

Abstract
The activity of the eukaryotic translation initiation factor eIF4E is modulated through conformational response to its ligands. For example, eIF4G and eIF4E-binding proteins (4E-BPs) modulate cap affinity, and thus physiological activity of eIF4E, by binding a site distal to the 7-methylguanosine cap-binding site. Further, cap binding substantially modulates eIF4E's affinity for eIF4G and the 4E-BPs. To date, only cap-bound eIF4E structures were reported. In the absence of structural information on the apo form, the molecular underpinnings of this conformational response mechanism cannot be established. We report here the first cap-free eIF4E structure. Apo-eIF4E exhibits structural differences in the cap-binding site and dorsal surface relative to cap-eIF4E. Analysis of structure and dynamics of apo-eIF4E, and changes observed upon ligand binding, reveal a molecular basis for eIF4E's conformational response to these ligands. In particular, alterations in the S4-H4 loop, distal to either the cap or eIF4G binding sites, appear key to modulating these effects. Mutation in this loop mimics these effects. Overall, our studies have important implications for the regulation of eIF4E.
Keywords: apo-eIF4E, conformational regulation, eIF4G, NMR, relaxation
Introduction
Normal development, differentiation and cellular growth rely on post-transcriptional control of gene expression. The eukaryotic translation initiation factor eIF4E regulates gene expression post-transcriptionally at multiple levels, including mRNA translation and mRNA export (Pestova et al, 2001; von der Haar et al, 2004). Through these activities, eIF4E promotes cellular proliferation, growth and survival. Even moderate overexpression of eIF4E leads to dysregulated cellular proliferation +and malignant transformation (Graff and Zimmer, 2003). eIF4E associates with the 5′ end of mRNAs via the 7-methyl guanosine dinucleotide cap structure, m7GpppN (7-methyl-5′-guanosinetriphosphate-5′-N) (where N is any nucleotide) (Sonenberg and Gingras, 1998). The association of eIF4E with mRNA via the cap is linked to its biochemical and subsequent growth promoting and transforming activities. eIF4E overexpression does not uniformly increase synthesis for all proteins; targeting those with complex 5′ untranslated regions (UTRs) (Sonenberg and Gingras, 1998).
The Sonenberg group first showed that eIF4E is organized into nuclear bodies in addition to its cytoplasmic localization (Rousseau et al, 1996). eIF4E forms nuclear bodies in a variety of cell types and organisms including yeast, Drosophila, Xenopus, mouse and human (Iborra and Cook, 2002). Up to 68% of total cellular eIF4E is found in the nucleus (Iborra et al, 2001; Strudwick and Borden, 2002; Culjkovic et al, 2005). The best described nuclear function involves nuclear mRNA export of selected transcripts (Rousseau et al, 1996). This level of regulation requires an ∼100 nucleotide sequence denoted the eIF4E sensitivity element (4E-SE) in the 3′UTR of sensitive transcripts (Culjkovic et al, 2005). Importantly, eIF4E requires its cap binding activity for its nuclear mRNA export function. For instance in the nucleus, the promyelocytic leukemia (PML) protein directly binds eIF4E, suppresses its mRNA export and transformation functions (Cohen et al, 2001). Mutagenesis studies indicate that both its mRNA export as well as its translation functions contribute to the ability of eIF4E to transform cells (Cohen et al, 2001; Topisirovic et al, 2003a, 2003b; Culjkovic et al, 2005).
eIF4E requires its cap binding activity to act in mRNA translation, mRNA nuclear export and in order to promote oncogenic transformation (Sonenberg and Gingras, 1998; Cohen et al, 2001; Topisirovic et al, 2003a, 2003b). The functions of eIF4E are tightly regulated in the cell through interactions with partner proteins. In translation, eIF4E binds the RNA through the 7-methylguanosine (m7G) cap structure and subsequently other components of the eIF4F complex including eIF4G (Gingras et al, 1999). eIF4G binds the dorsal surface of eIF4E, which is located distal to the cap binding site (Figure 1). The same site used for eIF4G is also bound by the translational inhibitors, the eIF4E binding proteins (4E-BPs). The 4E-BPs inhibit translation by sterically blocking eIF4G binding (Ptushkina et al, 1999). Importantly, both eIF4G and 4E-BP association with eIF4E leads to enhanced association of eIF4E with the m7G cap (von der Haar et al, 2004). This ligand binding leads to a conformational response by eIF4E. In this way, 4E-BP/eIF4E complexes can trap mRNAs making 4E-BP a potent inhibitor of translation by, in essence, sequestering the transcripts in inactive complexes (von der Haar et al, 2004). Theoretically, this effect on cap binding leads to eIF4G more effectively promoting translation. Importantly, many other regulators of eIF4E bind its dorsal surface including up to 200 homeoproteins many of which regulate nuclear and cytoplasmic activities of eIF4E (Topisirovic et al, 2003a). These proteins, like eIF4G and the 4E-BPs, contain a conserved eIF4E binding site defined as YXXXXLΦ where X is any residue and Φ is any hydrophobic. PML, a potent negative regulator of eIF4E in the nucleus, uses its RING domain to bind the dorsal surface. This leads to a reduction in the affinity of eIF4E for the cap by over 100-fold (Cohen et al, 2001; Kentsis et al, 2001). Thus, eIF4E is regulated through conformational rearrangements by a wide variety of protein partners and this results in modulation of the biological activities of eIF4E.
Figure 1.
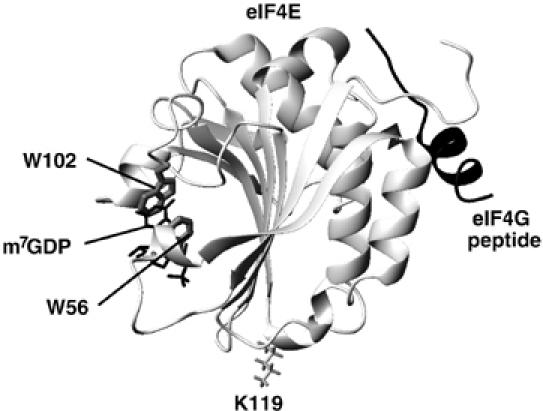
Crystal structure of the murine eIF4E/7-methyl-GDP–eIF4G peptide ternary complex (PDB 1EJH).
Detailed structural studies on eIF4E complexed to various cap analogs and to eIF4G and the 4E-BPs have led to important advances in our molecular understanding of eIF4E function (von der Haar et al, 2004). High-resolution crystal studies delineate the residues important to cap binding (Marcotrigiano et al, 1997; Niedzwiecka et al, 2002; Tomoo et al, 2002). It is well established that the specificity of cap recognition is imparted by the m7G moiety, which is stacked between two tryptophan residues, W56 and W102 (Sonenberg and Gingras, 1998). Also, the delocalization of the positive charge on m7G, due to the methylation of N7, promotes interaction with an acidic cavity (formed by E103) in eIF4E (Marcotrigiano et al, 1997). While the m7G moiety alone binds to eIF4E, addition of phosphate groups significantly enhances affinity (Zuberek et al, 2004).
Several biophysical studies highlight the conformational linkage between cap binding and the association of partner proteins on the distal dorsal surface of eIF4E. We will refer to this as conformational response or rearrangement. For example, a 100 residue eIF4G fragment increases the affinity of yeast eIF4E for the cap by 10-fold (Ptushkina et al, 1999) whereas studies with a minimal eIF4G peptide for human and mouse eIF4E increase affinity by about two-fold (Friedland et al, 2005). Conversely, cap binding enhances affinity of eIF4E for the eIF4G peptide and 4E-BPs (Shen et al, 2001; von der Haar et al, 2004; Tomoo et al, 2005). Recent solution NMR studies indicate that in the ternary complex, eIF4G binding only causes slight chemical shift perturbations in the cap binding site relative to the yeast eIF4E-cap complex (Gross et al, 2003). Using short eIF4G and 4E-BP1 peptides, crystallographic studies of mouse N-terminal truncated eIF4E indicated that there is no significant structural change between eIF4E-cap and ternary complexes containing either peptide (Marcotrigiano et al, 1999). Given the similarity of the cap binding site in the cap bound and ternary complexes, we hypothesize that eIF4G and the 4E-BPs alter affinity of eIF4E for the cap by modulating the structure of the cap-free form of eIF4E rather than causing substantial structural alterations to the cap bound form of the protein.
