

Abstract
Fluid and HCO3− secretion are vital functions of the pancreatic duct and other secretory epithelia. CFTR and Cl−/HCO3− exchange activity at the luminal membrane are required for these functions. The molecular identity of the Cl−/HCO3− exchangers and their relationship with CFTR in determining fluid and HCO3− secretion are not known. We show here that the Cl−/HCO3− exchanger slc26a6 controls CFTR activity and ductal fluid and HCO3− secretion. Unexpectedly, deletion of slc26a6 in mice and measurement of fluid and HCO3− secretion into sealed intralobular pancreatic ducts revealed that deletion of slc26a6 enhanced spontaneous and decreased stimulated secretion. Remarkably, inhibition of CFTR activity with CFTRinh-172, knock-down of CFTR by siRNA and measurement of CFTR current in WT and slc26a6−/− duct cells revealed that deletion of slc26a6 resulted in dis-regulation of CFTR activity by removal of tonic inhibition of CFTR by slc26a6. These findings reveal the intricate regulation of CFTR activity by slc26a6 in both the resting and stimulated states and the essential role of slc26a6 in pancreatic HCO3− secretion in vivo.
Keywords: CFTR, fluid and HCO3 − secretion, pancreatic duct, slc26a6
Introduction
Most epithelia secrete fluid containing a high concentration of HCO3−; chief among them is the pancreatic duct, which secretes a fluid containing up to 140 mM HCO3− while absorbing most of the Cl− from the pancreatic juice (Steward et al, 2005). In addition to serving as the biological buffer, HCO3− is a chaotropic anion that facilitates solubilization of macromolecules, such as digestive enzymes and mucins, in biological fluids. HCO3− also reduces the binding of pathogenic bacteria to mucins (Choi et al, 2001). The essential role of epithelial Cl− absorption and HCO3− secretion is best exemplified in cystic fibrosis (CF), in which aberrant Cl− absorption and HCO3− secretion (Wilschanski and Durie, 1998; Sokol, 2001) leads to destruction of the airway epithelia, the pancreas and the vas deferens (Wilschanski and Durie, 1998). Aberrant CFTR-dependent HCO3− secretion is likely to contribute to the development of chronic pancreatitis (Lee et al, 2003; Cohn, 2005).
The mechanism of epithelial Cl− absorption and HCO3− secretion, including by the pancreas is poorly understood (Cook et al, 1994; Kunzelmann and Mall, 2002; Melvin et al, 2005; Steward et al, 2005). A breakthrough was made with the discovery of the new family of luminal Cl−/HCO3− exchangers, the SLC26 transporters (Mount and Romero, 2004). Members of the family play an important role in many epithelial functions. Several diseases have been linked to mutations in members of the family, including Dystrophic Dysplasia (SLC26A2) (Superti-Furga et al, 1996), Congenital Chloride Diarrhea (SLC26A3) (Makela et al, 2002), Pendred's syndrome (SLC26A4) (Everett et al, 1997) and hearing loss (SLC26A5) (Liu et al, 2003). The absence of slc26a6 can lead to calcium-oxalate urolithiasis (Jiang et al, 2006).
Several members of the family show restricted expression, while others, including SLC26A6, are ubiquitous (Mount and Romero, 2004). Their role in epithelial HCO3− secretion was proposed by their ability to mediate Cl−/HCO3− exchange. In a recent work, we showed that slc26a3 and slc26a6 are electrogenic Cl−/HCO3− exchangers and that slc26a6 functions as a 1Cl−/2HCO3− exchanger (Ko et al, 2002; Shcheynikov et al, 2006). Another notable finding is the prominent mutual activation of the SLC26 transporters and CFTR, which is mediated by the CFTR R domain and the SLC26 transporters STAS domain (Ko et al, 2004), suggesting that the SLC26 transporters participate in and regulate epithelia Cl− absorption and HCO3− secretion. This possibility is further suggested by the reduced PGE2-stimulated intestinal HCO3− secretion in the slc26a6−/− mouse (Tuo et al, 2006). However, a caveat in this work is that deletion of slc26a6 had no effect on forskolin-stimulated HCO3− secretion (Tuo et al, 2006), although both forskolin and PGE2 act by increasing cellular cAMP. In the kidney and intestine, deletion of slc26a6 reduced oxalate absorption (Wang et al, 2005; Freel et al, 2006; Jiang et al, 2006). However, how slc26a6 affects and regulates HCO3− secretion and what its role is in pancreatic HCO3− secretion is not known. Also unknown is the role of slc26a6 in fluid secretion.
