

Abstract
Receptor interacting protein 140 (RIP140), a ligand-dependent corepressor for nuclear receptors, can be modified by arginine methylation. Three methylated arginine residues, at Arg-240, Arg-650, and Arg-948, were identified by mass spectrometric analysis. Site-directed mutagenesis studies demonstrated the functionality of these arginine residues. The biological activity of RIP140 was suppressed by protein arginine methyltransferase 1 (PRMT1) due to RIP140 methylation, which reduced the recruitment of histone deacetylases to RIP140 and facilitated its nuclear export by enhancing interaction with exportin 1. A constitutive negative (Arg/Ala) mutant of RIP140 was resistant to the effect of PRMT1, and a constitutive positive (Arg/Phe) mutation mimicked the effect of arginine methylation. The biological activities of the wild type and the mutant proteins were examined in RIP140-null MEF cells. This study uncovered a novel means to inactivate, or suppress, RIP140, and demonstrated protein arginine methylation as a critical type of modification for corepressor.
Keywords: CRM1, HDACs, nuclear export, protein arginine methylation, RIP140
Introduction
Post-translational modification is a hallmark of proteins involved in signal transduction, allowing the proteins to rapidly respond to extracellular signals by triggering a cascade of cellular responses. As early as 1967, arginine was first shown to be modified by methylation (Paik and Kim, 1967), but the biological consequence of most arginine methylation events remains unclear (reviewed by Bedford and Richard, 2005). Currently, eight protein-arginine methyltransferases (PRMTs) are known (see review by Bedford and Richard, 2005; Lee et al, 2005a). However, the exact roles for most of the PRMTs in physiology and gene regulation are not known. In genetic studies, knockout of PRMT 1 results in embryonic lethality, and CARM1 (PRMT4)-deficient mice usually die at later stages of embryogenesis or in the perinatal period (Pawlak et al, 2000; Yadav et al, 2003).
PRMTs have been reported to act directly as coactivators for nuclear receptors (Qi et al, 2002), transcription factors such as p53 (An et al, 2004), YY1 (Rezai-Zadeh et al, 2003), NF-κB (Covic et al, 2004). Further, they can activate coactivators including PGC-1α (Teyssier et al, 2005) and CBP/p300 (Xu et al, 2001; Chevillard-Briet et al, 2002; Lee et al, 2005b) by protein arginine methylation. However, it is not known whether any corepressor could be modified by protein arginine methylation, and if so, what would be the effect of methylation.
Receptor interacting protein 140 (RIP140) is a corepressor for most, if not all, nuclear receptors (Cavailles et al, 1995; L'Horset et al, 1996; Wei et al, 2000, 2001). RIP140-null mice showed defects in female reproduction (White et al, 2000), and fat accumulation and energy expenditure in adipose tissues and adipocytes (Leonardsson et al, 2004; Christian et al, 2005; Powelka et al, 2006). Recently, we have reported that RIP140 is extensively modified by post-translational modification including phosphorylation (Huq et al, 2005; Gupta et al, 2005) and acetylation (Huq and Wei, 2005), both of which positively regulate the biological activity of RIP140. However, it is unclear how its biological activity is negatively regulated, or suppressed, in order to achieve a homeostatic status in gene regulation involving RIP140.
In this study, we uncovered protein arginine methylation as an important means to inactivate RIP140 in vivo. We identified arginine methylation on endogenous RIP140, and determined the sites of methylation by liquid chromatography-tandem mass spectrometry (LC-ESI-MS/MS). Site-directed mutagenesis studies confirmed the functionality of these methylatable arginine residues in vivo. Methylated RIP140 was inactivated due to its reduced ability to recruit histone deacetylases (HDACs) and facilitated nuclear export through enhanced interaction with exportin1 (CRM1).
Results
Protein arginine methylation of RIP140 in vivo and in vitro
In vivo metabolic labeling experiments using 3H-SAM (tritiated S-adenosyl methionine) as methyl donors were conducted in differentiated 3T3-L1 cells, which expressed a high level of RIP140. The cell lysate was immunoprecipitated with anti-RIP140 antibody and resolved on SDS–PAGE (Figure 1A). The endogenous RIP140 was methylated (lane 2), which was completely blocked by adenosine-2,3-dialdehyde, a global methyltransferase inhibitor (MTI) (lane 3), supporting the specificity of RIP140 methylation in 3T3-L1 cells. We also determined the methylation status of ectopically expressed flag-tagged RIP140 in COS-1 cells by metabolic labeling experiments (Figure 1B, top panel). Similarly, the level of methylation on the ectopically expressed RIP140 was significantly reduced by MTI. To confirm methylation at arginine residues, immunoprecipitated flag-RIP140 was monitored using a specific antibody against methylated arginine (Figure 1B, bottom panel). RIP140 was efficiently detected by this antibody, which was also dramatically reduced by MTI, confirming the arginine methylation on RIP140.
Figure 1.
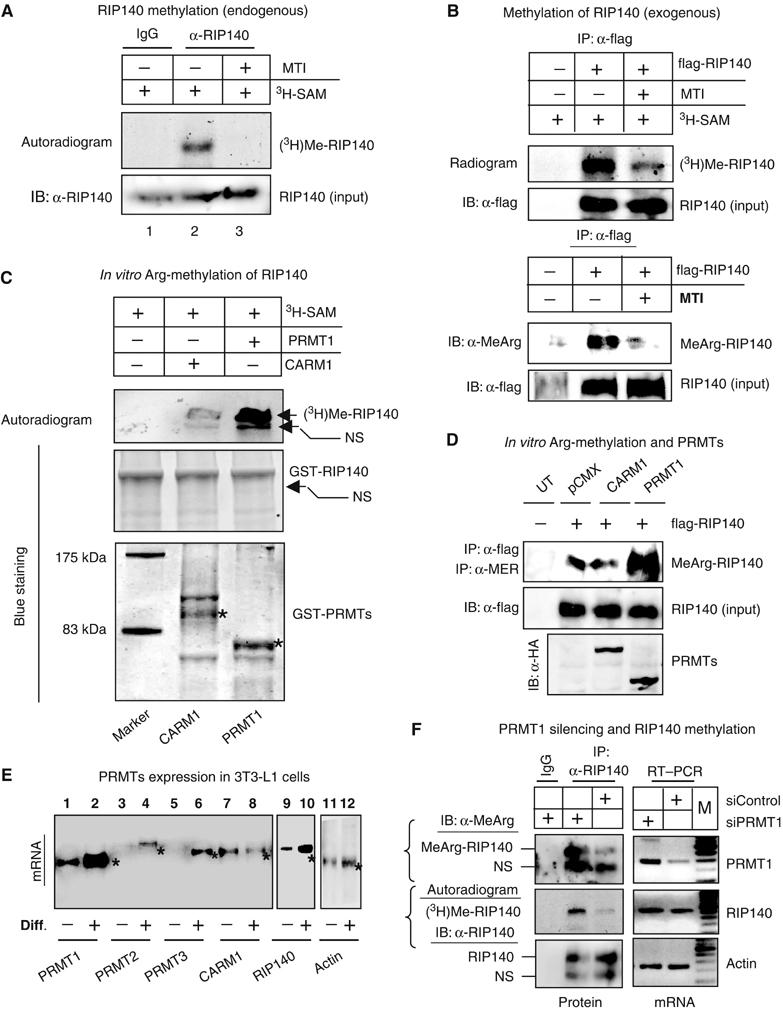
In vitro and in vivo methylation of RIP140. (A) Endogenous RIP140 is methylated in differentiated 3T3-L1 cells. MTI (adenosine 2,3-dialdehyde, 10 μM) blocked the incorporation of (3H)methyl group to RIP140. (B) In vivo global methylation (top panel) and arginine methylation (bottom panel) status of ectopically expressed flag-RIP140 in COS-1 in the presence or absence of MTI. (C) Protein arginine methyltransferase, PRMT1, but not CARM1, could methylate RIP140. (D) In vivo hypermethylation was induced by PRMT1, but not by CARM1. All experiments were conducted at least twice. (E) Differential expression patterns of PRMTs in pre- (minus sign) and post-differentiated (plus sign) 3T3-L1 cells determined by RT–PCR. * indicates the specific bands for PRMTs. (F) PRMT1 silencing demonstrated 60–70% knockdown as compared to the control (control siRNA) in differentiated 3T3-L1 cells (right panel, top). The level of protein arginine methylation of endogenous RIP140 was detected with IP (anti-RIP140 antibody) followed by Western blot (anti-methylated arginine antibody) (left panel, top), as well as 3H-SAM incorporation followed by autoradiography of specific immunoprecipitated proteins (left panel, middle). NS, indicates nonspecific bands.