The biophysical and biological data are clear, cells can modulate proliferation via conformational rearrangements of eIF4E. In order to understand any structurally based process, it is critical to know the relevant structural features of each state. To date, no structure has been solved for any cap free form of eIF4E. Here, we report the first apo-eIF4E structure. Our studies reveal the structural rearrangements that eIF4E undergoes in order to bind the m7G cap. Further, our data provide the first structural basis for the modulation of eIF4E activity via conformational rearrangements. In essence, the conformational response to eIF4G is mediated by partially structuring the apo-eIF4E in a state between apo- and cap-bound eIF4E; we refer to this as prestructuring. Similarly, cap addition modulates the conformation of the dorsal surface. Further, we discovered that a mutation (residue K119 mutated to alanine) distal to either the cap binding site or the dorsal surface (Figure 1) also contains structural features consistent with partial prestructuring of the apo mutant relative to apo-wild type eIF4E. Mutation in this location causes conformational changes in both the cap binding site and dorsal surface, consistent with its higher affinity for both cap and eIF4G.
Results and discussion
Production of apo-eIF4E
Full-length, untagged, soluble human eIF4E was produced by bacterial overexpression and purified using m7GDP affinity chromatography. To elute eIF4E from the column, m7G was used and subsequently removed by extensive dialysis. To ensure that the eIF4E protein was indeed cap-free after dialysis, we carried out mass spectrometry analysis. As can be seen in Figure 2A, the molecular mass of eIF4E is 24964.3±3.2 Da slightly less than the predicted mass 25097.2 Da. This mass difference (132.9 Da) corresponds to the loss of methionine (131.2 Da) at the N-terminus, which is a common event (Huang et al, 1987). The absence of Met1 in the NMR data corroborates this hypothesis. Clearly, there is no peak corresponding to eIF4E-m7G (which would be +297.2 Da higher), indicating that the eIF4E used in our studies is indeed cap free. To further ensure that the apo-eIF4E form was produced, we compared 1H-15N heteronuclear single quantum correlation (HSQC) of protein purified by elution of eIF4E from the cap resin with either high salt or the m7G cap. The 1H-15N HSQC for samples produced in these conditions were identical (Supplementary Figure 1). Consistently, the dissociation constant (Kd) for the cap for our apo-eIF4E is similar to previous studies, which purified eIF4E using a GST tag instead of using cap chromatography (Kd eIF4E-GST purified=1.2±0.2 μM, see Kentsis et al, 2001; Kd for cap chromatography followed by extensive dialysis=1.90±0.38 μM, see Figure 2E and Supplementary Figure 2). This further supports that eIF4E produced here is cap free. Further, addition of m7GDP to the cap-free form led to widespread alterations in chemical shifts and heteronuclear nuclear overhauser effect (hNOE) as compared to the apo form (Figures 2C and 4A). Finally, cap addition led to readily detectable cap-bound form (mass 25422.5±2.1 Da) by mass spectrometry (Figure 2B). Note that there are differences in ionization efficiencies of apo- versus cap-eIF4E (see figure legend in Supplementary Figure 3).
Figure 2.
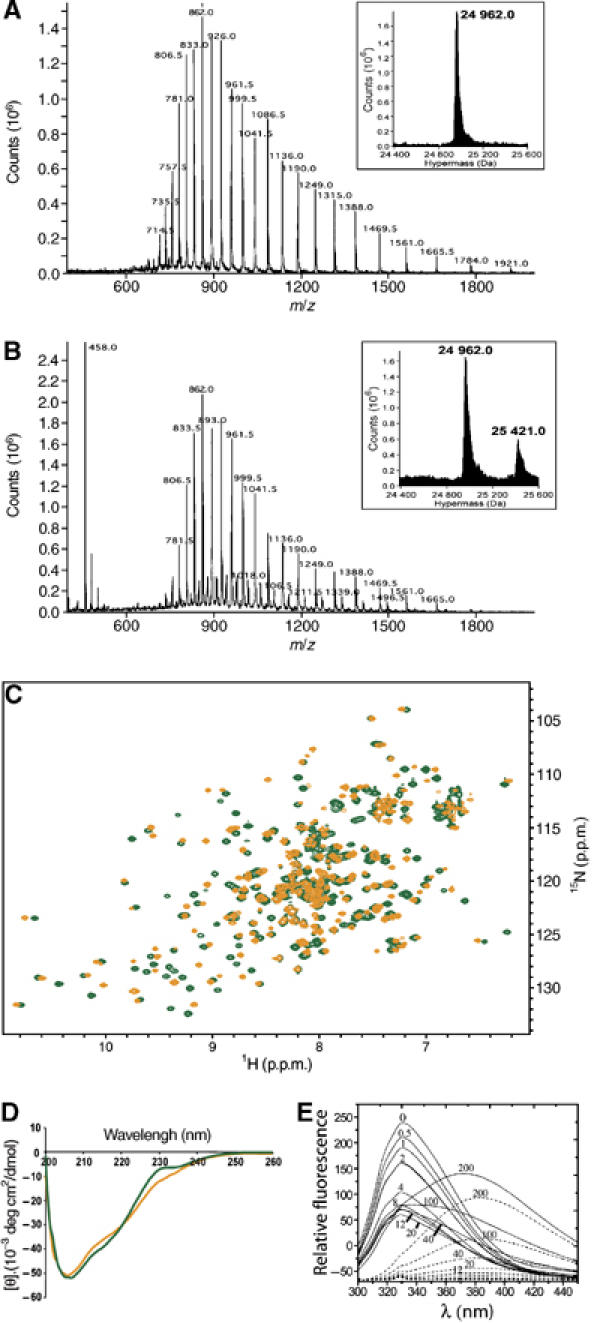
Mass spectrum of the apo- (A) and m7GDP-bound (B) forms of wild-type eIF4E. ES-MS spectra plotting ion abundance as a function of the mass/charge ratio. Insets: hypermass reconstruction of the spectrum. The low molecular weight peak at 458 is consistent with the presence of the m7GDP (457.23 Da). For more details, see figure legend in Supplementary Figure 3. Clearly, no cap-bound eIF4E is present in the apo sample. (C) Superposition of the 1H-15N HSQC spectrum of the apo-eIF4E (orange) and the m7GDP-bound eIF4E (green). (D) The far-UV CD spectra of apo-eIF4E (orange) and m7GDP–eIF4E (green). (E) Fluorescence emission of wild-type eIF4E in the presence of increasing concentrations of m7GDP (continuous) and intrinsic fluorescence of m7GDP in the absence of eIF4E (dashed). The different m7GDP concentrations (μM) are shown on the curves and fit is shown in Supplementary Figure 2.
Structure determination
For many years, apo-eIF4E has eluded attempts at high-resolution structural studies. Recent studies on yeast eIF4E, which has ∼30% identity to the human eIF4E studied here (Figure 3A), suggested that the apo form of eIF4E was unfolded (Niedzwiecka et al, 2004; von der Haar et al, 2006). To find appropriate conditions for NMR, we screened and characterized a wide variety of solution conditions. Significantly, we were able to obtain well-resolved NMR spectra for the apo-eIF4E for a narrow range of conditions indicating that eIF4E can exist as a folded protein in the apo state. The final solution conditions used for structure determination were 0.3 mM protein in 50 mM phosphate buffer (pH 7.4), 100 mM NaCl, 1 mM dithiothreitol (DTT), 0.02% NaN3. In these conditions, translational diffusion coefficients from NMR indicate that eIF4E was monomeric (see Supplementary Figure 4). This was confirmed by relaxation data, which estimated an overall correlation time of 19.1 ns from R2/R1 ratios, close to the expected value for a 217 residue protein at 20°C of ∼18 ns. This value is also close to that estimated from R2/R1 ratios for the cap form (18.5 ns). However, under some other conditions examined, and particularly in N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES), apo-eIF4E aggregated substantially (see Supplementary Figure 4). Addition of cap partially reversed this aggregation as previously shown (von der Haar et al, 2004; Niedzwiecka et al, 2005). It is also important to note here that cap-bound eIF4E was much less susceptible to aggregation and conformational changes associated with altering solution conditions (discussed below). CD analysis indicates that the apo form of eIF4E is folded (Figure 2D) consistent with previous studies (McCubbin et al, 1988; von der Haar et al, 2000; Cohen et al, 2001).
Figure 3.