Epithelial Cl− absorption and HCO3− secretion is intimately regulated by CFTR (Cook et al, 1994; Wilschanski and Durie, 1998; Kunzelmann and Mall, 2002; Irokawa et al, 2004; Melvin et al, 2005; Steward et al, 2005). To understand the role of slc26a6 in pancreatic Cl− absorption and HCO3− secretion, we deleted slc26a6 from mice and measured fluid and HCO3− secretion into sealed intralobular pancreatic ducts in primary culture. Unexpectedly, deletion of slc26a6 resulted in both enhanced spontaneous and suppressed stimulated fluid and HCO3− secretion. Remarkably, this was due to removal of tonic inhibition of CFTR by slc26a6 in the resting duct and by reduced activation of CFTR by slc26a6 in stimulated ducts. These findings reveal for the first time the intricate regulation of CFTR activity by slc26a6 and the essential role of slc26a6 in pancreatic HCO3− secretion in vivo.
Results
Slc26a6 and CFTR are essential for ductal fluid secretion
The pancreatic and salivary ducts secrete a fluid containing only 20 mM Cl− and as much as 140 mM HCO3− (Cook et al, 1994; Melvin et al, 2005; Steward et al, 2005). How the ducts can generate such a fluid is not known. It is clear that CFTR plays an important role in this secretory process, as the absence of CFTR, either in CF (Wilschanski and Durie, 1998) or in CF mouse models (Clarke and Harline, 1998; Lee et al, 1999), markedly reduces Cl− absorption and HCO3− secretion. However, CFTR is unlikely to directly mediate HCO3− secretion by the WT ducts as the HCO3− permeability of CFTR is low in the presence of normal luminal Cl− (Shcheynikov et al, 2004; Wright et al, 2004), and under physiological conditions, CFTR alone cannot produce a fluid containing 140 mM HCO3− (Sohma et al, 2000; Steward et al, 2005). On the other hand, CFTR activates (Ko et al, 2002) and is activated by members of the SLC26A Cl− and HCO3− transporters, including slc26a6 (Ko et al, 2004), which is expressed at high levels in the pancreatic duct (Lohi et al, 2000). Slc26a6 is an electrogenic Cl−/HCO3− exchanger that mediates a 1Cl−/2HCO3− exchange (Xie et al, 2002; Shcheynikov et al, 2006), and is likely to be essential for generation of the low Cl− high HCO3− content of the pancreatic juice (Ko et al, 2004; Steward et al, 2005). Therefore, to reveal the role of slc26a6 in pancreatic duct fluid and HCO3− secretion, we generated slc26a6−/− mice by deletion of exons 6–9 that code for the predicted membrane spanning domains of slc26a6. Figure 1A shows the strategy used to produce the ES cells that were used to generate the slc26a6−/− mice and genotyping of WT, heterozygous and slc26a6−/− mice.
Figure 1.

Deletion of slc26a6 in mice, knock down of CFTR and images of sealed duct. (A) Shows the vector used to delete the slc26a6 gene in mice, Southern blot of ES cells used to generate the mice and PCR with DNA prepared from WT and slc26a6−/− mice with the primers marked p1, p2 and p3. The two bands in the Southern blot of the ES cells are the WT (7.7 kb) and mutant (4.3 kb) alleles. In (B), mRNA was extracted from sealed WT and slc26a6−/− ducts that were treated with scrambled (control) and three CFTR-specific dicer siRNA and was used to evaluate the level of CFTR and actin mRNA by RT–PCR. Actin mRNA was used to verify that the same amount of mRNA was used in each sample. (C) Shows the results of Q-PCR of WT ducts treated with scrabbled or CFTR-specific dicer siRNA2. The results show the mean±s.e. from three duct preparations. (D) Shows the images of sealed WT and slc26a6−/− ducts incubated in HEPES and then HCO3−-buffered media and stimulated with 30 nM secretin.