Among the known PRMTs, PRMT1 and CARM1 have been widely studied and are known to be expressed abundantly in different tissues. Therefore, we further conducted in vitro protein arginine methylation on purified RIP140 from bacterial expression using PRMT1 or CARM1 enzymes in the presence of 3H-SAM (Figure 1C). RIP140 was significantly methylated at arginine residues by PRMT1. Interestingly, CARM1 was not an effective enzyme for arginine methylation on RIP140 (Figure 1C). To confirm the enzyme specificity in vivo, we examined arginine methylation status of RIP140 in cells expressing either PRMT1 or CARM1 (Figure 1D). The result was consistent with the in vitro finding that PRMT1 was more efficient for protein arginine methylation of RIP140. Together, the in vitro and in vivo data strongly supported the notion that the endogenous RIP140 could be modified by protein arginine methylation, and it was preferentially methylated by PRMT1, but not CARM1.
Given the role of RIP140 in adipose tissues, we then examined the methylation of RIP140 in differentiated 3T3-L1 adipocytes. The analysis of mRNA from pre- and postdifferentiated 3T3-L1 cells revealed that PRMT1 was more abundantly expressed in 3T3-L1 cells, as compared to other known PRMTs, including PRMT2, PRMT3, and PRMT4 (CARM1) (Figure 1E). It was interesting that the expression of PRMT1 robustly increased in post-differentiated 3T3-L1 cells where RIP140 was also elevated to a much higher level (Figure 1E). Therefore, it is likely that PRMT1 could be the major PRMT that was responsible for arginine methylation of endogenous RIP140 in post-differentiated 3T3-L1 cells (Figure 1A). To validate a direct, causal role for PRMT1 in arginine methylation of endogenous RIP140 in these cells, we conducted RNA silencing of PRMT1 in differentiated 3T3-L1 cells (Figure 1F). The endogenous PRMT1 was knockdown for 60–70% at the mRNA level (Figure 1F, right panel, top). The status of arginine methylation of endogenous RIP140 was then monitored by measuring 3H-SAM incorporation followed by immunoprecipitation (IP) using anti-RIP140 antibody, and detected by the specific antibody against methylated arginine (Figure 1F, left panel, top), as well as autoradiography (Figure 1F, left panel, middle). The depletion of endogenous RIP140 led to a dramatic reduction in the level of arginine methylation of endogenous RIP140 (Figure 1F, left panel). These data strongly support that PRMT1 is the primary enzyme for arginine methylation of endogenous RIP140 in 3T3-L1 adipocytes.
Mapping of arginine methylation sites on RIP140
To provide direct evidence for arginine methylation on RIP140, LC-ESI-MS/MS analysis was conducted for RIP140 expressed in either Sf21 insect cells or Escherichia coli (Huq et al, 2005; Huq and Wei, 2005). The information-dependent acquisition (IDA) was used to acquire MS/MS data. The MS/MS data from IDA experiments were subjected to MASCOT search (http://www.matrixscience.com). The result revealed several methylated tryptic peptides from RIP140 expressed in insect cells, but not that expressed in E. coli. The MS/MS data of the methylated peptides were analyzed manually in order to identify the methylated sites of RIP140. Finally, three mono-methylated arginine residues, at Arg-240, Arg-650 and Arg-948, were confirmed from the analysis of MS/MS data (Supplementary Figure S1).
Effects of arginine methylation on the interaction of RIP140 with HDACs
RIP140 is known to exert its corepressive activity by directly recruiting HDACs through its N-terminal domain (Wei et al, 2001; Hu et al, 2004) or by interaction with another corepressor CtBP through its central domain (Vo et al, 2001; Christian et al, 2004). We tested the interaction between RIP140 and HDAC3 in cells under an enhanced arginine methylation condition using mammalian two-hybrid interaction assay (Figure 2A). It appeared that in the presence of PRMT1, but not CARM1, interaction between RIP140 and HDAC3 was abrogated. This was further supported by the results of trans-repressive studies (Supplementary Figure 2A). Trans-repression by Gal4-RIP140 was significantly enhanced by exogenously expressed HDACs, but this was abrogated by the expression of PRMT1. To test whether the effect was mediated by arginine methylation of RIP140, site-specific mutation on the methylatable arginine residues was conducted. Since a single methylated arginine, Arg-240, is located in the N-terminal domain (1–495 aa) that directly interacts with HDACs, we started with R240A mutation in the context of the N-terminal domain. As expected, the wild-type N-terminal domain interacted with HDAC3, and the interaction was suppressed by PRMT1, but not by CARM1 (Figure 2B, wild type). Mutation of Arg-240 into Ala retained the ability to interact with HDAC, which, in contrast to the wild type, was not affected by PRMTs (Figure 2B, R240A). In fact, the mutant R240A N-terminal domain was resistant to the suppressive effects of PRMT1. This confirmed a functional role of Arg-240 methylation in modulating the interaction of RIP140 with HDACs.
Figure 2.