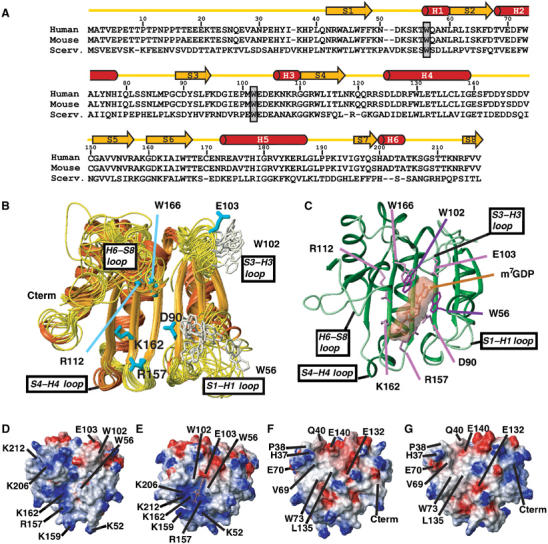
Structural comparison between apo- and cap-bound eIF4E. (A) Sequence alignments of eIF4E (Scerv., Saccharomyces cerevisiae). The secondary structural elements were assigned from the apo structure. W56 and W102 residues are boxed. (B) Superposition of the 10 lowest-energy NMR structures. Side chains of W56 and W102 are shown. (C) Crystal structure of human eIF4E bound to m7GDP (PDB 1EJ1). This structure contains density for residues 36–207 and 213–217. The side chains of residues that interact with the cap are shown. (D–G) Potential map of the surface of apo-eIF4E (D, F) and cap-eIF4E (E, G) calculated with MOLMOL. The orientation of (D) and (E) are the same as that of (B) and (C) (cap-binding site) while (F) and (G) represent a 180° rotation along a vertical axis compared with (D) and (E) (eIF4G binding site). A number of residues of interest are indicated on the structures (see text).
We determined the first 3D structure of the apo form of eIF4E using standard triple resonance NMR techniques (see Materials and methods). A total of 1631 nonredundant interproton distance restraints (65% of them being medium- and long-range NOEs) and 276 dihedral restraints were used in the final round of calculations. Restraint information and structural analysis are summarized in Table I. Using the Crystallography and NMR System (CNS) software, 50 structures were calculated, and the 10 lowest energy structures were chosen to represent the solution structure of apo-eIF4E (Figure 3B). Secondary structure elements identified from characteristic backbone NOE connectivities, deviations from random coil chemical shifts and H/D exchange experiments indicated apo-eIF4E is comprised of three long α-helices (68–78 H2, 125–139 H4 and 173–187 H5) and three short one-turn helices (56–60 H1, 106–110 H3 and 200–204 H6), plus eight β-strands (42–48 S1, 60–67 S2, 89–94 S3, 110–117 S4, 151–157 S5, 160–167 S6, 196–199 S7 and 215–217 S8) (Figure 3A). In this NMR ensemble of the apo-eIF4E, the loops S1–S2, H2–S3, H4–S5 and S7–S8 are less defined while the first 35 residues at the N-terminus are completely disordered. This correlates with the lack of structural restraints in these regions and is confirmed by relaxation data and significantly the 15N hNOE values (Figure 4A), which are distinctly lower for these regions with weighted mean values of 0.62±0.02, 0.70±0.03 and 0.55±0.05 for the S1–S2, H4–S5 and S7–S8 loops, respectively. Consistent results are obtained from T1 and T2 relaxation experiments (Supplementary Figure 5). We also note that no reliable NOE's or R1 and R2 values could be obtained from residues in the H2–S3 loop and at the centre of the S7–S8 loop, due to line broadening, most likely a consequence of motions on the ms timescales at these sites. Indeed, there is evidence for elevated R2 values in the H4–S5 loop for both the cap and apo forms of eIF4E (Supplementary Figure 6). In contrast, residues comprising the long helices and sheets are relatively non-flexible on the ns timescales, yielding an average NOE value of 0.77.
Table 1.
Structure statistics of apo-eIF4E
| Field and variable(s) | Value |
|---|---|
| Restraints for final structure calculations | |
| Total restraints used | 1973 |
| Total NOE restraints | 1697 |
| Intraresidue | 145 |
| Sequential (∣i−j∣=1) | 438 |
| Medium range (1<∣i−j∣=4) | 394 |
| Long range (∣i−j∣>4) | 560 |
| Hydrogen bond restraintsa | 160 |
| φ dihedral angles restraints | 138 |
| ψ dihedral angles restraints | 138 |
| Statistics for structure calculations (〈SA〉b) | |
| RMSD from idealized covalent geometry | |
| Bonds (Å) | 0.0018±0.0001 |
| Angles (deg) | 0.350±0.011 |
| Improper (deg) | 0.230±0.019 |
| RMSD from experimental restraints: distances (Å)c | 0.012±0.001 |
| Ramachandran plot statistics (%)d | |
| Residues in most favored regions | 78.7±1.7 |
| Residues in additional allowed regions | 15.7±1.5 |
| Residues in generously allowed regions | 3.7±1.3 |
| Residues in disallowed regions | 1.9±0.7 |
| a The amide protons implicated in all these hydrogen bonds were found to slowly exchange with D2O. | |
| b 〈SA〉 refers to the ensemble of the 10 structures with the lowest energy from 50 calculated structures. | |
| c No distance restraint in any of the structures included in the ensemble was violated by more than 0.2 Å. | |
| d Generated using PROCHECK on the ensemble of the 10 lowest-energy structures, residues 36–217. The residues located in the two latter regions are mostly located in flexible regions or connect secondary structural regions. | |
Figure 4.
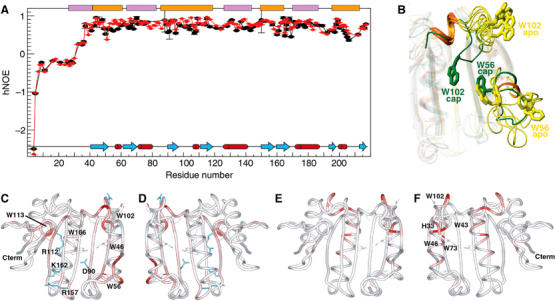
(A) hNOEs for backbone amide nitrogens of the apo-eIF4E (black) and m7GDP–eIF4E (red) measured at a proton frequency of 600 MHz. The cap (orange) and eIF4G (purple) binding sites on eIF4E are shown. (B) Motions associated with cap binding in the W56 and W102 loops. Four structures of the NMR ensemble of the apo-eIF4E (yellow) were superimposed with the cap structure (green), and loops containing W56 (S1–S2) and W102 (S3–S4) were highlighted for clarity. (C–F) Chemical shift perturbation of backbone 1HN and 15N resonances (color-coded) between the apo-eIF4E and m7GDP–eIF4E (C, D), and between the apo-eIF4E and apo-eIF4E-eIF4G peptide (E, F). These perturbations were mapped onto the apo-eIF4E structure. The orientation of (D) and (F) represent a 180° rotation along a vertical axis compared with panels (C) and (E), respectively. Analysis of ligand-induced shifts was performed by applying the Pythagorean theorem to weighted chemical shifts: Δδ (1H,15N)={Δδ(1H)2+0.2 × Δδ(15N)2}1/2, where Δδ(1H) and Δδ(15N) are the chemical-shift differences of the amide proton and nitrogen, respectively (Grzesiek et al, 1996; Pellecchia et al, 1999). These deviations are also shown for the indole Hɛ1 of the Trp residues.
The root mean square deviation of this family with respect to the mean coordinate positions is 1.26 and 2.25 Å for backbone and heavy atoms, respectively, for regular secondary structure elements. These values are close to those obtained for the yeast eIF4E/m7GDP/eIF4G fragment complex (1.24 and 2.08 Å, respectively) (Gross et al, 2003).
Comparison to cap-bound eIF4E structures
The cap bound structure is often described as a cupped hand (Figure 3C). As a comparison, we describe the apo-form as an open hand. Our apo structure determined in solution is in many ways very similar to the crystal eIF4E-cap structures reported for human and mouse eIF4E, the latter being 98% identical to the human protein. The apo form, like the cap bound form, contains an 8-stranded anti-parallel β-sheet on the cap binding side, and three long helices on the convex face that binds other protein partners (Figure 3B). The core β-strands are positioned nearly identically. Differences between the two structures are observed in the loops, the helical content and for some beta strands (S4–S8). In particular, structural differences are observed for the loops S1–S2, H1–S3, S3–S4, H2–S5, S5–S6, H3–S7 and S7–S8. Extent of structural changes are consistent with previous CD data that predicted ∼20% changes (Cai et al, 1999; von der Haar et al, 2000; Cohen et al, 2001). However, in contrast to all other reported crystal structures of binary and tertiary eIF4E complexes, the apo structure differs substantially at the cap binding surface (Figure 3D and E), at the dorsal surface important for eIF4G/4E-BPs binding and adjacent regions (Figure 3F and G). For the cap binding site, significant rearrangements are observed for the tryptophan residues crucial for m7G binding. Additionally, the positively charged pocket in the cap binding site formed by the side chains of residues K52, R157, K159, K162, K206, K212 in the cap-bound structure is dispersed in the apo structure, as alluded to by its open-hand conformation. Conformational rearrangements of the loops S1–S2 and S7–S8 largely contribute to this dispersion, while R157, K159 and K162 side chains adopt a slightly different position. In terms of previously reported conformational regulation of eIF4E, cap binding results in alterations in the electrostatic potential on the dorsal surface, consistent with increased affinity of the cap-bound form for eIF4G (von der Haar et al, 2004; Figure 3F and G).
