans A, Stylli SS. The treatment of established intracranial tumors by in situ retroviral IFN-gamma transfer. Gene Ther. 2000;7(20):1715–24. doi: 10.1038/sj.gt.3301273. [DOI] [PubMed] [Google Scholar]
- [125].Fathallah-Shaykh HM, Zhao LJ, Kafrouni AI, Smith GM, Forman J. Gene transfer of IFN-gamma into established brain tumors represses growth by antiangiogenesis. J. Immunol. 2000;164(1):217–22. doi: 10.4049/jimmunol.164.1.217. [DOI] [PubMed] [Google Scholar]
- [126].Ehtesham M, Samoto K, Kabos P, Acosta FL, Gutierrez MA, Black KL, Yu JS. Treatment of intracranial glioma with in situ interferon-gamma and tumor necrosis factor-alpha gene transfer. Cancer Gene Ther. 2002;9(11):925–34. doi: 10.1038/sj.cgt.7700516. [DOI] [PubMed] [Google Scholar]
- [127].Chang JF, Thomas CA, 3rd., Kung JT. Mitogen-induced IL-production and proliferation at defined stages of T helper cell development. J. Immunol. 1991;147(3):860–6. [PubMed] [Google Scholar]
- [128].Karasuyama H, Tohyama N, Tada T. Autocrine growth and tumorigenicity of interleukin 2-dependent helper T cells transfected with IL-2 gene. J. Exp. Med. 1989;169(1):13–25. doi: 10.1084/jem.169.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Maraskovsky E, Chen WF, Shortman K. IL-2 and IFN-gamma are two necessary lymphokines in the development of cytolytic T cells. J. Immunol. 1989;143(4):1210–4. [PubMed] [Google Scholar]
- [130].Iwadate Y, Tagawa M, Namba H, Oga M, Kawamura K, Tasaki K, Sakiyama S, Yamaura A. Immunological responsiveness to interleukin-2-producing brain tumors can be restored by concurrent subcutaneous transplantation of the same tumors. Cancer Gene Ther. 2000;7(9):1263–9. doi: 10.1038/sj.cgt.7700223. [DOI] [PubMed] [Google Scholar]
- [131].Iwadate Y, Yamaura A, Sato Y, Sakiyama S, Tagawa M. Induction of immunity in peripheral tissues combined with intracerebral transplantation of interleukin-2-producing cells eliminates established brain tumors. Cancer Res. 2001;61(24):8769–74. [PubMed] [Google Scholar]
- [132].Lichtor T, Glick RP. Cytokine immuno-gene therapy for treatment of brain tumors. J. Neurooncol. 2003;65(3):247–59. doi: 10.1023/b:neon.0000003654.83272.4a. [DOI] [PubMed] [Google Scholar]
- [133].Chen B, Timiryasova TM, Andres ML, Kajioka EH, Dutta-Roy R, Gridley DS, Fodor I. Evaluation of combined vaccinia virus-mediated antitumor gene therapy with p53, IL-2, and IL-12 in a glioma model. Cancer Gene Ther. 2000;7(11):1437–47. doi: 10.1038/sj.cgt.7700252. [DOI] [PubMed] [Google Scholar]
- [134].Furtado GC, Curotto de Lafaille MA, Kutchukhidze N, Lafaille JJ. Interleukin 2 signaling is required for CD4(+) regulatory T cell function. J. Exp. Med. 2002;196(6):851–7. doi: 10.1084/jem.20020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Almeida AR, Legrand N, Papiernik M, Freitas AA. Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J. Immunol. 2002;169(9):4850–60. doi: 10.4049/jimmunol.169.9.4850. [DOI] [PubMed] [Google Scholar]
- [136].Malek TR. The main function of IL-2 is to promote the development of T regulatory cells. J. Leukoc. Biol. 2003;74(6):961–5. doi: 10.1189/jlb.0603272. [DOI] [PubMed] [Google Scholar]
- [137].Ebner S, Hofer S, Nguyen VA, Furhapter C, Herold M, Fritsch P, Heufler C, Romani N. A novel role for IL-3: human monocytes cultured in the presence of IL-3 and IL-4 differentiate into dendritic cells that produce less IL-12 and shift Th cell responses toward a Th2 cytokine pattern. J. Immunol. 2002;168(12):6199–207. doi: 10.4049/jimmunol.168.12.6199. [DOI] [PubMed] [Google Scholar]
- [138].Lorentz A, Wilke M, Sellge G, Worthmann H, Klempnauer J, Manns MP, Bischoff SC. IL-4-induced priming of human intestinal mast cells for enhanced survival and Th2 cytokine generation is reversible and associated with increased activity of ERK1/2 and c-Fos. J. Immunol. 2005;174(11):6751–6. doi: 10.4049/jimmunol.174.11.6751. [DOI] [PubMed] [Google Scholar]