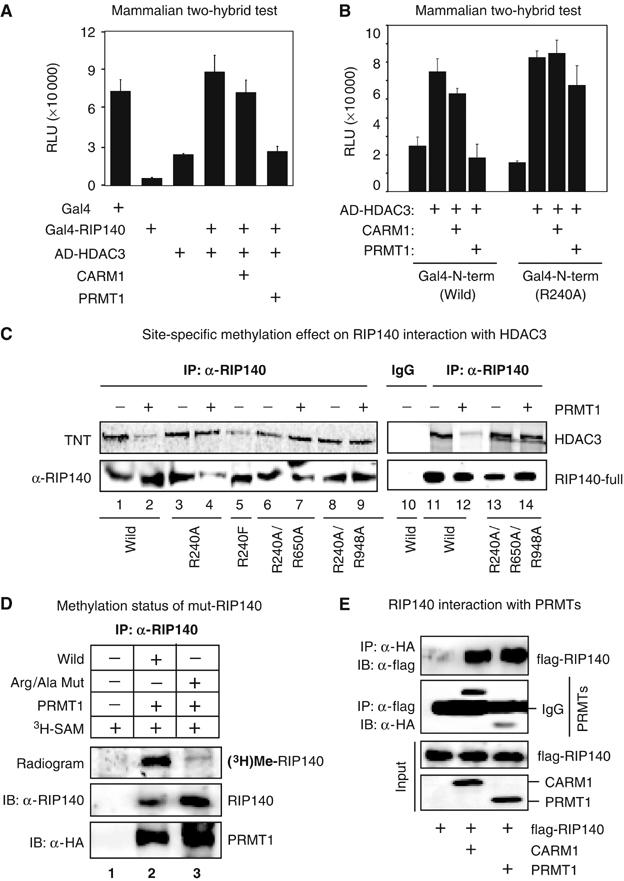
Effect of arginine methylation on the interaction of RIP140 with HDAC3. (A) In a mammalian two-hybrid test on Gal4-Luc reporter in COS-1 cells, Gal4BD-RIP140 (bait) and VP16-ADHADC3 (prey) were expressed alone, or together, in the presence or absence of PRMTs. Hypermethylation induced by PRMT1, but not CARM1, reduced the interaction between RIP140 and HDAC3. (B) In COS-1 cells, hypermethylation reduced the interaction of the wild N-terminal domain (1–495 aa) with HDAC3. The Gal4-R240A mutant of the N-terminal domain constitutively interacted with VP16AD-HDAC3, which was not affected by PRMT1. (C) Hypermethylation induced by PRMT1 in COS-1 cells reduced the direct interaction of RIP140 with in vitro translated HDAC3 as depicted by in vitro IP experiment, whereas the interactions of the Arg/Ala mutants of RIP140 were not affected. (D) Triple-Arg/Ala mutation attenuated the methylation level of RIP140, confirming the specificity of the methylated sites determined from the LC-ESI-MS/MS analysis. (E) In vivo interaction of RIP140 with PRMT1, or CARM1 in a co-IP experiment conducted in COS-1 cells.
We then constructed a single-Arg/Ala (R240A) mutant, two double-Arg/Ala (R240A/R650A and R240A/R948A) mutants, and a triple-Arg/Ala (R240A/R650A/R948A) mutant by sequential replacement of all three methylatable arginine residues, each with an alanine in the context of the full-length protein. The wild type and mutants were each co-expressed with PRMT1 in COS-1 cells and tested for the interaction with HDAC3 by IP (Figure 2C). It appeared that the interaction of wild-type RIP140 with HDAC3 was dramatically reduced in the presence of PRMT1 (lanes 1 and 2 or lanes 11 and 12). On the other hand, the interaction of HDAC3 with any of the RIP140 mutants was not significantly affected. Remarkably, the single-Arg/Ala (R240A) mutation on the N-terminus was sufficient to block the effect exerted by PRMT1 (lanes 3 and 4). The sequential incorporation of mutation, on other methylatable arginine residues, into the single-Arg/Ala (R240A) mutant failed to significantly enhance the interaction with HDAC3 (lanes 6–9 and lanes 13–14). This would support the notion that Arg-240 methylation plays a crucial role in modulating the interaction of RIP140 with HDAC3. To provide further evidence, the wild-type RIP140 full-length and the single-Arg/Ala (R240A) mutant were also tested for their ability to repress RAR-β2 reporter in the presence or absence of PRMT1 (Supplementary Figure S2B). The result was consistent with the result of HDAC3 interaction shown above (Figure 2C). Together, these data confirmed the notion that protein arginine methylation of RIP140 abrogated its interaction with HDAC3.
To evaluate whether mutation on all methylatable arginine residues could block RIP140 methylation by PRMT1, the triple-Arg/Ala (R240A/R650A/R948A) mutant and the wild-type RIP140 full-length proteins were examined using the in vivo metabolic labeling (Figure 2D) in COS-1 cells. As predicted, the level of global methylation of triple-Arg/Ala mutant of RIP140 (lane 3) was dramatically reduced as compared to the wild-type RIP140 (lane 2), but not completely blocked. Therefore, it is likely that other unidentified methylation sites are present on RIP140. To this end, we have identified several non-arginine methylation sites on RIP140 by mass spectrometric analysis (data not shown), but the biological significance of those sites needs to be further investigated.
To clarify the specificity of enzymatic activity of PRMT1 versus CARM1 to RIP140, we determined the ability of each enzyme to interact with RIP140 in a co-IP experiment (Figure 2E). Interestingly, while RIP140 is not a preferred substrate of CARM1, it can be associated with CARM1 in vivo as efficiently as with PRMT1. It is possible that CARM1 interacts with RIP140 for some other reasons, rather than using RIP140 as a substrate (see Discussion).
Effects of arginine methylation on nuclear export of RIP140
Ectopically expressed RIP140 is known to reside, primarily, in the nucleus with many fine punctate foci (Tazawa et al, 2003). To examine if arginine methylation affected its cellular localization, GFP fusions of RIP140 were generated and tested as shown in Figure 3. We first examined Arg-240 mutant in the context of the dissected N-terminal domain (1–495 aa) that is also known to be located primarily in the nucleus (Tazawa et al, 2003). In the presence of PRMT1, the wild-type N-terminal domain migrated to the cytoplasm efficiently (Figure 3A), which did not occur in the presence of CARM1. Interestingly, the mutated N-terminal domain R240A resided exclusively in the nucleus even in the presence of PRMTs. This revealed that Arg-240 methylation played a role in the transport of RIP140 from the nucleus to the cytoplasm.
Figure 3.
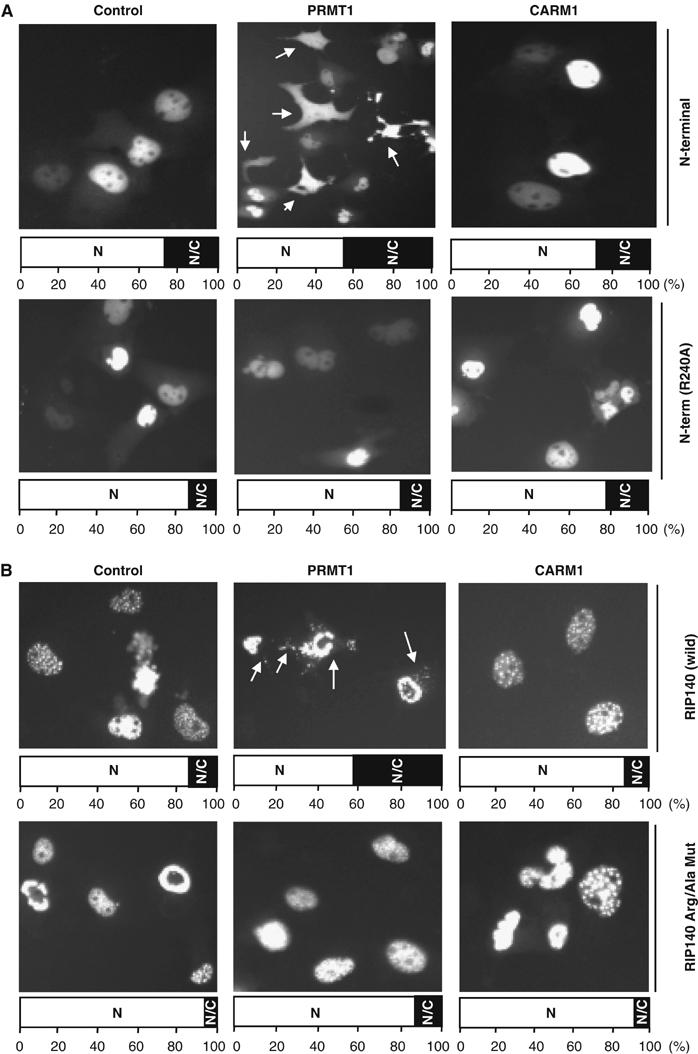
Effect of arginine methylation on subcellular localization of RIP140. (A) In COS-1 cells, the wild-type RIP140 N-terminal domain (1–495 aa) migrated to the cytoplasm in the presence of PRMT1. The R240A mutant exclusively resided in the nucleus and was not affected by PRMT1. (B) PRMT1, but not CARM1, enhanced the migration of GFP-RIP140 full-length protein from the nucleus to the cytoplasm in COS-1 cells (top panel). The triple-Arg/Ala mutant of the full-length protein was retained in the nucleus, but lost its typical pattern of fine punctate foci, which was not responsive to PRMT1. (The bar diagram at the bottom of each image represents the subcellular distribution (N, nuclear; N/C, both nuclear and cytosolic; C, cytosolic) in percentage from the differential counting of 400 GFP positive cells. All images were taken at the same time with a magnification of × 20 by a constant exposure for 10 s.)