We also investigated the importance of the N-terminus in stabilizing eIF4E and in cap binding. For instance, deletion of residues 1–20 from eIF4E reduces the Kd of eIF4E for eIF4G by three-fold, while it is reduced by 16- and 60-fold by deleting residues 1–30 and 1–35, respectively (Gross et al, 2003). Interestingly, our own deletion studies demonstrate that deletion of the first 36 residues leads to substantial chemical shift alterations throughout helix 2 (H2) (data not shown), suggesting that residues in this helix may play important roles in stabilizing the apo-eIF4E form as well as making critical contacts with eIF4G.
Structural rearrangements associated with cap binding: locking a hinge
In order to compare the cap bound and apo structures in solution, we analyzed chemical shift perturbations and dynamics in the apo and cap bound forms of eIF4E (Figures 4 and 5). 1H and 15N chemical shift perturbation arise from alterations in the local chemical environment. This can be due to conformational rearrangements and/or to proximity to the ligand. In contrast, 13Cα chemical shift perturbations result from alterations in the backbone structure usually implying differences in secondary structure. A summary of cap binding induced chemical shift perturbations is shown in Figure 5. The major 1H and 15N chemical shifts perturbations between apo and cap bound forms of eIF4E involved residues known to bind the cap including the main chain amide and the indoles of W56 and W102. In the apo structure, W56 is present on a flexible loop (residues 49–60). Overlaying just this part of the apo-structure reveals that loop is relatively well-defined structurally and is likely moving as a hinge relative to the rest of apo-eIF4E. Consistently, there are only small changes in the αC chemical shifts for this loop upon cap binding, indicating that the W56 loop does not undergo substantial changes in secondary structure in the apo and cap bound forms of eIF4E. In contrast, W102 goes through substantial alterations in its backbone conformation as can be seen by observing the large differences in αC chemical shifts for W102 and nearby residues including E103 (Figure 5C). In fact upon cap binding, this region undergoes the largest secondary structural changes in the protein. Consistent with chemical shift perturbation studies, we observe NOEs between the cap and eIF4E in the cap-bound complex (Supplementary Figure 6). In summary, upon cap binding, eIF4E undergoes two distinct motions: (1) locking the W56 hinge and (2) rotating W102 and nearby residues into the cap binding site (Figure 4B). The order of these events, or if they are concerted, has yet to be determined. The 15N hNOE data (Figure 4A) and R1 data confirm the presence of mobility on the ns–ps timescales for the loops containing W56 and W102 residues that become abrogated upon cap binding (especially for the W56 containing loop). T1 values that, together with hNOEs, are sensitive to high-frequency motions also show similar tendencies (Supplementary Figure 5). While for T2, which is a function of much slower processes, this phenomenon appears less pronounced in these two loops. All together, these data confirm the reduction of the rapid large amplitude motions around W56 upon cap binding.
Figure 5.
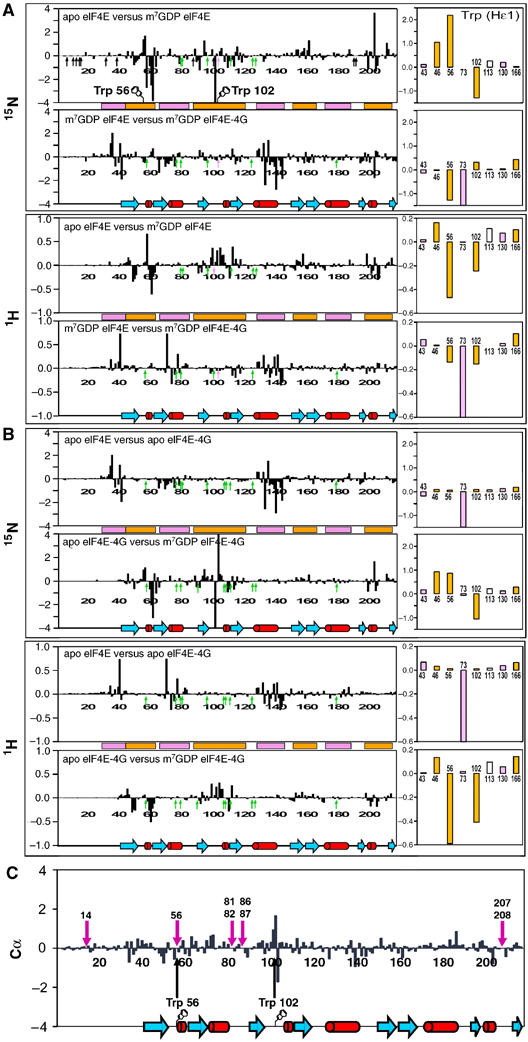
Chemical shift differences between the apo and the ternary complex with either the cap-eIF4E (A), or the eIF4E-4G peptide (B) as binary complexes. The right panels represent the chemical shift differences for the eight Trp indoles HN. Positions indicated with arrows show proline residues (black), E103 which is not assigned in cap-eIF4E (purple) or peaks from which no data were collected due to overlap (green). Regions located on the cap (orange) and eIF4G (purple) binding sides on eIF4E are shown. (C) Differences in chemical shift index of α-protons between the apo and the cap-bound form of eIF4E. The secondary-structure elements of the apo structure are indicated below.
Crystallographic data indicate that several residues make direct contacts with the cap including E103, D90, W166, R112, R157 and K162. Like W102, the αC chemical shift data indicate that E103 also undergoes large-scale alterations in secondary structure upon cap binding, consistent with this region rotating into the cap binding site. Other residues involved in cap binding also undergo substantial changes in 1H and/or 15N chemical shift as observed in the HSQCs. For instance, residues in the S5–S6 loop around 155–160 are altered as is the indole and main chain resonances for W166 (Figure 5A and B).
Earlier crystallographic studies suggested that K159, in the S5–S6 loop, may play an important role in clamping the cap into position once S209, in the H6–S8 loop, is phosphorylated (Tomoo et al, 2002). As discussed above, K159 is in a region of eIF4E that undergoes alteration in chemical shift, and presumably alterations in conformation and/or dynamics upon cap binding. Interestingly, the C-terminal S209 region was not observed due to poor electron density in the crystal structures. In our studies, this region is clearly dynamic in the apo-eIF4E form as observed by poor structural resolution and dynamics studies (Figure 4A). This region undergoes substantial chemical shift perturbation upon cap binding and reduced motion as observed by hNOE studies. Our studies indicate that eIF4E undergoes conformational changes mainly involving the loops and short helical stretches as described above. Clearly, we demonstrate that the apo form of eIF4E is structured in the absence of the cap.
Prestructuring and eIF4E conformational rearrangements
Determining the apo-eIF4E structure puts us in a unique position to investigate the structural underpinnings of the ligand-induced effects observed for eIF4E. Two key observations are relevant: binding apo-eIF4E to eIF4G enhances its affinity for the cap (Ptushkina et al, 1999; von der Haar et al, 2004; Friedland et al, 2005). Second, binding the cap enhances its affinity for eIF4G and 4E-BP (Shen et al, 2001; von der Haar et al, 2004; Tomoo et al, 2005). It has been suggested that these effects are important to translational control and further provides a model system for other cases of conformational regulation of eIF4E, as discussed in the introduction. We reasoned that eIF4G or the m7G cap pre-structure eIF4E into conformational states in between the apo form and cap bound or eIF4G bound structures, respectively. This is despite the fact that the cap and eIF4G binding sites are on opposite faces of eIF4E (Figure 1). In order to investigate the structural underpinnings of these biophysical observations, we monitored 1H and 15N chemical shift changes between apo-eIF4E, cap-eIF4E, eIF4E–eIF4G peptide and the cap-eIF4E–eIF4G ternary complex (Figure 6). Studies were carried out with the peptide rather than the 100 residue fragment corresponding to yeast eIF4G because of problems finding suitable solution conditions using the human version of this larger fragment.
Figure 6.