To assay the role of CFTR in fluid and HCO3− secretion in sealed pancreatic ducts in primary culture, we downregulated CFTR with dicer siRNA. The dicer siRNA approach was used to study the effect of acute removal of CFTR and avoid the potential complications that may be encountered and the long time needed to generate mice that are deficient in both the CFTR and slc26a6 genes. The effectiveness of siRNA was evaluated by RT–PCR because the small amount of material from the cultured duct is not sufficient for Western blot analysis, and for an unknown reason the sealed ducts showed very poor staining in several immunofluorescence procedures tested. Figure 1B shows that siRNA2 most effectively downregulated CFTR mRNA in ducts from WT and slc26a6−/− mice. Real-time quantitative PCR (Q-PCR) in three separate duct preparation showed that dicer siRNA2 reduced CFTR mRNA by 65±13% (Figure 1C). This dicer siRNA was used in all subsequent experiments.
Fluid secretion into the lumen of the sealed ducts results in expansion of the lumen, and measurement of the lumen volume before and after stimulation can be used to assay fluid secretion (Szalmay et al, 2001). This procedure was validated by injection of a known amount of fluorescently labeled fluid into non-secreting ducts and obtaining excellent correlation between the expected and measured change in lumen volume (Ishiguro et al, 1998). Figure 1D shows selective images of WT and slc26a6−/− sealed ducts incubated in HEPES-buffered media. After a control period of 10 min, the ducts were incubated in HCO3−-buffered media. The gain in lumen volume in response to exposure to HCO3− is considered as spontaneous secretion. Finally, the ducts were stimulated with the pancreatic secretagogue secretin. The time course of fluid secretion was measured by recording the images at 2 or 5 min intervals and the results are expressed as the ratio of volume at time t/volume at t=0 (Vt/V0). This is possible as secretion before cell stimulation was similar in WT and slc26a6−/− ducts, as indicated by their similar diameter when incubated in HEPES-buffered media. Thus, the diameter of WT ducts treated with control and dicer siRNA averaged (n⩾18) 81±7 and 78±8 μm, respectively, and the diameter of slc26a6−/− ducts treated with control and dicer siRNA averaged (n⩾15) 82±5 and 79±5 μm, respectively.
Figure 2 shows the spontaneous and stimulated fluid secretion in WT and slc26a6−/− ducts and the obligatory role of CFTR in both activities. Incubation of WT ducts in HCO3−-buffered media resulted in a small spontaneous fluid secretion (Figure 2B and C, gray circles). Stimulation of the duct with the physiological concentration of 30 nM secretin (Figure 2A, close circles and B) or with the supramaximal concentration of 10 μM forskolin (Figure 2C, close circles) markedly stimulated fluid secretion by the WT ducts. Notably, fluid secretion was nearly abolished by downregulation of CFTR with siRNA (Figure 2A, open circles and Figure 2B). A strikingly different pattern was observed with the slc26a6−/− ducts. First, deletion of slc26a6 increased spontaneous fluid secretion (Figure 2A and C, close squares) that was most apparent on exposing the ducts to HCO3−-buffered media. Second, deletion of slc26a6 reduced secretin-stimulated fluid secretion by 77±11% (Figure 2A and B, n=8) and forskolin-stimulated fluid secretion by 63±9% (Figure 2B, n=6). Notably, the spontaneous and residual stimulated fluid secretions were inhibited by knock-down of CFTR (Figure 2A and C, gray open squares).
Figure 2.
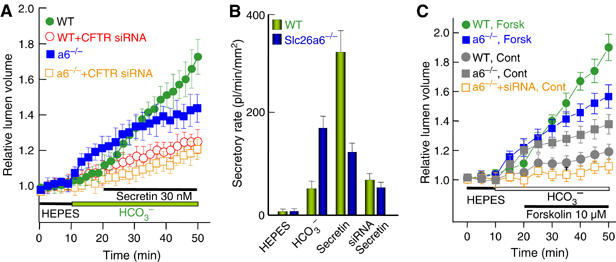
Fluid secretion by sealed intralobular pancreatic duct. In (A, C), WT (circles) and slc26a6−/− ducts (squares) were treated with scrambled (filled symbols) and siRNA2 (open symbols) and cultured for 36–48 h. The ducts were incubated in HEPES-buffered media for 10 min and then in HCO3−-buffered media for an additional 10 min before stimulation with 30 nM secretin (A) or 10 μM forskolin (C). Images were captured at a resolution of 2 (A) or 5 min/image (C). The means±s.e. of 9–12 ducts in (A) and 4–6 ducts in (C) from four (A) and three (C) mice of each line are shown. The average diameter and length of the ducts in (A) were used to calculate the secretory rates in terms of pl/min/mm2 (B). Secretory rates were calculated for the 10 min intervals in which the ducts were incubated in HEPES-, then HCO3−-buffered media and the first 10 min of stimulation with secretin for WT (green columns) and slc26a6−/− ducts (blue columns). (For colour figure see online version.)