- [139].Pace L, Pioli C, Doria G. IL-4 modulation of CD4+CD25+ T regulatory cell-mediated suppression. J. Immunol. 2005;174(12):7645–53. doi: 10.4049/jimmunol.174.12.7645. [DOI] [PubMed] [Google Scholar]
- [140].Stack RM, Lenschow DJ, Gray GS, Bluestone JA, Fitch FW. IL-4 treatment of small splenic B cells induces costimulatory molecules B7-1 and B7-2. J. Immunol. 1994;152(12):5723–33. [PubMed] [Google Scholar]
- [141].Noelle RJ, Daum J, Bartlett WC, McCann J, Shepherd DM. Cognate interactions between helper T cells and B cells. V. Reconstitution of T helper cell function using purified plasma membranes from activated Th1 and Th2 T helper cells and lymphokines. J. Immunol. 1991;146(4):1118–24. [PubMed] [Google Scholar]
- [142].Lohning M, Richter A, Stamm T, Hu-Li J, Assenmacher M, Paul WE, Radbruch A. Establishment of memory for IL-10 expression in developing T helper 2 cells requires repetitive IL-4 costimulation and does not impair proliferation. Proc. Natl. Acad. Sci. USA. 2003;100(21):12307–12. doi: 10.1073/pnas.2035254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Benedetti S, Pirola B, Pollo B, Magrassi L, Bruzzone MG, Rigamonti D, Galli R, Selleri S, Di Meco F, De Fraja C, Vescovi A, Cattaneo E, Finocchiaro G. Gene therapy of experimental brain tumors using neural progenitor cells. Nat. Med. 2000;6(4):447–50. doi: 10.1038/74710. [DOI] [PubMed] [Google Scholar]
- [144].Okada H, Villa L, Attanucci J, Erff M, Fellows WK, Lotze MT, Pollack IF, Chambers WH. Cytokine gene therapy of gliomas: effective induction of therapeutic immunity to intracranial tumors by peripheral immunization with interleukin-4 transduced glioma cells. Gene Ther. 2001;8(15):1157–66. doi: 10.1038/sj.gt.3301496. [DOI] [PubMed] [Google Scholar]
- [145].Sampson JH, Archer GE, Ashley DM, Fuchs HE, Hale LP, Dranoff G, Bigner DD. Subcutaneous vaccination with irradiated, cytokine-producing tumor cells stimulates CD8+ cell-mediated immunity against tumors located in the “immunologically privileged” central nervous system. Proc. Natl. Acad. Sci. USA. 1996;93(19):10399–404. doi: 10.1073/pnas.93.19.10399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Okada H, Pollack IF, Lieberman F, Lunsford LD, Kondziolka D, Schiff D, Attanucci J, Edington H, Chambers W, Kalinski P, Kinzler D, Whiteside T, Elder E, Potter D. Gene therapy of malignant gliomas: a pilot study of vaccination with irradiated autologous glioma and dendritic cells admixed with IL-4 transduced fibroblasts to elicit an immune response. Hum. Gene Ther. 2001;12(5):575–95. doi: 10.1089/104303401300042528. [DOI] [PubMed] [Google Scholar]
- [147].Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, Murphy M, Wolf SF, Gately MK. Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J. Exp. Med. 1993;178(4):1223–30. doi: 10.1084/jem.178.4.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Nastala CL, Edington HD, McKinney TG, Tahara H, Nalesnik MA, Brunda MJ, Gately MK, Wolf SF, Schreiber RD, Storkus WJ, et al. Recombinant IL-12 administration induces tumor regression in association with IFN-gamma production. J. Immunol. 1994;153(4):1697–706. [PubMed] [Google Scholar]
- [149].Liu Y, Ehtesham M, Samoto K, Wheeler CJ, Thompson RC, Villarreal LP, Black KL, Yu JS. In situ adenoviral interleukin 12 gene transfer confers potent and long-lasting cytotoxic immunity in glioma. Cancer Gene Ther. 2002;9(1):9–15. doi: 10.1038/sj.cgt.7700399. [DOI] [PubMed] [Google Scholar]
- [150].Yamanaka R, Zullo SA, Ramsey J, Yajima N, Tsuchiya N, Tanaka R, Blaese M, Xanthopoulos KG. Marked enhancement of antitumor immune responses in mouse brain tumor models by genetically modified dendritic cells producing Semliki Forest virus-mediated interleukin-12. J. Neurosurg. 2002;97(3):611–8. doi: 10.3171/jns.2002.97.3.0611. [DOI] [PubMed] [Google Scholar]