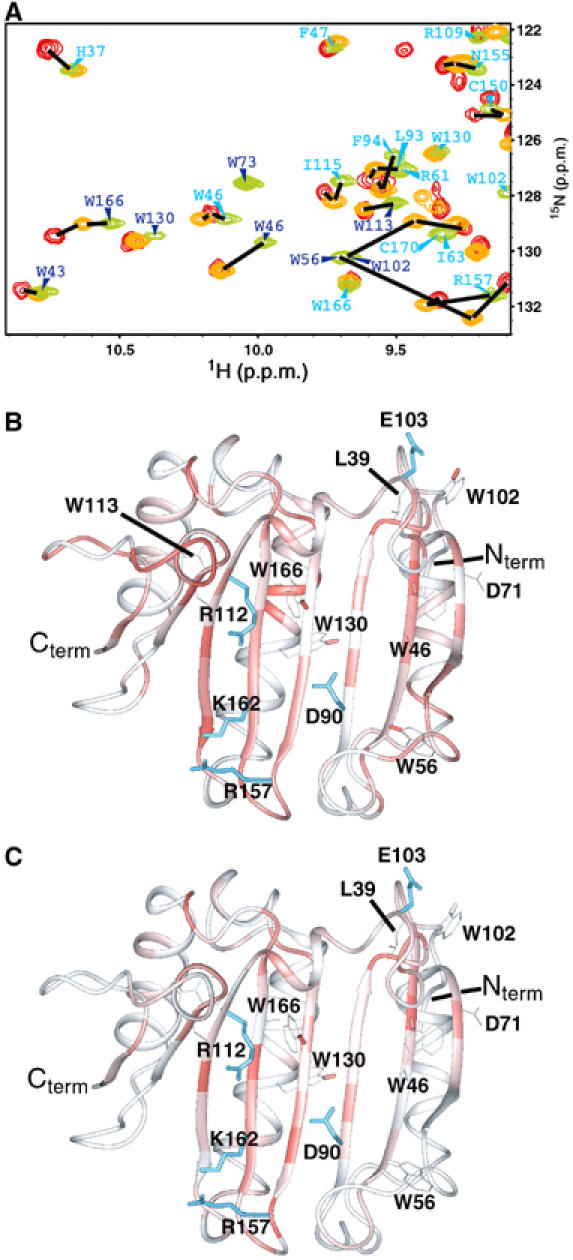
Prestructuring during cap and eIF4G binding. (A) Comparison of a section of the HSQC spectra of the apo-eIF4E (green), binary cap-eIF4E (orange) and ternary cap-eIF4E–eIF4G peptide (red). Assignments are indicated for the backbone HN (cyan) and the Trp indoles HN (blue). Chemical shifts for peaks were scored most highly (red, e.g. indole of W166 in part A) when their chemical shifts were intermediate between apo and ternary complexes upon addition of either the cap (B) or eIF4G (C).
Analyses of our apo-eIF4E structure and previously reported cap-bound eIF4E structures reveal structural differences at the dorsal surface (Figure 3F and G). Our analysis of chemical shift differences revealed that cap binding leads to widespread perturbations, including residues 71 (H2), 132 (H4) and the side chain indole of residue 130 (Figure 4), on the dorsal surface. Importantly, NOEs between eIF4E and the cap analog were limited to the cap binding site of eIF4E (Supplementary Figure 6). Consistently, our dynamic studies indicate that the changes observed in the ns–ps motions between the apo and cap forms are not restricted to the cap binding sites (Figure 4A). Significantly, regions on the dorsal surface involved in eIF4G/4E-BP binding exhibit less flexibility upon cap binding. Most notably are residues 138–141 that interact directly with eIF4G and 4E-BPs and also adjacent regions. We extended our studies to assess the extent to which cap binding leads to structural rearrangements in eIF4E intermediate between the apo form and ternary complexes. To visualize this, we determined which residues had chemical shifts in the cap bound form intermediate between the apo form and the ternary cap-eIF4E–eIF4G complex (see Figure 6A) and mapped these onto the apo-eIF4E structure (Figure 6B). We refer to this as prestructuring. This analysis clearly shows prestructuring throughout the β-sheet and also for some residues located in the long helices, notably the region comprising residues 130–134 and around residues 39 and 71, known to bind eIF4G and far removed from the cap binding site. In summary, binding of the cap results in structural rearrangements and localized modulation in dynamics in both the cap binding site and on the dorsal surface providing a mechanism for the enhanced affinity of eIF4G and 4E-BP for cap bound eIF4E relative to the apo form.
Conversely, eIF4G and 4E-BP binding enhances affinity of eIF4E for the cap (Ptushkina et al, 1999; von der Haar et al, 2004; Friedland et al, 2005). Consistently, our studies indicate that addition of eIF4G resulted in 1H and 15N chemical shift perturbations and thus structural rearrangements not only on the dorsal surface (Figure 4F) but also in the cap binding site (Figure 4E). Analysis of NOEs in the eIF4E–4G complex indicated that NOEs were limited to the dorsal surface of eIF4E, consistent with previous ternary structures (Supplementary Figure 6). We analyzed prestructuring in the presence of eIF4G as described above (Figure 6C). The β-sheet together with the side chain indole of W166 in the cap binding site were modulated. Taken together (Figures 4 and 6), our studies show there are chemical shift perturbations in S7 and H6 and in the S5–S6 loop including R157 and K159 upon eIF4G addition. Note that this region is also altered upon formation of the eIF4E-cap complex. An example of the different 1H/15N HSQC peak positions of residue R157, which directly contacts the phosphate backbone of 7-methylguanosine 5′-triphosphate (m7GTP), is shown in Figure 7A. In contrast, W56 and W102 do not move substantially indicating that the part of the site that contacts the m7G moiety is not formed in the apo-eIF4E/eIF4G peptide complex. Specifically, the eIF4G peptide alters the conformation around the region of the cap binding site that makes contacts with the phosphate group more than the region that makes contacts with the m7G moiety such as R157 and W166 but not W56 or W102. This is interesting as the m7G moiety is what allows the cap to bind specifically to eIF4E. Further, m7G binds eIF4E but with a 100-fold lower affinity than m7GTP (Niedzwiecka et al, 2002). Taken together, these observations provide physical explanations for the increased cap affinity of eIF4E–eIF4G or eIF4E–4E-BP complexes relative to eIF4E alone.
Figure 7.
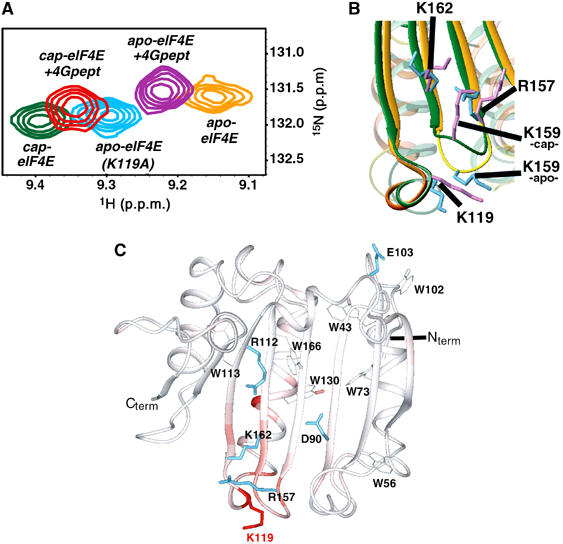
The eIF4E (K119A) mutant. (A) Superposition of 2D HSQC spectra showing residue R157 in the different complexes, together with the apo-eIF4E (K119A) mutant. (B) Structural differences between the apo- (orange) and the cap-eIF4E (green) around the residue K119 (see text for details). (C) Chemical shift perturbation of backbone 1HN and 15N resonances (color-coded) between apo-eIF4E and apo-eIF4E K119A. The deviations (in p.p.m.) were quantified with the same method as in Figure 4. K119 is shown in red.