The remaining two methylated arginine residues, Arg-650 and Arg-948, were located in the central domain of RIP140. To explore their possible roles in nuclear export, we generated two single-Arg/Ala mutants (R650A and R948A), and a double-Arg/Ala mutant (R650A/R948A) in the context of the central domain (336–1006 aa). It appeared that both Arg-650 and Arg-948 were also important for nuclear export of the central domain (data not shown). The individual methylated site in the context of full-length protein was also examined by image analysis (not shown). It appeared that all three methylatable arginine residues play some roles in the export of RIP140. However, the effect is most drastic when all three residues were mutated (the triple-Arg/Ala mutant). Therefore, we presented only the subcellular redistribution pattern of the triple-Arg/Ala mutant for a comparison to the wild-type full-length protein (Figure 3B). With additional PRMT1, the wild-type RIP140 migrated efficiently from the nucleus to the cytoplasm. As predicted, CARM1 exerted no effect. However, the triple-Arg/Ala mutant resided exclusively in the nucleus, but lacked the pattern of fine foci exhibited by the wild-type protein. Further, this mutant was completely unresponsive to PRMT1.
To examine the molecular target that preferentially facilitated the nuclear export of methylated RIP140, we determined the in vivo interaction of RIP140 with the endogenous exportin 1 (CRM1) in the presence or absence of PRMT1 (Figure 4). It appeared that PRMT1 facilitated the interaction of the wild-type RIP140, but not the triple-Arg/Ala mutant RIP140, with CRM1. This further confirmed the role of arginine methylation in modulating the interaction of RIP140 with the nuclear export machinery. We also tested its interaction with one of the key components in the nuclear import machinery, importin-β1 (karyopherin β1). It appeared that neither the wild type nor the mutant was affected by PRMT1 in terms of interaction with importin-β1.
Figure 4.
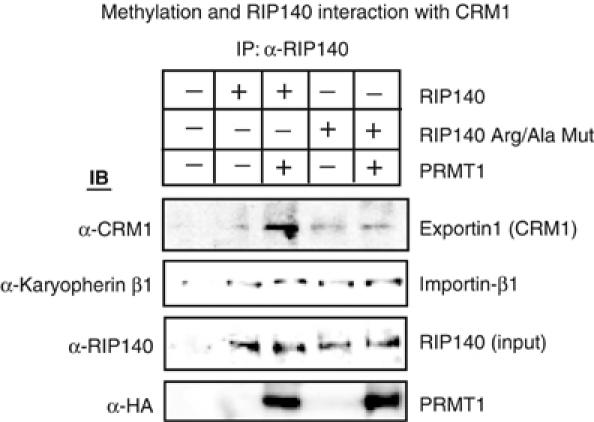
Methylation effects on RIP140 interaction with the transport machinery. In the presence of PRMT1, RIP140 preferentially interacted with CRM1 in COS-1 cells in a co-IP experiment. Interaction of the triple-Arg/Ala mutant with CRM1 was not affected by PRMT1. Interaction with importin-β1 was not affected by PRMT1. The experiment was repeated twice.
Together, these data confirmed that methylation on Arg-240, Arg-650, and Arg-948 of RIP140 enhanced its nuclear export, at least in part, through its increased interaction with the export machinery that contained exportin 1 (CRM1).
Effect of arginine methylation on the repressive activity of RIP140
To determine whether the global methylation status ultimately affects the biological activity of RIP140, we first utilized a Gal4-RIP140 system to determine the effect of methylation by employing the methylase inhibitor. As shown in Figure 5A, the trans-repressive activity of RIP140 was potentiated under a hypomethylated condition. The effects of PRMT1 and CARM1 on the trans-repressive activity of RIP140 were then examined using this system (Figure 5B). In the presence of PRMT1, but not CARM1, the trans-repressive activity of RIP140 was abrogated. Therefore, the specificity of PRMT1 to the biological activity of RIP140 was attributed to its ability to methylate RIP140, but not to its interaction with RIP140.
Figure 5.
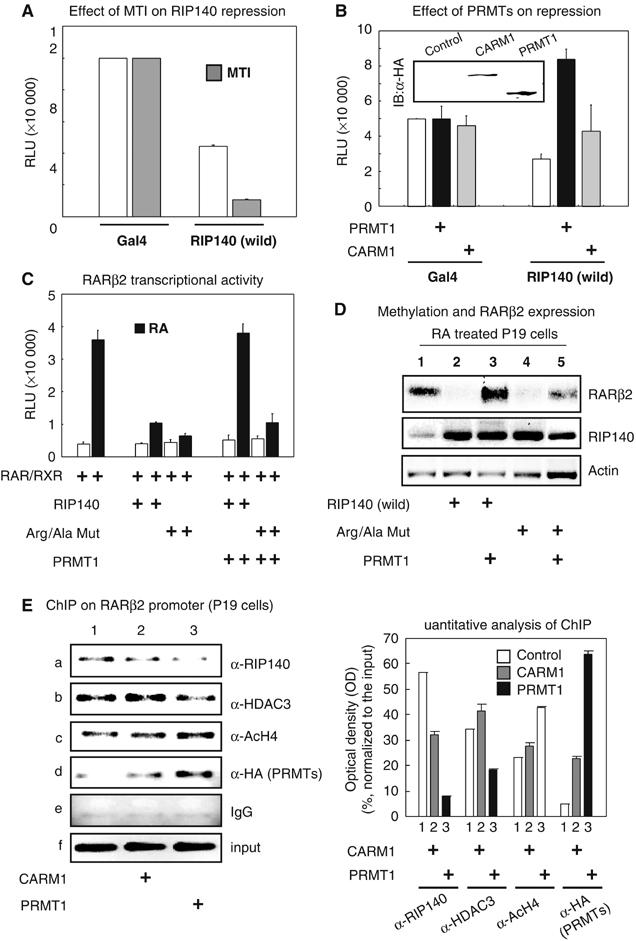
Effect of RIP140 methylation on its corepressor activities. (A) Trans-repressive activities of RIP140 full-length fused to Gal4BD in the presence of MTI in COS-1 cells. (B) Hypermethylation induced by PRMT1 abrogated the repressive activities of RIP140. (C) Hypermethylation induced by PRMT1 abolished RIP140 mediated repression on retinoic acid (RA) induced RAR-β2 reporter gene transcription, while the triple-Arg/Ala mutant was not affected by PRMT1. (D) In P19 cells, PRMT1 abolished the repressive activity of the wild-type RIP140, but not the triple-Arg/Ala mutant, on the endogenous RAR-β2 expression at mRNA level as determined by RT–PCR followed by southern blot analyses. (E) ChIP analyses of the endogenous RAR-β2 promoter in P19 cells. Quantitation of ChIP by densitometric analyses of amplified genomic DNA normalized to the input DNA was shown on the right panel. Both RIP140 (subpanel a) and HDAC3 (subpanel b) were effectively recruited to the endogenous RAR-β2 gene promoter even in the presence of CARM1 (lanes 1 and 2), but their recruitment was substantially reduced in the presence of PRMT1 (lane 3, subpanel d). As a result, the acetylation of histone H4 on the chromatin target (lane 3, subpanel c) was increased and RAR-β2 gene expression was enhanced (Figure 5D). While a significant amount of PRMT1 was recruited to the genomic target (lane 3, subpanel d) less RIP140 was associated with this region (lane 3, subpanel a), suggesting that RIP140 methylation might occur on the chromatin target by PRMT1, and that the methylated RIP140 was released from the chromatin target, which could also contribute to the reduction of its co-repressive activity.