- [151].Ando H, Saio M, Ohe N, Tamakawa N, Yu H, Nakayama T, Yoshimura S, Kaku Y, Iwama T, Shinoda J, Sakai N, Takami T. B7.1 immunogene therapy effectively activates CD(4+) tumor-infiltrating lymphocytes in the central nervous system in comparison with B7.2 gene therapy. Int. J. Oncol. 2002;20(4):807–12. [PubMed] [Google Scholar]
- [152].Morioka J, Kajiwara K, Yoshikawa K, Ideguchi M, Uchida T, Ohmoto Y, Suzuki M. Adenovirus-mediated gene transfer of B7.1 induces immunological anti-tumor effects in a murine brain tumor. J. Neurooncol. 2002;60(1):13–23. doi: 10.1023/a:1020260822669. [DOI] [PubMed] [Google Scholar]
- [153].Paul DB, Barth RF, Yang W, Shen GH, Kim J, Triozzi PL. B7.1 expression by the weakly immunogenic F98 rat glioma does not enhance immunogenicity. Gene Ther. 2000;7(12):993–9. doi: 10.1038/sj.gt.3301209. [DOI] [PubMed] [Google Scholar]
- [154].Gitlitz BJ, Belldegrun AS, Zisman A, Chao DH, Pantuck AJ, Hinkel A, Mulders P, Moldawer N, Tso CL, Figlin RA. A pilot trial of tumor lysate-loaded dendritic cells for the treatment of metastatic renal cell carcinoma. J. Immunother. 2003;26(5):412–9. doi: 10.1097/00002371-200309000-00004. [DOI] [PubMed] [Google Scholar]
- [155].Hernando JJ, Park TW, Kubler K, Offergeld R, Schlebusch H, Bauknecht T. Vaccination with autologous tumour antigenpulsed dendritic cells in advanced gynaecological malignancies: clinical and immunological evaluation of a phase I trial. Cancer Immunol. Immunother. 2002;51(1):45–52. doi: 10.1007/s00262-001-0255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Mackensen A, Herbst B, Chen JL, Kohler G, Noppen C, Herr W, Spagnoli GC, Cerundolo V, Lindemann A. Phase I study in melanoma patients of a vaccine with peptide-pulsed dendritic cells generated in vitro from CD34(+) hematopoietic progenitor cells. Int. J. Cancer. 2000;86(3):385–92. doi: 10.1002/(sici)1097-0215(20000501)86:3<385::aid-ijc13>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- [157].Dillman R, Barth N, Selvan S, Beutel L, de Leon C, DePriest C, Peterson C, Nayak S. Phase I/II trial of autologous tumor cell line-derived vaccines for recurrent or metastatic sarcomas. Cancer Biother. Radiopharm. 2004;19(5):581–8. doi: 10.1089/cbr.2004.19.581. [DOI] [PubMed] [Google Scholar]
- [158].Vuylsteke RJ, Molenkamp BG, Gietema HA, van Leeuwen PA, Wijnands PG, Vos W, van Diest PJ, Scheper RJ, Meijer S, de Gruijl TD. Local administration of granulocyte/macrophage colony-stimulating factor increases the number and activation state of dendritic cells in the sentinel lymph node of early-stage melanoma. Cancer Res. . 2004;64(22):8456–60. doi: 10.1158/0008-5472.CAN-03-3251. [DOI] [PubMed] [Google Scholar]
- [159].Ribas A, Glaspy JA, Lee Y, Dissette VB, Seja E, Vu HT, Tchekmedyian NS, Oseguera D, Comin-Anduix B, Wargo JA, Amarnani SN, McBride WH, Economou JS, Butterfield LH. Role of dendritic cell phenotype, determinant spreading, and negative costimulatory blockade in dendritic cell-based melanoma immunotherapy. J. Immunother. 2004;27(5):354–67. doi: 10.1097/00002371-200409000-00004. [DOI] [PubMed] [Google Scholar]
- [160].Maraskovsky E, Brasel K, Teepe M, Roux ER, Lyman SD, Shortman K, McKenna HJ. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: multiple dendritic cell subpopulations identified. J. Exp. Med. 1996;184(5):1953–62. doi: 10.1084/jem.184.5.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Saunders D, Lucas K, Ismaili J, Wu L, Maraskovsky E, Dunn A, Shortman K. Dendritic cell development in culture from thymic precursor cells in the absence of granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1996;184(6):2185–96. doi: 10.1084/jem.184.6.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Lyman SD, James L, Johnson L, Brasel K, de Vries P, Escobar SS, Downey H, Splett RR, Beckmann MP, McKenna HJ. Cloning of the human homologue of the murine flt3 ligand: a growth factor for early hematopoietic progenitor cells. Blood. 1994;83(10):2795–801. [PubMed] [Google Scholar]