High affinity cap binding mutant, K119A, traps eIF4E in a higher affinity state
Recent reports indicated that mutation of residues in the S4–H4 loop enhances the affinity of eIF4E ∼11 fold for the m7GpppG and three-fold for m7GTP (Spivak-Kroizman et al, 2002; Friedland et al, 2005). It was also found that the mutant binds the eIF4G peptide with nearly five-fold increased affinity relative to wild type. Furthermore, addition of the eIF4G peptide prior to the m7GTP leads to an increase in cap affinity by 10-fold, whereas addition of the same peptide only increased wild-type eIF4E's affinity for the cap by two- to three-fold. The best-studied mutation in the S4–H4 loop is K119A. As previously shown by Spivak-Kroizman et al (2002) and in agreement with our studies (see Supplementary Figure 7), this mutation does not lead to global folding transitions as monitored by circular dichroism. Further, this loop is placed far from either the cap or eIF4G binding sites. Thus, it is unclear how this mutation increases the affinity of eIF4E for either of these ligands. We reasoned that there was prestructuring in the apo form of the K119A mutant relative to apo wild-type eIF4E, which likely increases its affinity for the cap and for the eIF4G peptide. We engineered the mutation in the same construct we used to generate human eIF4E. The mutant protein was prepared as the wild type and mass spectrometry methods confirmed that the apo form of the mutant was indeed cap free (Supplementary Figure 3). NMR assignments were generated using the wild-type assignments as a starting point and 15N-edited nuclear overhauser effect spectroscopy (NOESY) was used to unambiguously assign K119 and surrounding residues. Additionally, we confirmed that K119A bound cap with a higher affinity than the wild type in our solution conditions (Supplementary Figure 2) as demonstrated by Friedland et al (2005).
To determine whether there were conformational changes in the K119A mutant relative to the wild-type protein, we compared 1H/15N chemical shifts between the two forms (Figure 7). As expected, the largest alterations in chemical shift were observed in the mutated residue and adjacent residues in the S4–H4 loop. However, our analysis revealed that there were perturbations throughout the eIF4E structure. Most interestingly were alterations in residues in the S5–S6 loop including R157 and K159, which is adjacent to the S4–H4 loop (Figure 7C), consistent with our model of prestructuring. Importantly, these same residues were altered as part of conformational changes in the apo wild-type eIF4E form upon addition of either eIF4G peptide or the m7GDP cap. Interestingly for the apo-K119 form, the chemical shift for the amide of R157 is midway between the apo and ternary complexes (Figure 7A), suggesting that the conformation of the S5–S6 loop, and its recognition of the phosphate backbone of the m7G cap, is important for increasing the affinity of the K119A mutant for the cap. Similar to the prestructuring described above, no substantial perturbations were observed for W56 and W102 indole or main chain chemical shifts indicating that alterations are localized around the phosphate binding region of the cap site rather than the m7G recognition part.
Importantly, the apo-K119A mutant also has a higher affinity for eIF4G than wild-type apo-eIF4E. We observed chemical shift perturbations on the dorsal surface of the apo-K119A mutant where the eIF4G peptide binds. Some 1H/15N chemical shift perturbations are therefore observed for the amide backbone of residues located all along H4 and for the side chain indoles W113 and W130 (Figure 7C). Consistent with alterations in both the cap binding site and the dorsal surface, these two tryptophans are in the hydrophobic core of apo-eIF4E, between the cap and eIF4G binding sites. These data indicate that there is likely a small but significant conformational rearrangement within the core of the protein leading to a more optimized surface for association with eIF4G and the cap.
Addition of the eIF4G peptide to apo-K119A increases its affinity for m7GTP by 10-fold relative to apo-K119A (Friedland et al, 2005). Even more strikingly, the K119A–eIF4G complex binds m7GTP with 15-fold higher affinity than the wild-type eIF4E–eIF4G complex. Comparison of chemical shifts between K119A–eIF4G and wild-type eIF4E–eIF4G reveals that structural differences involve the S4–H4 loop, which includes the K119A mutation and the S5–S6 loop which includes K159 and R157 (Supplementary Figure 8).
Given our above analysis of the K119A mutant, we examined the apo- and cap-bound structures to determine how this mutation could alter the conformation in the adjacent S5–S6 loop. In the apo structure, K162 and R157, residues that directly contact the phosphate backbone of the cap, are far from K119 (Figure 7B). The only residue that is near to K119 in the S5–S6 loop is K159 where these two residues appear to pack. Importantly, K119A binds m7GpppG about 10-fold more tightly than wild-type eIF4E (Supplementary Figure 2). In the cap bound form of eIF4E, these two residues do not appear to pack, with the angles between these loops altered. Mutation of K119 to alanine presumably reduces the interactions between the S5–S6 and S4–H4 loops. In this way, the S5–S6 loop can more readily undergo conformational motions linked to cap binding.
Previous biophysical studies also indicate that mutation of K159 to alanine leads to little change for the binding of m7GMP (7-methylguanosine 5′-monophosphate) or m7GDP but as the phosphate chain is lengthened, the affinity of K159A for those analogs is dramatically reduced relative to wild-type eIF4E (Niedzwiecka et al, 2002). For instance, the affinity of K159A for m7GTP relative to wild type is reduced three-fold, but for m7Gp4 (7-methylguanosine 5′-tetraphosphate) this is reduced by seven-fold. Given our results, one might predict that mutation of K159A would lead to tighter binding due to reduced interactions with the S4–H4 loop. However, it is clear that the side chain of K159 plays an important functional role in recognition of the phosphate backbone and thus cannot be mutated.
Conformational dependence of apo-eIF4E on solution conditions
The affinity of eIF4E for the cap can vary over several orders of magnitude depending on the buffer, pH, salt and temperature in which the experiments are carried out (Carberry et al, 1989; Zuberek et al, 2004). Detailed biophysical studies indicate that electrostatic steering of the positively charged cap to the negatively charged cleft in eIF4E is in large part responsible for these differences in affinity (Niedzwiecka et al, 2002). However given the above findings, we reasoned that apo-eIF4E may also undergo conformational changes, which could contribute to solution condition differences in cap affinity. In other words, the apo form may be more dependent on solution conditions and therefore more susceptible to conformational changes than cap-bound eIF4E. We analyzed chemical shift perturbations as a function of solution conditions. Interestingly, there were very large changes in chemical shift simply by substituting HEPES for phosphate, even if the rest of the conditions was unchanged. In fact, most of the residues located on the cap-binding side are clearly affected and in particular in S4, S7, and in the loops S5–S6, S7–S8 (Figure 8). Our studies also indicate that apo-eIF4E more readily aggregates in HEPES than phosphate buffer while the cap-bound form has similar NMR characteristics in both buffers. The different HSQCs and their corresponding diffusion coefficients are provided in Supplementary Figure 4. Interestingly, higher affinities for eIF4E-cap interaction were reported in HEPES versus phosphate conditions (30-fold; see Kentsis et al, 2001; Zuberek et al, 2004). The conformational alterations between these two conditions appear to lead to conformational states of eIF4E, which are more likely to aggregate and may alter other ligand binding properties of eIF4E such as specificity for cap analogs. Previous studies showed that the affinity of eIF4E for the cap is modulated upon ordered assembly into bodies (Kentsis et al, 2002). Thus, affinity for eIF4E for the cap could have a polyphasic component and thereby be enhanced due to aggregation states observed in HEPES. However, other conformational issues in this aggregated state may alter ligand specificity (Wyman and Gill, 1980). These possibilities and their biological effects will be investigated in future. Importantly, the conformation of cap-bound eIF4E was much less sensitive to changes in solution conditions. HSQCs spectra in the same conditions as studied above for apo-eIF4E (HEPES, pH 8.0) indicate that only minor structural changes occur relative to phosphate buffer (pH 7.4). Observed changes in cap-eIF4E largely relate to ionization of histidines.
Figure 8.
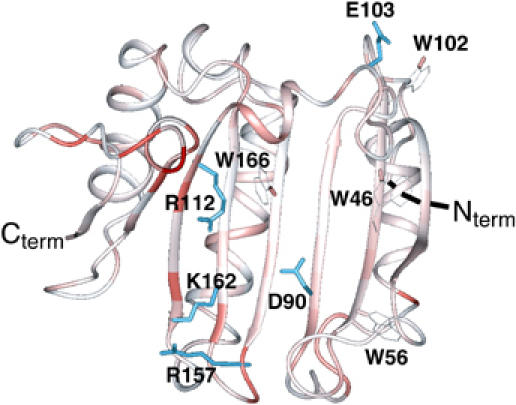
Buffer effect on apo-eIF4E. Chemical shift perturbation of backbone 1HN and 15N resonances (color-coded) between apo-eIF4E in 50 mM phosphate pH 7.4, 100 mM NaCl and 20 mM HEPES pH 7.4, 100 mM NaCl. The deviations were quantified as in Figure 4. Side chains of some residues involved in cap binding are shown in blue.