We then determined the effects of arginine methylation on the repressive activity of RIP140 using a previously established RIP140 target model gene, RAR-β2 (Farooqui et al, 2003). As shown in Figure 5C, both the wild type and the triple-Arg/Ala mutant efficiently repressed RA activation of this reporter. However, in the presence of PRMT1, the repressive activity of the wild type, but not the triple-Arg/Ala mutant, was dramatically reduced (Figure 5C). We then examined the effect of RIP140 on the endogenous RAR-β2 gene expression in P19 cells, which normally expressed both genes (Figure 5D). Consistently, both the wild type and the mutant proteins reduced RA-activated expression of the endogenous RAR-β2 gene (lanes 2 and 4). In the presence of PRMT1, the repression of the endogenous RAR-β2 gene by the wild-type RIP140 was very effectively rescued (lanes 2 and 3), while the repression by the triple-Arg/Ala-mutant was partially affected by the expression of PRMT1 (lanes 4 and 5). It was somewhat surprising that PRMT1 was also able to partially reverse the repressive effect obtained in the presence of the triple-Arg/Ala mutant. This could be due to the depression of the endogenous RIP140 (middle panel, lane 1). In addition, the unidentified methylated residues (see methylation status of Arg/Ala mutant; Figure 2D) and/or some other roles of PRMT1 as a co-activator (Qi et al, 2002) may also contribute to this phenomenon.
To determine if arginine methylation affected the binding of RIP140, as well as its partner HDAC3, to the endogenous chromatin target, a chromatin immunoprecipitation (ChIP) assay was conducted on RAR-β2 gene promoter (Figure 5E). In normal condition, both RIP140 and HDAC3 were recruited to the endogenous RAR-β2 gene promoter, which was considerably reduced by the expression of PRMT1 (Figure 5E, lanes 1 and 3). It appeared that when PRMT1 was recruited to this genomic target (Figure 5E, subpanel d, lane 3), less RIP140 and HDAC3 could occupy this region. This suggested that arginine methylation might have also affected RIP140 interaction with HDAC3 on the chromatin target. As a result, RAR-β2 transcription was enhanced, which was supported by increased acetylated histone 4 on this region (Figure 5E, subpanel c, lanes 1 and 3).
Hydrophobicity mimics constitutive arginine methylation of RIP140
It is known that arginine residues in a protein increase the positive charge of a protein because of its guanidine group (Bedford et al, 2000). Modification of arginine by methylation makes the arginine residue more hydrophobic, which can produce steric hindrance due to the bulky methyl group. Alternatively, it can enhance van der Waals or London forces interaction. We generated a mutation that should mimic constitutive arginine methylation by replacing arginine with phenylalanine that has a similar size of hydrocarbon chain and carries a bulky hydrophobic moiety. To test the applicability of this strategy, we initially examined the subcellular distribution of such a mutant in the context of the N-terminal domain, the R240F mutant. This mutant was found to indeed exhibit a pattern of cytoplasmic distribution, similar to that triggered by arginine methylation (data not shown). In contrast, a null-mutant generated by replacing a non-methylatable Arg-251 by phenylalanine (R251F null-mutant) exclusively resided in the nucleus (not shown). This suggested that the hydrophobicity induced by phenylalanine mutation was specific to the methylatable arginine residues. We then applied this strategy to replace all three methylatable arginine residues each with a phenylalanine in the GFP fusion of the full-length protein (triple-Arg/Phe mutant). This mutant RIP140 behaved just like the hypermethylated wild-type RIP140, and was effectively exported (Figure 6A, bottom panel). This constitutive hypermethylated mutant was shown to preferentially interact with exportin 1, as compared to the wild-type RIP140 (Figure 6B). These data verified the efficiency of our strategy in producing mutant proteins mimicking constitutive arginine methylation, and supported our prediction that the arginine methylation-induced hydrophobicity enhanced the export of RIP140.
Figure 6.
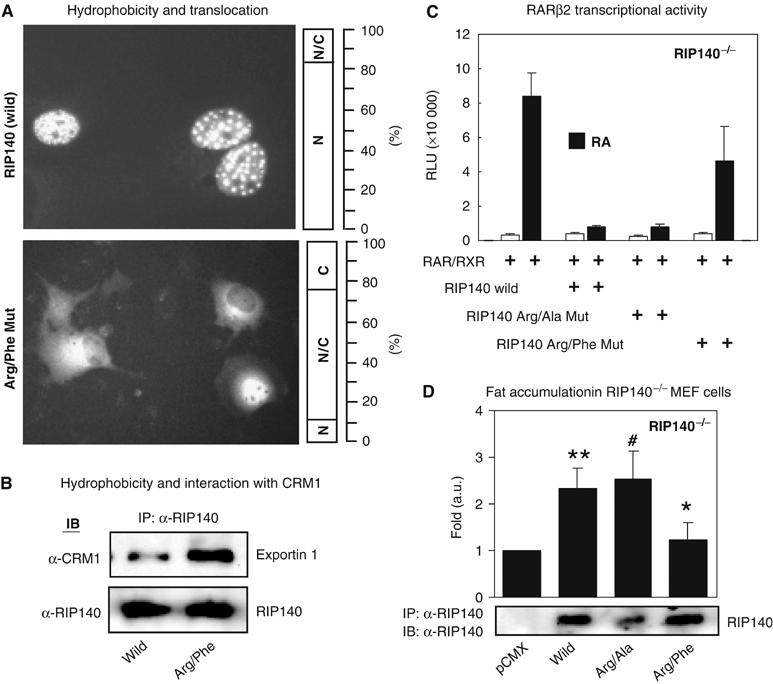
Hydrophobicity mimics arginine methylation on RIP140 and its physiological significance. (A) The hydrophobic triple-Arg/Phe mutant mimicking constitutive hypermethylation was exclusively distributed to the cytoplasm (bottom panel), as compared to the wild-type RIP140 (top panel). (The bar graph next to each image represents the sub-cellular distribution (N, nuclear; N/C, both nuclear and cytosolic; C, cytosolic) in percentage from the counting of 400 GFP positive cells.) All images were taken at the same time with a magnification of × 20 by a constant time of exposure for 10 s. (B) The triple-Arg/Phe mutant preferentially interacts with exportin 1 (CRM1) in co-IP. (C) The wild-type RIP140 and the triple-Arg/Ala mutant repressed RA induced RAR-β2 expression in a reporter assay conducted in RIP140-null MEF cells, while the triple-Arg/Phe mutant failed to effectively repress RA induction of RAR-β2 expression. (D) Gain of RIP140 function in the mouse embryonic fibroblast differentiation model. Fat accumulation in RIP140 knockout cells by re-expressing various forms of RIP140. ** versus * (P<0.05); # versus * (P<0.01).