The results in Figure 2 have several important implications. CFTR is essential for all forms of ductal fluid secretion. Similarly, slc26a6 modulates both forms of ductal fluid secretion. It suppresses the activity of the transporter mediating spontaneous secretion and it is required for stimulated ductal fluid secretion as stimulation of CFTR with secretin or forskolin in slc26a6−/− ducts did not elicit maximal fluid secretion. As CFTR is involved in both activities (Figure 2) and slc26a6 interacts with and regulates CFTR channel activity (Ko et al, 2004), it is likely that slc26a6 regulates the activity of CFTR at the resting and stimulated state, resulting in the phenotype of the slc26a6−/− ducts. In addition, slc26a6 may regulate the expression of CFTR and deletion of slc26a6 may have resulted in upregulation of another pancreatic duct Cl−/HCO3− exchange. RT–PCR analysis revealed that the pancreatic duct expresses slc26a2, slc26a3, slc26a6 and slc26a11. However, expression in oocytes showed that slc26a2 and slc26a11 are active as Cl− transporters but do not function as Cl−/HCO3− exchangers (not shown). Therefore, we tested the effect of deletion of slc26a6 on expression of CFTR and slc26a3. Figure 3A shows that expression level and localization of CFTR was the same in WT and slc26a6−/− ducts. As antibodies that reliably detect slc26a3 in the pancreatic duct are not available, we evaluated the level of slc26a3 mRNA by Q-PCR. Figure 3B shows that slc26a3 mRNA was not upregulated in the pancreatic duct. To further verify this finding, we measured slc26a3 mRNA in the submandibular (SMG) duct, another duct that secretes a high amount of HCO3− and expresses the same SLC26 transporters as the pancreatic duct. Figure 3B shows that slc26a3 mRNA was not altered in the SMG duct of slc26a6−/− mice.
Figure 3.
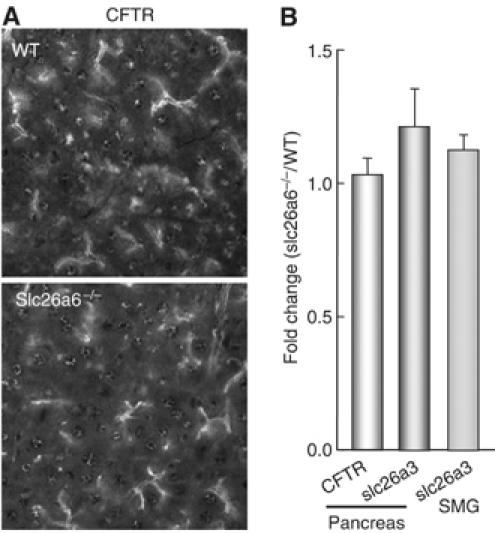
Expression of CFTR and slc26a3 mRNA in WT and slc26a6−/− ducts. CFTR expression was evaluated by immunolocalization (A). Images captured randomly from slides prepared from the pancreas of two WT and two slc26a6−/− mice were used to determine the intensity of duct staining. Images were captured under identical image capture setting and thresholded before measurement of intensity of individual ducts (at least 10 in each image). The intensities were averaged and the ratio of staining intensity in slc26a6−/−/WT ducts is recorded in the first column in (B). mRNA was extracted from the pancreatic ducts of three WT and three slc26a6−/− mice and the SMG of two of these WT and slc26a6−/− mice. The mRNA was used to determine slc26a6 mRNA level by Q-PCR and the means±s.e. or the slc26a6−/−/WT ratio is shown in (B).