- [163].Lynch DH, Andreasen A, Maraskovsky E, Whitmore J, Miller RE, Schuh JC. Flt3 ligand induces tumor regression and antitumor immune responses in vivo. Nat. Med. 1997;3(6):625–31. doi: 10.1038/nm0697-625. [DOI] [PubMed] [Google Scholar]
- [164].Ali S, Curtin JF, Zirger JM, Xiong W, King GD, Barcia C, Liu C, Puntel M, Goverdhana S, Lowenstein PR, Castro MG. Inflammatory and anti-glioma effects of an adenovirus expressing human soluble Fms-like tyrosine kinase 3 ligand (hsFlt3L): treatment with hsFlt3L inhibits intracranial glioma progression. Mol. Ther. 2004;10(6):1071–84. doi: 10.1016/j.ymthe.2004.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Culver KW, Ram Z, Wallbridge S, Ishii H, Oldfield EH, Blaese RM. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science. 1992;256(5063):1550–2. doi: 10.1126/science.1317968. [DOI] [PubMed] [Google Scholar]
- [166].Oldfield EH, Ram Z, Culver KW, Blaese RM, DeVroom HL, Anderson WF. Gene therapy for the treatment of brain tumors using intra-tumoral transduction with the thymidine kinase gene and intravenous ganciclovir. Hum. Gene Ther. 1993;4(1):39–69. doi: 10.1089/hum.1993.4.1-39. [DOI] [PubMed] [Google Scholar]
- [167].Moolten FL. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res. 1986;46(10):5276–81. [PubMed] [Google Scholar]
- [168].Rainov NG, Kramm CM, Banning U, Riemann D, Holzhausen HJ, Heidecke V, Burger KJ, Burkert W, Korholz D. Immune response induced by retrovirus-mediated HSV-tk/GCV pharmacogene therapy in patients with glioblastoma multiforme. Gene Ther. 2000;7(21):1853–8. doi: 10.1038/sj.gt.3301311. [DOI] [PubMed] [Google Scholar]
- [169].Ali S, King GD, Curtin JF, Candolfi M, Xiong W, Liu C, Puntel M, Cheng Q, Prieto J, Ribas A, Kupiec-Weglinski J, van Rooijen N, Lassmann H, Lowenstein PR, Castro MG. Combined immunostimulation and conditional cytotoxic gene therapy provide long-term survival in a large glioma model. Cancer Res. 2005;65(16):7194–204. doi: 10.1158/0008-5472.CAN-04-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Steinman RM, Turley S, Mellman I, Inaba K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J. Exp. Med. 2000;191(3):411–6. doi: 10.1084/jem.191.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Feng H, Zeng Y, Graner MW, Katsanis E. Stressed apoptotic tumor cells stimulate dendritic cells and induce specific cytotoxic T cells. Blood. 2002;100(12):4108–15. doi: 10.1182/blood-2002-05-1389. [DOI] [PubMed] [Google Scholar]
- [172].Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425(6957):516–21. doi: 10.1038/nature01991. [DOI] [PubMed] [Google Scholar]
- [173].Sampath P, Hanes J, DiMeco F, Tyler BM, Brat D, Pardoll DM, Brem H. Paracrine immunotherapy with interleukin-2 and local chemotherapy is synergistic in the treatment of experimental brain tumors. Cancer Res. 1999;59(9):2107–14. [PubMed] [Google Scholar]
- [174].Benedetti S, Pirola B, Poliani PL, Cajola L, Pollo B, Bagnati R, Magrassi L, Tunici P, Finocchiaro G. Dexamethasone inhibits the anti-tumor effect of interleukin 4 on rat experimental gliomas. Gene Ther. 2003;10(2):188–92. doi: 10.1038/sj.gt.3301863. [DOI] [PubMed] [Google Scholar]
- [175].Wheeler CJ, Das A, Liu G, Yu JS, Black KL. Clinical responsiveness of glioblastoma multiforme to chemotherapy after vaccination. Clin. Cancer Res. 2004;10(16):5316–26. doi: 10.1158/1078-0432.CCR-04-0497. [DOI] [PubMed] [Google Scholar]
- [176].Hall WA, Rustamzadeh E, Asher AL. Convection-enhanced delivery in clinical trials. Neurosurg. Focus. 2003;14(2):e2. doi: 10.3171/foc.2003.14.2.3. [DOI] [PubMed] [Google Scholar]