The effect of increased hydrophobicity of RIP140 on its ability to corepress RAR-β2 gene was also examined. In order to reduce the background repression by RIP140, we used mouse embryonic fibroblasts devoid of this gene. As expected, the wild type and the triple-Arg/Ala mutant exerted a potent corepressor activity to this reporter, but the triple-Arg/Phe (phenylalanine) mutant failed to repress this reporter (Figure 6C), which was consistent with the inefficient interaction of the hydrophobic Arg/Phe mutant with HDAC3 (Figure 2C, lane 5 and Supplementary Figure S2B). These data demonstrated the biological significance of arginine methylation in modulating the activity of RIP140, which was due to, at least in part, its increased hydrophobicity.
Physiological relevance of arginine methylation of RIP140 to fat accumulation
In a physiological context, RIP140 plays an important role in the regulation of fat accumulation and energy expenditure in adipose tissues and adipocytes (Leonardsson et al, 2004; Christian et al, 2005; Powelka et al, 2006). To address the physiological significance of protein arginine methylation of RIP140 in a system where this protein is highly expressed, gain-of-function approach was employed in the RIP140-null MEF cells. The various forms of RIP140, including the wild type, the triple-Arg/Ala mutant that cannot be methylated on arginine, and the triple-Arg/Phe mutant that mimics constitutive arginine methylation, were each introduced into the RIP140-null MEF cells. The cells were then induced to differentiate into adipocytes, and their fat accumulation was monitored by determining the triglyceride content in the cells. As shown in Figure 6D, re-expression of wild-type RIP140 in this mutant cell culture enhanced its fat accumulation by 2.5-fold. The triple-Arg/Ala mutant was also able to induce a similar level of fat accumulation. Importantly, the triple-Arg/Phe mutant failed to exert a significant effect (Figure 6D). This result confirmed a physiological significance of protein arginine methylation of RIP140 in terms of its role in modulating fat accumulation in adipocytes, which was attributed to the increased hydrophobicity of the protein.
Discussion
In this study, we report a novel means to suppress or inactivate the biological activity of corepressor RIP140 by protein arginine methylation. This modification reduces its interaction with HDACs and facilitates its nuclear export through interaction with exportin 1. We further demonstrate that the hydrophobicity induced by arginine methylation significantly alters the behavior, property and activity of RIP140. These effects are demonstrated in a physiological context, such as accumulation of fat during adipocytes differentiation.
Most of previous protein arginine methylation studies have been conducted in vitro. While methylated residues of RIP140 were identified on proteins expressed in insect cells in this study, each residue has been individually tested and confirmed in the mammalian system. It is known that the consensus target sequences of PRMTs, from yeast to mammalian cells, are very close and they exert very similar catalytic activities for arginine methylation (see review by Lee et al, 2005a). Therefore, the insect system seems to be appropriate for systematic identification of protein modifications in vivo. Whether all potentially modifiable residues in mammalian cells could be identified from proteins expressed in insect cells needs further investigation.
Protein arginine methylation has been shown to potentiate the activity of coactivators, such as PGC-1α (Teyssier et al, 2005) and CBP/p300 (Xu et al, 2001; Chevillard-Briet et al, 2002). In this study, we found that the repressive activity of RIP140 was suppressed through protein arginine methylation. This is the first study to demonstrate a novel mechanism to suppress the activity of a corepressor mediated through specific protein arginine methylation. It remains to be determined whether the effects of arginine methylation of corepressors and coactivators are fundamentally different.
We generated a mutant RIP140 mimicking constitutive hypermethylation by replacing the methylatable arginine residues with bulky phenylalanine residues. The hydrophobicity was specific to methylated arginine residues in terms of the biological effects because a mutation of a nonmethylated arginine, Arg-251, to phenylalanine did not alter the behavior of RIP140 (data not shown). This could be an efficient strategy to generate constitutive, specific arginine-methylated proteins for studying the biological effects of protein arginine methylation. We showed that hypermethylated (or increased site-specific hydrophobicity) form of RIP140 interacted more effectively with the export machinery for protein translocation. It was shown that methylation of RNA binding proteins enhanced their nuclear export based on image analysis, but the molecular basis was unknown (Lukong and Richard, 2004; Smith et al, 2004). It would be interesting to test whether the interaction of RNA binding proteins with the export machinery is enhanced by protein arginine methylation due to increased hydrophobicity as demonstrated here with RIP140.
The contribution of arginine methylation as a means to suppress the repressive activity of RIP140 was demonstrated by analyzing the expression of the endogenous RAR-β2 gene in P19 cells. The expression of PRMT1 was able to reverse the inhibitory effect of the wild-type RIP140, but not a mutant version that could not be methylated. The physiological significance of arginine methylation was analyzed in adipocytes differentiation where RIP140 has been shown to regulate networks of genes involved in triglyceride synthesis, fatty acid oxidation and oxidative phosphorylation (Christian et al, 2005). The observation that re-expression of wild-type RIP140 in RIP140-null MEF cells promoted triglyceride accumulation was consistent with the reported ability of RIP140 to suppress triglyceride utilization in adipocytes. The failure of the triple-Arg/Phenylalanine mutant to promote triglyceride accumulation confirmed the physiological relevance of the state of arginine methylation. To this end, we also found that endogenous RIP140 could be modified by methylation in differentiated 3T3-L1 cells (Figure 1A). It is tempting to speculate that the expression or recruitment of PRMTs to RIP140 is regulated according to the physiological needs for adipogenesis.
Expression of PRMT1, but not CARM1, reduced the biological activity of RIP140 as a corepressor. We also tested several other PRMTs (Class I PRMTs) including PRMT2 and PRMT3. All the tested PRMTs were able to interact with RIP140 in vivo (data not shown). In addition, PRMT2 and PRMT3, but not CARM1, could also modulate the corepressive activity of RIP140 (data not shown). However, it awaits further investigation into whether other PRMTs can directly methylate RIP140 in vitro. It is clear that although CARM1 strongly interacted with RIP140 in vivo, it could not use RIP140 as a substrate for arginine methylation. It has been reported that both CARM1 and PRMT1 could methylate histones and other proteins (see review by Bedford and Richard, 2005; Lee et al, 2005a). However, recent studies of PGC-1α (Teyssier et al, 2005) and CBP/p300 (Xu et al, 2001; Chevillard-Briet et al, 2002) methylation demonstrated that these enzymes have different substrate preferences. Therefore, it is also important to examine how these PRMTs differentially recognize RIP140 as a substrate, and what is the substrate of CARM1 when it is associated with RIP140.