The standard buffer used in the majority of translation assays is 20 mM HEPES, pH 8.0 and thus these solution-dependent conformational changes could impact on the results observed by different groups. Further, chemical shifts are perturbed throughout the molecule upon varying salt concentrations (50–400 mM NaCl) in both HEPES and phosphate (data not shown). This is consistent with studies demonstrating a reduction in cap affinity of ∼15-fold varying KCl from 100 to 300 mM (Blachut-Okrasinska et al, 2000; Zuberek et al, 2004). We also observe a clear pH effect between 6.5 and 8.0, which appears to go beyond local effects due to the presence of titratable groups in this pH range (data not shown). In summary, although clear effects for pH and salt for cap binding are definitely electrostatic in nature, solution conditions also alter the conformation of apo-eIF4E and thereby contribute to modulating cap affinity.
Conclusions
Cap binding is governed by a conformational switch made up of a concerted hinge lock mechanism. Here, W102 and the adjacent region rotate into the cap site while the 50–60 loop moves in a concerted hinge-like fashion in order to lock W56 onto the cap. Further, our studies reveal the structural basis for ligand induced conformational rearrangements of eIF4E. These structural alterations are localized to R157 and K159 in the S5–S6 loop. Surprisingly, mutation of a single residue (K119A) in the adjacent S4–H4 loop enhances affinity of eIF4E for both the m7G cap and the eIF4G by altering conformation on both the dorsal surface and the phosphate binding region of the cap site. The K119A mutation even alters the environment of the W113 and W130, two residues in the hydrophobic core of the protein, providing a means by which to effect both the dorsal and cap binding surfaces. It is important to note that the conformational changes induced by the mutation or ligand-induced interactions are localized to specific regions of eIF4E. In support of this, addition of cap to eIF4E or mutation at K119A does not result in large differences in molar ellipticity in the CD. In this way, the chemical shift perturbations observed here likely reflect fine-tuning motions, which lead to optimized interactions rather than a global folding transition. In summary, determination of the apo-eIF4E structure has yielded new molecular insights into ligand-induced conformational regulation of eIF4E.
Materials and methods
The plasmid containing the full-length cDNA for human eIF4E (gift from Dr N Shimma, Chugai Pharmaceutical Co., Japan) was expressed in Escherichia coli BL21 (DE3) (Miura et al, 2003). The eIF4E K119A mutant was generated by PCR-directed mutagenesis and cloned into similar E. coli expression vectors as the wild-type protein and sequenced. Both proteins were purified using an m7GDP-sepharose (cap column), and eluted with 8 mM m7G in phosphate buffer, or by high salt (2 M NaCl). The protein was extensively dialyzed against phosphate buffer and applied to an anion-exchange (Mono-Q, Amersham Biosciences) column by FPLC. Backbone NMR assignments for cap-bound eIF4E, which were recently reported (Miura et al, 2003), were confirmed in our lab and served as a starting point for apo-eIF4E assignment. NMR assignment of the 1H side chain resonances was carried out using the 3D HCCH-TOCSY experiment and the 15N- and 13C-edited NOESY. About 60% of the protons were assigned. Chemical shift data are available from the BioMagResBank under Accession No. 7115. Distance restraints were obtained from 3D 15N-edited and 13C-edited NOESY spectra (100 ms mixing time) acquired at 800 MHz equipped with a cold probe using the standard sequences in Biopack. Additional longer–range NH–NH distance restraints were obtained from a 200 ms mixing time 3D 15N-edited NOESY spectrum acquired on a uniformly 15N-2H labeled eIF4E sample to help overcome the effects of spin-diffusion. Structures were calculated using the CNS protocol (Brunger et al, 1998) starting with an extended molecule. The quality of structures obtained was assessed with PROCHECK (Laskowski et al, 1993) without the unstructured residues 1–35. Coordinates were deposited in the Protein Data Bank under PDB ID code 2GPQ. Cap titration and fluorescence measurements were carried out as described previously (Kentsis et al, 2001). Briefly, 2 μM protein was incubated with the increasing concentrations of m7GDP, m7GTP or m7GpppG (0–100 μM) in 50 mM NaH2PO4 pH 7.4, 100 mM NaCl, 1 mM DTT. Fluorescence measurements were performed in a 0.3 × 0.3 cm2 fluorescence cuvette (Hellma, Forest Hills, NY), using Jasco FP6500 fluorimeter. Far-UV CD spectra were collected using Jasco-810 spectropolarimeter with 0.847 cm tandem cuvette (Hellma) at room temperature as reported previously (Kentsis et al, 2001). Finally, samples for mass spectrometry were electrosprayed directly into a PE Sciex API-III triple quadrapole mass spectrometer. Experimental masses were calculated by Hypermass and Hypermass Reconstruct programs within Biomultiview 1.0 (supplied by the instrument's manufacturer). Detailed descriptions of procedures are provided in Supplementary data.
Supplementary Material
Supplementary Figure 1
{kind=link}
Supplementary Figure 2
{kind=link}
Supplementary Figure 3
{kind=link}
Supplementary Figure 4
{kind=link}
Supplementary Figure 5
{kind=link}
Supplementary Figure 6
{kind=link}
Supplementary Figure 7
{kind=link}
Supplementary Figure 8
{kind=link}
Supplementary Methods
Acknowledgments
We particularly thank the Chugai pharmaceutical Co., Ltd for providing the full-length human eIF4E plasmid and for helpful discussions with Takaaki Miura. We would like to thank the Quebec/Eastern Canada High Field NMR facility (QANUC) and the Canadian National High Field NMR Centre (NANUC) for their assistance and use of their 800 MHz spectrometers. We are indebted to Drs Nahum Sonenberg, Curt H Hagedorn and David Rovnyak for helpful discussions. We are grateful to Dr Bernard Gibbs and Mike Aguiar (Sheldon Biotechnology Centre, Montreal) for use of the mass spectrometer and their expertise. Research was supported by NIH R01 80728. LV acknowledges support from the MCETC/FRSQ/CIHR Strategic training program in Cancer Experimental Therapeutics. KLBB holds a Canada Research Chair.
References
- Blachut-Okrasinska E, Bojarska E, Niedzwiecka A, Chlebicka L, Darzynkiewicz E, Stolarski R, St pinski J, Antosiewicz JM (2000) Stopped-flow and Brownian dynamics studies of electrostatic effects in the kinetics of binding of 7-methyl-GpppG to the protein eIF4E. Eur Biophys J 29: 487–498 [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54 (Part 5): 905–921 [DOI] [PubMed] [Google Scholar]
- Cai A, Jankowska-Anyszka M, Centers A, Chlebicka L, Stepinski J, Stolarski R, Darzynkiewicz E, Rhoads RE (1999) Quantitative assessment of mRNA cap analogues as inhibitors of in vitro translation. Biochemistry 38: 8538–8547 [DOI] [PubMed] [Google Scholar]
- Carberry SE, Rhoads RE, Goss DJ (1989) A spectroscopic study of the binding of m7GTP and m7GpppG to human protein synthesis initiation factor 4E. Biochemistry 28: 8078–8083 [DOI] [PubMed] [Google Scholar]
- Cohen N, Sharma M, Kentsis A, Perez JM, Strudwick S, Borden KL (2001) PML RING suppresses oncogenic transformation by reducing the affinity of eIF4E for mRNA. EMBO J 20: 4547–4559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culjkovic B, Topisirovic I, Skrabanek L, Ruiz-Gutierrez M, Borden KL (2005) eIF4E promotes nuclear export of cyclin D1 mRNAs via an element in the 3′UTR. J Cell Biol 169: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland DE, Wooten WN, LaVoy JE, Hagedorn CH, Goss DJ (2005) A mutant of eukaryotic protein synthesis initiation factor eIF4E(K119A) has an increased binding affinity for both m7G cap analogues and eIF4G peptides. Biochemistry 44: 4546–4550 [DOI] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Sonenberg N (1999) eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem 68: 913–963 [DOI] [PubMed] [Google Scholar]