- [177].Weber F, Asher A, Bucholz R, Berger M, Prados M, Chang S, Bruce J, Hall W, Rainov NG, Westphal M, Warnick RE, Rand RW, Floeth F, Rommel F, Pan H, Hingorani VN, Puri RK. Safety, tolerability, and tumor response of IL4-Pseudomonas exotoxin (NBI-3001) in patients with recurrent malignant glioma. J. Neurooncol. 2003;64(12):125–37. doi: 10.1007/BF02700027. [DOI] [PubMed] [Google Scholar]
- [178].Rich JN, Bigner DD. Development of novel targeted therapies in the treatment of malignant glioma. Nat. Rev. Drug Discov. 2004;3(5):430–46. doi: 10.1038/nrd1380. [DOI] [PubMed] [Google Scholar]
- [179].Mori T, Abe T, Wakabayashi Y, Hikawa T, Matsuo K, Yamada Y, Kuwano M, Hori S. Up-regulation of urokinase-type plasminogen activator and its receptor correlates with enhanced invasion activity of human glioma cells mediated by transforming growth factor-alpha or basic fibroblast growth factor. J. Neurooncol. 2000;46(2):115–23. doi: 10.1023/a:1006339717748. [DOI] [PubMed] [Google Scholar]
- [180].Hall WA, Godal A, Juell S, Fodstad O. In vitro efficacy of transferrin-toxin conjugates against glioblastoma multiforme. J. Neurosurg. 1992;76(5):838–44. doi: 10.3171/jns.1992.76.5.0838. [DOI] [PubMed] [Google Scholar]
- [181].Liu TF, Tatter SB, Willingham MC, Yang M, Hu JJ, Frankel AE. Growth factor receptor expression varies among high-grade gliomas and normal brain: epidermal growth factor receptor has excellent properties for interstitial fusion protein therapy. Mol. Cancer Ther. 2003;2(8):783–7. [PubMed] [Google Scholar]
- [182].Martell LA, Agrawal A, Ross DA, Muraszko KM. Efficacy of transferrin receptor-targeted immunotoxins in brain tumor cell lines and pediatric brain tumors. Cancer Res. 1993;53(6):1348–53. [PubMed] [Google Scholar]
- [183].Debinski W, Gibo DM. Molecular expression analysis of restrictive receptor for interleukin 13, a brain tumor-associated cancer/testis antigen. Mol. Med. 2000;6(5):440–9. [PMC free article] [PubMed] [Google Scholar]
- [184].Debinski W, Gibo DM, Hulet SW, Connor JR, Gillespie GY. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin. Cancer Res. 1999;5(5):985–90. [PubMed] [Google Scholar]
- [185].Debinski W, Obiri NI, Pastan I, Puri RK. A novel chimeric protein composed of interleukin 13 and Pseudomonas exotoxin is highly cytotoxic to human carcinoma cells expressing receptors for interleukin 13 and interleukin 4. J. Biol. Chem. 1995;270(28):16775–80. doi: 10.1074/jbc.270.28.16775. [DOI] [PubMed] [Google Scholar]
- [186].Joshi BH, Leland P, Asher A, Prayson RA, Varricchio F, Puri RK. In situ expression of interleukin-4 (IL-4) receptors in human brain tumors and cytotoxicity of a recombinant IL-4 cytotoxin in primary glioblastoma cell cultures. Cancer Res. 2001;61(22):8058–61. [PubMed] [Google Scholar]
- [187].Liu TF, Willingham MC, Tatter SB, Cohen KA, Lowe AC, Thorburn A, Frankel AE. Diphtheria toxin-epidermal growth factor fusion protein and Pseudomonas exotoxin-interleukin 13 fusion protein exert synergistic toxicity against human glioblastoma multiforme cells. Bioconjug. Chem. 2003;14(6):1107–14. doi: 10.1021/bc034111+. [DOI] [PubMed] [Google Scholar]
- [188].Puri RK, Leland P, Kreitman RJ, Pastan I. Human neurological cancer cells express interleukin-4 (IL-4) receptors which are targets for the toxic effects of IL4-Pseudomonas exotoxin chimeric protein. Int. J. Cancer. 1994;58(4):574–81. doi: 10.1002/ijc.2910580421. [DOI] [PubMed] [Google Scholar]
- [189].Liu TF, Hall PD, Cohen KA, Willingham MC, Cai J, Thorburn A, Frankel AE. Interstitial diphtheria toxin-epidermal growth factor fusion protein therapy produces regressions of subcutaneous human glioblastoma multiforme tumors in athymic nude mice. Clin. Cancer Res. 2005;11(1):329–34. [PubMed] [Google Scholar]
- [190].Phillips PC, Levow C, Catterall M, Colvin OM, Pastan I, Brem H. Transforming growth factor-alpha-Pseudomonas exotoxin fusion protein (TGF-alpha-PE38) treatment of subcutaneous and intracranial human glioma and medulloblastoma xenografts in athymic mice. Cancer Res. 1994;54(4):1008–15. [PubMed] [Google Scholar]