Materials and methods
Plasmids
The following plasmids were described previously: the Gal4BD and GFP fusions of RIP140 full-length and its N-terminal (1–495 aa) domains, GST-RIP140 and his6-RIP140 (Huq et al, 2005; Huq and Wei, 2005), flag-RIP140 and VP16-ADHDAC3 (Gupta et al, 2005), Gal4-tk-Luc and RAR-β2 reporter plasmids (Lee et al, 1998; Farooqui et al, 2003). GST and HA-tagged PRMTs were obtained from Michael R Stallcup (University of Southern California, Los Angeles, CA, USA). Site-directed mutagenesis at Arg-240, Arg-251, Arg-650, and Arg-948 was conducted using QuickChange™ mutagenesis kits (Stratagene). Primer sequences are available upon request.
Expression, purification of RIP140, and mass spectrometry
His6-RIP140 was expressed in Sf21 insect cells and GST-RIP140 was expressed in E. coli (BL21), and purified as described (Huq et al, 2005). Detailed experimental procedures for mass spectrometric analysis by LC-ESI-MS/MS were as described (Huq et al, 2005).
Cell culture, transfection, and reporter assays
COS-1, HEK293, 3T3-L1, and RIP140-nul MEF cells were cultured in DMEM supplemented with 10% FBS. P19 cells were maintained in MEM supplemented with 7.5% CS and 2.5% FBS. Trans-repression was examined in transient transfection experiments using Lipofectamine™-2000 (Invitrogen). Mammalian two-hybrid interaction tests were conducted in COS-1 cells on a Gal4 reporter in using Gal4BD-RIP140 as bait and VP16AD-HDAC3 as prey in the presence or absence of PRMT1. All reporter assays were carried out at least three times and data were presented as mean±s.d.
In vitro and in vivo methylation
In vitro methylation on purified GST-RIP140 protein using PRMTs was conducted using 3H-SAM as described (Teyssier et al, 2005). In vivo methylation on endogenous RIP140 in differentiated 3T3-L1 cells and ectopically expressed RIP140 protein in COS-1 cells was conducted by metabolic labeling using 3H-SAM (25 μCi/ml). Briefly, the confluent cells were washed with methionine-free DMEM (Gibco) twice and 3H-SAM (Sigma) was added directly to the medium and incubated for 4 h in presence or absence of MTI (10 μM). The cells were washed twice with PBS and harvested. The cell lysates were prepared in Co-IP buffer by freeze–thaw cycles for IP experiment.
IP, Western blot, and ChIP experiments
IP was conducted in a Co-IP buffer (50 mM Tris, 150 mM NaCl, 20% glycerol, 0.1% NP-40, pH 8.0). Anti-HA (Santa Cruz Biotechnology, Inc.) antibody for PRMTs, or anti-flag antibody (Sigma) for RIP140 (2 μg each) was used. Anti-RIP140 antibody was generated in house. The immunocomplexes were detected on Western blots. The in vivo methylation status of RIP140 was detected using a methylated arginine-specific antibody (Abcam Ltd). Interaction with endogenous exportin1 or importin-β1 was detected using anti-CRM1 (Abcam Ltd) or anti-karyopherin β1 (importin-β1) (Santa Cruz) antibodies. The expression of PRMTs was monitored on Western blot using anti-HA antibody. ChIP analyses for RIP140 and HDAC3 on endogenous RAR-β2 promoter were conducted in P19 cells in the absence or presence of PRMTs (PRMT1 and CARM1) using specific antibodies against RIP140, HDAC3 (Santa Cruz Biotechnology), and aetylated histone 4 (Santa Cruz), as well as anti-HA for HA-tagged PRMTs. Immunoprecipitated DNA was analyzed with PCR amplification of the proximal promoter containing the retinoic acid response element using the specific primers (Hu et al, 2004) flanking a region of 100 bp in size.
RT–PCR, Southern blot, and microscopy
Total mRNA was isolated using TRIzol® (Invitrogen) and RT was conducted using Superscript™ (Invitrogen) reverse transcriptase. The expression of RAR-β2 mRNA was monitored by RT–PCR followed by Southern blot analysis using 32P-labeled RAR-β2 probes (Hu et al, 2004). The image of subcellular distribution of RIP140 was captured under an inverted fluorescence microscope with a × 20 magnification (see Supplementary data).
PRMT1 silencing, differentiation, and fat accumulation in adipocytes
The RIP140-null MEF cells were generated (Christian et al, 2005). RIP140-null MEF cells and 3T3-L1 cells were differentiated to adipocytes by a differentiation mixture containing insulin (170 nM), IBMX (250 μM), thyroid hormone (2 nM), and dexamethasone (2 μM). Silencing of endogenous PRMT1 in 3T3-L1 cells was conducted using siGENOME SMART pool siRNA for PRMT1 (Dharmacon, Catalogue No. M049497-00). As a control, ON-TARGETplus siCONTROL Non-targeting siRNA (Dharmacon, Catalogue No. D-001810-01) was used. siRNAs (100 nM) (for 3T3-L1 cells) and RIP140 plasmids (for RIP140-null MEF cells) were introduced using Lipofectamine-2000 (Invitrogen). At 24-h post-transfection, the cells were subjected to the differentiation cocktail treatment. At 48-h post-differentiation, cells were harvested for the analyses of mRNAs and proteins, and the determination of triglyceride level (TG) using the enzymatic endpoint assay kit (Thermo DMA). The TG level was normalized either to the protein concentration or to the dried weight of the total cell content. Differential expression patterns of PRMTs in pre- and postdifferentiated cells were monitored by RT–PCR using specific primers to the 3′-region of the cNDAs encoded for the variable C-terminus region of different PRMTs. Primers sequences are available in the Supplementary data.
Supplementary Material
Supplementary Information
Acknowledgments
This work was supported by NIH Grants DA11190, DA11806, DK54733, DK60521, K02-DA13926 to L-N Wei. We thank Michael R Stallcup (University of Southern California, Los Angeles, CA 90089) for generous gifts of PRMTs constructs and proteins, and Ya-Ping Lin for her help to generate antibodies against RIP140. We also thank the staffs of the Mass Spectrometry Consortium for the Life Sciences, University of Minnesota, Department of Biochemistry, Molecular Biology and Biophysics at St Paul, USA for recording the mass spectra for the protein samples.