- Graff JR, Zimmer SG (2003) Translational control and metastatic progression: enhanced activity of the mRNA cap-binding protein eIF-4E selectively enhances translation of metastasis-related mRNAs. Clin Exp Metastasis 20: 265–273 [DOI] [PubMed] [Google Scholar]
- Gross JD, Moerke NJ, von der Haar T, Lugovskoy AA, Sachs AB, McCarthy JE, Wagner G (2003) Ribosome loading onto the mRNA cap is driven by conformational coupling between eIF4G and eIF4E. Cell 115: 739–750 [DOI] [PubMed] [Google Scholar]
- Grzesiek S, Stahl SJ, Wingfield PT, Bax A (1996) The CD4 determinant for downregulation by HIV-1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR. Biochemistry 35: 10256–10261 [DOI] [PubMed] [Google Scholar]
- Huang S, Elliott RC, Liu PS, Koduri RK, Weickmann JL, Lee JH, Blair LC, Ghosh-Dastidar P, Bradshaw RA, Bryan KM, Einarson B, Kendall RL, Kolacz KH, Saito K (1987) Specificity of cotranslational amino-terminal processing of proteins in yeast. Biochemistry 26: 8242–8246 [DOI] [PubMed] [Google Scholar]
- Iborra FJ, Cook PR (2002) The interdependence of nuclear structure and function. Curr Opin Cell Biol 14: 780–785 [DOI] [PubMed] [Google Scholar]
- Iborra FJ, Jackson DA, Cook PR (2001) Coupled transcription and translation within nuclei of mammalian cells. Science 293: 1139–1142 [DOI] [PubMed] [Google Scholar]
- Kentsis A, Dwyer EC, Perez JM, Sharma M, Chen A, Pan ZQ, Borden KL (2001) The RING domains of the promyelocytic leukemia protein PML and the arenaviral protein Z repress translation by directly inhibiting translation initiation factor eIF4E. J Mol Biol 312: 609–623 [DOI] [PubMed] [Google Scholar]
- Kentsis A, Gordon RE, Borden KL (2002) Control of biochemical reactions through supramolecular RING domain self-assembly. Proc Natl Acad Sci USA 99: 15404–15409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Marcotrigiano J, Gingras AC, Sonenberg N, Burley SK (1997) Cocrystal structure of the messenger RNA 5′ cap-binding protein (eIF4E) bound to 7-methyl-GDP. Cell 89: 951–961 [DOI] [PubMed] [Google Scholar]
- Marcotrigiano J, Gingras AC, Sonenberg N, Burley SK (1999) Cap-dependent translation initiation in eukaryotes is regulated by a molecular mimic of eIF4G. Mol Cell 3: 707–716 [DOI] [PubMed] [Google Scholar]
- McCubbin WD, Edery I, Altmann M, Sonenberg N, Kay CM (1988) Circular dichroism and fluorescence studies on protein synthesis initiation factor eIF-4E and two mutant forms from the yeast Saccharomyces cerevisiae. J Biol Chem 263: 17663–17671 [PubMed] [Google Scholar]
- Miura T, Shiratori Y, Shimma N (2003) Backbone resonance assignment of human eukaryotic translation initiation factor 4E (eIF4E) in complex with 7-methylguanosine diphosphate (m7GDP) and a 17-amino acid peptide derived from human eIF4GII. J Biomol NMR 27: 279–280 [DOI] [PubMed] [Google Scholar]
- Niedzwiecka A, Darzynkiewicz E, Stolarski R (2004) Thermodynamics of mRNA 5′ cap binding by eukaryotic translation initiation factor eIF4E. Biochemistry 43: 13305–13317 [DOI] [PubMed] [Google Scholar]
- Niedzwiecka A, Darzynkiewicz E, Stolarski R (2005) Deaggregation of eIF4E induced by mRNA 5′ cap binding. Nucleosides Nucleotides Nucleic Acids 24: 507–511 [DOI] [PubMed] [Google Scholar]
- Niedzwiecka A, Marcotrigiano J, Stepinski J, Jankowska-Anyszka M, Wyslouch-Cieszynska A, Dadlez M, Gingras AC, Mak P, Darzynkiewicz E, Sonenberg N, Burley SK, Stolarski R (2002) Biophysical studies of eIF4E cap-binding protein: recognition of mRNA 5′ cap structure and synthetic fragments of eIF4G and 4E-BP1 proteins. J Mol Biol 319: 615–635 [DOI] [PubMed] [Google Scholar]
- Pellecchia M, Sebbel P, Hermanns U, Wuthrich K, Glockshuber R (1999) Pilus chaperone FimC–adhesin FimH interactions mapped by TROSY-NMR. Nat Struct Biol 6: 336–339 [DOI] [PubMed] [Google Scholar]
- Pestova TV, Kolupaeva VG, Lomakin IB, Pilipenko EV, Shatsky IN, Agol VI, Hellen CU (2001) Molecular mechanisms of translation initiation in eukaryotes. Proc Natl Acad Sci USA 98: 7029–7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptushkina M, von der Haar T, Karim MM, Hughes JM, McCarthy JE (1999) Repressor binding to a dorsal regulatory site traps human eIF4E in a high cap-affinity state. EMBO J 18: 4068–4075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau D, Kaspar R, Rosenwald I, Gehrke L, Sonenberg N (1996) Translation initiation of ornithine decarboxylase and nucleocytoplasmic transport of cyclin D1 mRNA are increased in cells overexpressing eukaryotic initiation factor 4E. Proc Natl Acad Sci USA 93: 1065–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Tomoo K, Uchiyama S, Kobayashi Y, Ishida T (2001) Structural and thermodynamic behavior of eukaryotic initiation factor 4E in supramolecular formation with 4E-binding protein 1 and mRNA cap analogue, studied by spectroscopic methods. Chem Pharm Bull (Tokyo) 49: 1299–1303 [DOI] [PubMed] [Google Scholar]
- Sonenberg N, Gingras AC (1998) The mRNA 5′ cap-binding protein eIF4E and control of cell growth. Curr Opin Cell Biol 10: 268–275 [DOI] [PubMed] [Google Scholar]
- Spivak-Kroizman T, Friedland DE, De Staercke C, Gernert KM, Goss DJ, Hagedorn CH (2002) Mutations in the S4-H2 loop of eIF4E which increase the affinity for m7GTP. FEBS Lett 516: 9–14 [DOI] [PubMed] [Google Scholar]
- Strudwick S, Borden KL (2002) The emerging roles of translation factor eIF4E in the nucleus. Differentiation 70: 10–22 [DOI] [PubMed] [Google Scholar]
- Tomoo K, Matsushita Y, Fujisaki H, Abiko F, Shen X, Taniguchi T, Miyagawa H, Kitamura K, Miura K, Ishida T (2005) Structural basis for mRNA cap-binding regulation of eukaryotic initiation factor 4E by 4E-binding protein, studied by spectroscopic, X-ray crystal structural, and molecular dynamics simulation methods. Biochim Biophys Acta 1753: 191–208 [DOI] [PubMed] [Google Scholar]
- Tomoo K, Shen X, Okabe K, Nozoe Y, Fukuhara S, Morino S, Ishida T, Taniguchi T, Hasegawa H, Terashima A, Sasaki M, Katsuya Y, Kitamura K, Miyoshi H, Ishikawa M, Miura K (2002) Crystal structures of 7-methylguanosine 5′-triphosphate (m(7)GTP)- and P(1)-7-methylguanosine-P(3)-adenosine-5′,5′-triphosphate (m(7)GpppA)-bound human full-length eukaryotic initiation factor 4E: biological importance of the C-terminal flexible region. Biochem J 362: 539–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topisirovic I, Culjkovic B, Cohen N, Perez JM, Skrabanek L, Borden KL (2003a) The proline-rich homeodomain protein, PRH, is a tissue-specific inhibitor of eIF4E-dependent cyclin D1 mRNA transport and growth. EMBO J 22: 689–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topisirovic I, Guzman ML, McConnell MJ, Licht JD, Culjkovic B, Neering SJ, Jordan CT, Borden KL (2003b) Aberrant eukaryotic translation initiation factor 4E-dependent mRNA transport impedes hematopoietic differentiation and contributes to leukemogenesis. Mol Cell Biol 23: 8992–9002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Haar T, Ball PD, McCarthy JE (2000) Stabilization of eukaryotic initiation factor 4E binding to the mRNA 5′-Cap by domains of eIF4G. J Biol Chem 275: 30551–30555 [DOI] [PubMed] [Google Scholar]
- von der Haar T, Gross JD, Wagner G, McCarthy JE (2004) The mRNA cap-binding protein eIF4E in post-transcriptional gene expression. Nat Struct Mol Biol 11: 503–511 [DOI] [PubMed] [Google Scholar]
- von der Haar T, Oku Y, Ptushkina M, Moerke N, Wagner G, Gross JD, McCarthy JE (2006) Folding transitions during assembly of the eukaryotic mRNA cap-binding complex. J Mol Biol 356: 982–992 [DOI] [PubMed] [Google Scholar]
- Wyman J, Gill SJ (1980) Ligand-linked phase changes in a biological system: applications to sickle cell hemoglobin. Proc Natl Acad Sci USA 77: 5239–5242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuberek J, Jemielity J, Jablonowska A, Stepinski J, Dadlez M, Stolarski R, Darzynkiewicz E (2004) Influence of electric charge variation at residues 209 and 159 on the interaction of eIF4E with the mRNA 5′ terminus. Biochemistry 43: 5370–5379 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8
Supplementary Methods