- [191].Debinski W, Obiri NI, Powers SK, Pastan I, Puri RK. Human glioma cells overexpress receptors for interleukin 13 and are extremely sensitive to a novel chimeric protein composed of interleukin 13 and pseudomonas exotoxin. Clin. Cancer Res. . 1995;1(11):1253–8. [PubMed] [Google Scholar]
- [192].Todhunter DA, Hall WA, Rustamzadeh E, Shu Y, Doumbia SO, Vallera DA. A bispecific immunotoxin (DTAT13) targeting human IL-13 receptor (IL-13R) and urokinase-type plasminogen activator receptor (uPAR) in a mouse xenograft model. Protein Eng. Des. Sel. 2004;17(2):157–64. doi: 10.1093/protein/gzh023. [DOI] [PubMed] [Google Scholar]
- [193].Li C, Hall WA, Jin N, Todhunter DA, Panoskaltsis-Mortari A, Vallera DA. Targeting glioblastoma multiforme with an IL-13/diphtheria toxin fusion protein in vitro and in vivo in nude mice. Protein Eng. 2002;15(5):419–27. doi: 10.1093/protein/15.5.419. [DOI] [PubMed] [Google Scholar]
- [194].Husain SR, Joshi BH, Puri RK. Interleukin-13 receptor as a unique target for anti-glioblastoma therapy. Int. J. Cancer. 2001;92(2):168–75. doi: 10.1002/1097-0215(200102)9999:9999<::aid-ijc1182>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- [195].Kawakami K, Kawakami M, Kioi M, Husain SR, Puri RK. Distribution kinetics of targeted cytotoxin in glioma by bolus or convection-enhanced delivery in a murine model. J. Neurosurg. 2004;101(6):1004–11. doi: 10.3171/jns.2004.101.6.1004. [DOI] [PubMed] [Google Scholar]
- [196].Debinski W, Gibo DM, Obiri NI, Kealiher A, Puri RK. Novel anti-brain tumor cytotoxins specific for cancer cells. Nat. Biotechnol. 1998;16(5):449–53. doi: 10.1038/nbt0598-449. [DOI] [PubMed] [Google Scholar]
- [197].Madhankumar AB, Mintz A, Debinski W. Interleukin 13 mutants of enhanced avidity toward the glioma-associated receptor, IL13Ralpha2. Neoplasia. 2004;6(1):15–22. doi: 10.1016/s1476-5586(04)80049-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Obiri NI, Debinski W, Leonard WJ, Puri RK. Receptor for interleukin 13. Interaction with interleukin 4 by a mechanism that does not involve the common gamma chain shared by receptors for interleukins 2, 4, 7, 9, and 15. J. Biol. Chem. 1995;270(15):8797–804. doi: 10.1074/jbc.270.15.8797. [DOI] [PubMed] [Google Scholar]
- [199].Cicuttini FM, Hurley SF, Forbes A, Donnan GA, Salzberg M, Giles GG, McNeil JJ. Association of adult glioma with medical conditions, family and reproductive history. Int. J. Cancer. 1997;71(2):203–7. doi: 10.1002/(sici)1097-0215(19970410)71:2<203::aid-ijc13>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- [200].Obiri NI, Leland P, Murata T, Debinski W, Puri RK. The IL-13 receptor structure differs on various cell types and may share more than one component with IL-4 receptor. J. Immunol. 1997;158(2):756–64. [PubMed] [Google Scholar]
- [201].Maini A, Hillman G, Haas GP, Wang CY, Montecillo E, Hamzavi F, Pontes JE, Leland P, Pastan I, Debinski W, Puri RK. Interleukin-13 receptors on human prostate carcinoma cell lines represent a novel target for a chimeric protein composed of IL-13 and a mutated form of Pseudomonas exotoxin. J. Urol. 1997;158(3 Pt 1):948–53. doi: 10.1097/00005392-199709000-00077. [DOI] [PubMed] [Google Scholar]
- [202].Kornmann M, Kleeff J, Debinski W, Korc M. Pancreatic cancer cells express interleukin-13 and -4 receptors, and their growth is inhibited by Pseudomonas exotoxin coupled to interleukin-13 and -4. Anticancer Res. 1999;19(1A):125–31. [PubMed] [Google Scholar]
- [203].Kawakami K, Kawakami M, Puri RK. Overexpressed cell surface interleukin-4 receptor molecules can be successfully targeted for antitumor cytotoxin therapy. Crit. Rev. Immunol. . 2001;21(13):299–310. [PubMed] [Google Scholar]