References
- An W, Kim J, Roeder RG (2004) Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell 117: 735–748 [DOI] [PubMed] [Google Scholar]
- Bedford MT, Frankel A, Yaffe MB, Clarke S, Leder P, Richard S (2000) Arginine methylation inhibits the binding of proline-rich ligands to Src homology 3, but not WW, domains. J Biol Chem 275: 16030–16036 [DOI] [PubMed] [Google Scholar]
- Bedford MT, Richard S (2005) Arginine methylation an emerging regulator of protein function. Mol Cell 18: 263–272 [DOI] [PubMed] [Google Scholar]
- Cavailles V, Dauvois S, L'Horset F, Lopez G, Hoare S, Kushner PJ, Parker MG (1995) Nuclear factor RIP140 modulates transcriptional activation by the estrogen receptor. EMBO J 14: 3741–3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevillard-Briet M, Trouche D, Vandel L (2002) Control of CBP co-activating activity by arginine methylation. EMBO J 21: 5457–5466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian M, Kiskinis E, Debevec D, Leonardsson G, White R, Parker MG (2005) RIP140-targeted repression of gene expression in adipocytes. Mol Cell Biol 25: 9383–9391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian M, Tullet JMA, Parker MG (2004) Characterization of four autonomous repression domains in the corepressor receptor interacting protein 140. J Biol Chem 279: 15645–15651 [DOI] [PubMed] [Google Scholar]
- Covic M, Hassa PO, Saccani S, Buerki C, Meier NI, Lombardi CR, Imhof R, Bedford MT, Natoli G, Hottiger MO (2004) Arginine methyltransferase CARM1 is a promoter-specific regulator of NF-kappaB-dependent gene expression. EMBO J 24: 85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqui M, Franco PJ, Thompson J, Kagechika H, Chandraratna RA, Banaszak L, Wei LN (2003) Effects of retinoid ligands on RIP140: molecular interaction with retinoid receptors and biological activity. Biochemistry 42: 971–979 [DOI] [PubMed] [Google Scholar]
- Gupta P, Huq MD, Khan SA, Tsai NP, Wei LN (2005) Regulation of co-repressive activity of and HDAC recruitment to RIP140 by site-specific phosphorylation. Mol Cell Proteomics 4: 1776–1784 [DOI] [PubMed] [Google Scholar]
- Hu X, Chen Y, Farooqui M, Thomas MC, Chiang CM, Wei LN (2004) Suppressive effect of receptor-interacting protein 140 on coregulator binding to retinoic acid receptor complexes, histone-modifying enzyme activity, and gene activation. J Biol Chem 279: 319–325 [DOI] [PubMed] [Google Scholar]
- Huq MD, Khan SA, Park SW, Wei LN (2005) Mapping of phosphorylation sites of nuclear corepressor receptor interacting protein 140 by liquid chromatography-tandem mass spectroscopy. Proteomics 5: 2157–2166 [DOI] [PubMed] [Google Scholar]
- Huq MD, Wei LN (2005) Post-translational modification of nuclear co-repressor receptor-interacting protein 140 by acetylation. Mol Cell Proteomics 4: 975–983 [DOI] [PubMed] [Google Scholar]
- L'Horset F, Dauvois S, Heery DM, Cavailles V, Parker MG (1996) RIP-140 interacts with multiple nuclear receptors by means of two distinct sites. Mol Cell Biol 16: 6029–6036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Chinpaisal C, Wei LN (1998) Cloning and characterization of mouse RIP140, a corepressor for nuclear orphan receptor TR2. Mol Cell Biol 18: 6745–6755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DY, Teyssier C, Strahl BD, Stallcup MR (2005a) Role of protein methylation in regulation of transcription. Endocr Rev 26: 147–170 [DOI] [PubMed] [Google Scholar]
- Lee YH, Coonrod SA, Kraus WL, Jelinek MA, Stallcup MR (2005b) Regulation of coactivator complex assembly and function by protein arginine methylation and demethylimination. Proc Natl Acad Sci USA 102: 3611–3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardsson G, Steel JH, Christian M, Pocock V, Milligan S, Bell J, So PW, Medina-Gomez G, Vidal-Puig A, White R, Parker MG (2004) Nuclear receptor corepressor RIP140 regulates fat accumulation. Proc Natl Acad Sci USA 101: 8437–8442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukong KE, Richard S (2004) Arginine methylation signals mRNA export. Nat Struct Mol Biol 11: 914–915 [DOI] [PubMed] [Google Scholar]
- Paik WK, Kim S (1967) Enzymatic methylation of protein fractions from calf thymus nuclei. Biochem Biophys Res Commun 29: 14–20 [DOI] [PubMed] [Google Scholar]
- Pawlak MR, Scherer CA, Chen J, Roshon MJ, Ruley HE (2000) Arginine N-methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol Cell Biol 20: 4859–4869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powelka AM, Seth A, Virbasius JV, Kiskinis E, Nicoloro SM, Guilherme A, Tang X, Straubhaar J, Cherniack AD, Parker MG, Czech MP (2006) Suppression of oxidative metabolism and mitochondrial biogenesis by the transcriptional corepressor RIP140 in mouse adipocytes. J Clin Invest 116: 125–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi C, Chang J, Zhu Y, Yeldandi AV, Rao SM, Zhu YJ (2002) Identification of protein arginine methyltransferase 2 as a coactivator for estrogen receptor alpha. J Biol Chem 277: 28624–28630 [DOI] [PubMed] [Google Scholar]
- Rezai-Zadeh N, Zhang X, Namour F, Fejer G, Wen YD, Yao YL, Gyory I, Wright K, Seto E (2003) Targeted recruitment of a histone H4-specific methyltransferase by the transcription factor YY1. Genes Dev 17: 1019–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WA, Schurter BT, Wong-Staal F, David M (2004) Arginine methylation of RNA helicase a determines its subcellular localization. J Biol Chem 279: 22795–22798 [DOI] [PubMed] [Google Scholar]
- Tazawa H, Osman W, Shoji Y, Treuter E, Gustafsson JA, Zilliacus J (2003) Regulation of subnuclear localization is associated with a mechanism for nuclear receptor corepression by RIP140. Mol Cell Biol 12: 4187–4198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teyssier C, Ma H, Emter R, Kralli A, Stallcup MR (2005) Activation of nuclear receptor coactivator PGC-1alpha by arginine methylation. Genes Dev 19: 1466–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo N, Fjeld C, Goodman RH (2001) Acetylation of nuclear hormone receptor-interacting protein RIP140 regulates binding of the transcriptional corepressor CtBP. Mol Cell Biol 21: 6181–6188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei LN, Farooqui M, Hu X (2001) Ligand-dependent formation of retinoid receptors, receptor-interacting protein 140 (RIP140), and histone deacetylase complex is mediated by a novel receptor-interacting motif of RIP140. J Biol Chem 276: 16107–16112 [DOI] [PubMed] [Google Scholar]
- Wei LN, Hu X, Chandra D, Seto E, Farooqui M (2000) Receptor-interacting protein 140 directly recruits histone deacetylases for gene silencing. J Biol Chem 275: 40782–40787 [DOI] [PubMed] [Google Scholar]
- White R, Leonardsson G, Rosewell I, Jacobs MA, Milligan S, Parker MG (2000) The nuclear receptor co-repressor nrip1 (RIP140) is essential for female fertility. Nat Med 6: 1368–1374 [DOI] [PubMed] [Google Scholar]
- Xu W, Chen H, Du K, Asahara H, Tini M, Emerson BM, Montminy M, Evans RM (2001) A transcriptional switch mediated by cofactor methylation. Science 294: 2507–2511 [DOI] [PubMed] [Google Scholar]
- Yadav N, Lee J, Kim J, Shen J, Hu MC, Aldaz CM, Bedford MT (2003) Specific protein methylation defects and gene expression perturbations in coactivator-associated arginine methyltransferase 1-deficient mice. Proc Natl Acad Sci USA 100: 6464–6468 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information