- [204].Husain SR, Behari N, Kreitman RJ, Pastan I, Puri RK. Complete regression of established human glioblastoma tumor xenograft by interleukin-4 toxin therapy. Cancer Res. 1998;58(16):3649–53. [PubMed] [Google Scholar]
- [205].Kawakami M, Kawakami K, Puri RK. Interleukin-4-Pseudomonas exotoxin chimeric fusion protein for malignant glioma therapy. J. Neurooncol. 2003;65(1):15–25. doi: 10.1023/a:1026294416718. [DOI] [PubMed] [Google Scholar]
- [206].Rand RW, Kreitman RJ, Patronas N, Varricchio F, Pastan I, Puri RK. Intratumoral administration of recombinant circularly permuted interleukin-4-Pseudomonas exotoxin in patients with high-grade glioma. Clin. Cancer Res. 2000;6(6):2157–65. [PubMed] [Google Scholar]
- [207].Engebraaten O, Hjortland GO, Juell S, Hirschberg H, Fodstad O. Intratumoral immunotoxin treatment of human malignant brain tumors in immunodeficient animals. Int. J. Cancer. 2002;97(6):846–52. doi: 10.1002/ijc.10137. [DOI] [PubMed] [Google Scholar]
- [208].Laske DW, Youle RJ, Oldfield EH. Tumor regression with regional distribution of the targeted toxin TF-CRM107 in patients with malignant brain tumors. Nat. Med. 1997;3(12):1362–8. doi: 10.1038/nm1297-1362. [DOI] [PubMed] [Google Scholar]
- [209].Weaver M, Laske DW. Transferrin receptor ligand-targeted toxin conjugate (Tf-CRM107) for therapy of malignant gliomas. J. Neurooncol. 2003;65(1):3–13. doi: 10.1023/a:1026246500788. [DOI] [PubMed] [Google Scholar]
- [210].Barker FG, 2nd., Simmons ML, Chang SM, Prados MD, Larson DA, Sneed PK, Wara WM, Berger MS, Chen P, Israel MA, Aldape KD. EGFR overexpression and radiation response in glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 2001;51(2):410–8. doi: 10.1016/s0360-3016(01)01609-1. [DOI] [PubMed] [Google Scholar]
- [211].Chakravarti A, Delaney MA, Noll E, Black PM, Loeffler JS, Muzikansky A, Dyson NJ. Prognostic and pathologic significance of quantitative protein expression profiling in human gliomas. Clin. Cancer Res. 2001;7(8):2387–95. [PubMed] [Google Scholar]
- [212].Jones NR, Rossi ML, Gregoriou M, Hughes JT. Investigation of the expression of epidermal growth factor receptor and blood group A antigen in 110 human gliomas. Neuropathol. Appl. Neurobiol. 1990;16(3):185–92. doi: 10.1111/j.1365-2990.1990.tb01155.x. [DOI] [PubMed] [Google Scholar]
- [213].Sampson JH, Akabani G, Archer GE, Bigner DD, Berger MS, Friedman AH, Friedman HS, Herndon JE, 2nd., Kunwar S, Marcus S, McLendon RE, Paolino A, Penne K, Provenzale J, Quinn J, Reardon DA, Rich J, Stenzel T, Tourt-Uhlig S, Wikstrand C, Wong T, Williams R, Yuan F, Zalutsky MR, Pastan I. Progress report of a Phase I study of the intracerebral microinfusion of a recombinant chimeric protein composed of transforming growth factor (TGF)-alpha and a mutated form of the Pseudomonas exotoxin termed PE-38 (TP-38) for the treatment of malignant brain tumors. J. Neurooncol. . 2003;65(1):27–35. doi: 10.1023/a:1026290315809. [DOI] [PubMed] [Google Scholar]
- [214].Bosq J, Penault-Llorca F, Sabourin JC. [Expression of epidermal growth factor receptor and its ligands in other malignant tumor pathologies] Bull. Cancer. 2003;90:S241–4. Spec No. [PubMed] [Google Scholar]
- [215].Overwijk WW, Lee DS, Surman DR, Irvine KR, Touloukian CE, Chan CC, Carroll MW, Moss B, Rosenberg SA, Restifo NP. Vaccination with a recombinant vaccinia virus encoding a “self” antigen induces autoimmune vitiligo and tumor cell destruction in mice: requirement for CD4(+) T lymphocytes. Proc. Natl. Acad. Sci. USA. 1999;96(6):2982–7. doi: 10.1073/pnas.96.6.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang L, Feigenbaum AM, Kruisbeek SA, Rosenberg NP, Restifo LN. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J. Exp. Med. 2003;198(4):569–80. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, Becher B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat. Med. 2005;11(3):328–34. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- [218].McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat. Med. 2005;11(3):335–9. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- [219].Fujinami RS, Paterson PY, Day ED, Varitek VA. Myelin basic protein serum factor. An endogenous neuroantigen influencing development of experimental allergic encephalomyelitis in Lewis rats. J. Exp. Med. 1978;148(6):1716–21. doi: 10.1084/jem.148.6.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].McFarlin DE, Blank SE, Kibler RF. Recurrent experimental allergic encephalomyelitis in the Lewis rat. J. Immunol. 1974;113(2):712–5. [PubMed] [Google Scholar]
- [221].Massoud TF, Gambhir SS. Molecular imaging in living subjects: seeing fundamental biological processes in a new light. Genes Dev. 2003;17(5):545–80. doi: 10.1101/gad.1047403. [DOI] [PubMed] [Google Scholar]
- [222].Damadian R. Tumor detection by nuclear magnetic resonance. Science. 1971;171(976):1151–3. doi: 10.1126/science.171.3976.1151. [DOI] [PubMed] [Google Scholar]
- [223].Henson JW, Gaviani P, Gonzalez RG. MRI in treatment of adult gliomas. Lancet Oncol. 2005;6(3):167–75. doi: 10.1016/S1470-2045(05)01767-5. [DOI] [PubMed] [Google Scholar]
- [224].Liu RS. Clinical Application of. Clin. Positron Imaging. 2000;3(4):185. doi: 10.1016/s1095-0397(00)00097-2. [DOI] [PubMed] [Google Scholar]
- [225].Mineura K, Sasajima T, Kowada M, Ogawa T, Hatazawa J, Uemura K. Early delineation of cerebral glioma using amino acid positron tracers. Comput. Med. Imaging Graph. 1997;21(1):63–6. doi: 10.1016/s0895-6111(96)00051-1. [DOI] [PubMed] [Google Scholar]
- [226].Liang Q, Satyamurthy N, Barrio JR, Toyokuni T, Phelps MP, Gambhir SS, Herschman HR. Noninvasive, quantitative imaging in living animals of a mutant dopamine D2 receptor reporter gene in which ligand binding is uncoupled from signal transduction. Gene Ther. 2001;8(19):1490–8. doi: 10.1038/sj.gt.3301542. [DOI] [PubMed] [Google Scholar]
- [227].Liang Q, Nguyen K, Satyamurthy N, Barrio JR, Phelps ME, Gambhir SS, Herschman HR. Monitoring adenoviral DNA delivery, using a mutant herpes simplex virus type 1 thymidine kinase gene as a PET reporter gene. Gene Ther. 2002;9(24):1659–66. doi: 10.1038/sj.gt.3301899. [DOI] [PubMed] [Google Scholar]
- [228].Yaghoubi SS, Barrio JR, Namavari M, Satyamurthy N, Phelps ME, Herschman HR, Gambhir SS. Imaging progress of herpes simplex virus type 1 thymidine kinase suicide gene therapy in living subjects with positron emission tomography. Cancer Gene Ther. 2005;12(3):329–39. doi: 10.1038/sj.cgt.7700795. [DOI] [PubMed] [Google Scholar]
- [229].Wang Y, Iyer M, Annala AJ, Chappell S, Mauro V, Gambhir SS. Noninvasive monitoring of target gene expression by imaging reporter gene expression in living animals using improved bicistronic vectors. J. Nucl. Med. 2005;46(4):667–74. [PubMed] [Google Scholar]
- [230].Bhaumik S, Gambhir SS. Optical imaging of Renilla luciferase reporter gene expression in living mice. Proc. Natl. Acad. Sci. USA. 2002;99(1):377–82. doi: 10.1073/pnas.012611099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Bhaumik S, Lewis XZ, Gambhir SS. Optical imaging of Renilla luciferase, synthetic Renilla luciferase, and firefly luciferase reporter gene expression in living mice. J. Biomed. Opt. 2004;9(3):578–86. doi: 10.1117/1.1647546. [DOI] [PubMed] [Google Scholar]
- [232].De A, Lewis XZ, Gambhir SS. Noninvasive imaging of lentiviral-mediated reporter gene expression in living mice. Mol. Ther. 2003;7(5 Pt 1):681–91. doi: 10.1016/s1525-0016(03)00070-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Ray P, De A, Min JJ, Tsien RY, Gambhir SS. Imaging tri-fusion multimodality reporter gene expression in living subjects. Cancer Res. 2004;64(4):1323–30. doi: 10.1158/0008-5472.can-03-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]