Slc26a6 and CFTR determine resting and stimulated luminal HCO3− permeability of the pancreatic duct
Pancreatic ductal fluid secretion is coupled to Cl− absorption and HCO3− secretion. Both CFTR (Poulsen et al, 1994; Linsdell et al, 1997) and slc26a6 (Ko et al, 2002, 2004; Xie et al, 2002) were shown to function as HCO3− transporters. Therefore, it was of interest to measure ductal HCO3− transport and whether it is modified by slc26a6. In the first set of experiments, we measured Cl−/HCO3− exchange activity in the sealed ducts by monitoring the change in intracellular pH (pHi) in response to removal and re-addition of bath Cl−. Two experimental protocols were used to evaluate the role of CFTR in this activity: inhibition of CFTR by the selective CFTR inhibitor CFTRinh-172 (Ma et al, 2002) and downregulation of CFTR with dicer siRNA. To economize on the use of inhibitor, the effect of CFTRinh-172 was tested by preincubating the ducts in HCO3−- buffered medium, equilibrated with 5% CO2 and containing 5 μM CFTRinh-172 for 15 min. The preincubation is required for the maximal inhibition of CFTR by CFTRinh-172. Perfusion commenced after this incubation with media that did not contain inhibitor and the effect of Cl−o removal was tested within 1 min of the start of perfusion. This is possible as reversibility of inhibition of CFTR by CFTRinh-172 is slow, and after incubation with 1 μM CFTRinh-172, reversal of inhibition of CFTR required more than 10 min washout (see Ma et al, 2002). Figure 4A shows that CFTRinh-172 did not inhibit Cl−/HCO3− exchange activity in WT ducts. In fact, CFTRinh-172 consistently slightly increased the rate of Cl−/HCO3− exchange, although in six ducts from two mice the increase did not reach statistical significance. Unexpectedly, deletion of slc26a6 markedly increased basal Cl−/HCO3− exchange activity of the duct by about 72±11%. In contrast with its effect in WT ducts, CFTRinh-172 inhibited the enhanced basal Cl−/HCO3− exchange activity in slc26a6−/− ducts by about 36±6%.
Figure 4.

Properties of Cl−/HCO3− exchange activity in WT and slc26a6−/− ducts. (A) Sealed ducts prepared from WT (black and red traces and columns) and slc26a6−/− ducts (blue and green traces and columns) were incubated for 15 min in HCO3−-buffered media containing 5 μM CFTRinh-172 (red and green) or vehicles (black and blue-controls) before perfusion with Cl−-free and Cl−-containing medium for the indicated period of time. The mean±s.e. of the rates of pHi change obtained with 5–7 ducts prepared from two WT and two slc26a6−/− mice are shown in the columns. #P<0.01 relative to WT, *P<0.05 relative to untreated slc26a6−/− ducts. WT (B) and slc26a6−/− ducts (C) were treated with scrambled (black and blue traces and columns) or sense siRNA2 (red and green traces and columns) and cultured for 36–48 h. The ducts were equilibrated in HCO3−-buffered media and Cl−/HCO3− exchange activity was measured by alternately exposing the ducts to Cl−-free and Cl−-containing medium before and after stimulation with 30 nM secretin. The columns are the mean±s.e. of at least six experiments from three WT and three slc26a6−/− mice. *P<0.05 of the respective control, #P<0.01 relative to unstimulated WT, ‡better than P<0.001 relative to control slc26a6−/− ducts. (For colour figure see online version.)
The findings in Figure 4A provided the first evidence that CFTR activity was altered in the slc26a6−/− mice and that CFTR contributed to basal Cl−/HCO3− exchange activity in these ducts. To obtain independent evidence to support these findings, we tested the effect of downregulation of CFTR by siRNA on basal and stimulated Cl−/HCO3− exchange activity of WT and slc26a6−/− ducts. Figure 4B shows that stimulation of WT ducts with secretin enhanced Cl−/HCO3− exchange activity by 65±9%. Knock-down of CFTR by siRNA had no effect on basal exchange activity but largely prevented secretin-stimulated Cl−/HCO3− exchange activity. Stimulation of slc26a6−/− ducts with secretin slightly increased Cl−/HCO3− exchange activity over enhanced basal activity, but in eight experiments the stimulation did not reach statistical significance. Most notably, knock-down of CFTR almost completely inhibited Cl−/HCO3− exchange activity in slc26a6−/− ducts and stimulation of the ducts only slightly increased Cl−/HCO3− exchange activity (Figure 4C), probably due to incomplete knock-down of CFTR. Hence, siRNA was more effective in inhibiting Cl−/HCO3− exchange activity than CFTRinh-172, suggesting that Cl−/HCO3− exchange activity of the slc26a6−/− duct was mediated in part by CFTR and in part by a CFTR-dependent mechanism.
The protocol in Figure 4 assays the entire Cl−/HCO3− exchange activity of the ducts. To isolate the luminal membrane HCO3− permeability, we used the protocol developed by Szalmay et al (2001), in which the basolateral membrane HCO3− influx mechanisms (Na+-HCO3− cotransport by pNBC and Na+/H+ exchange by NHE1) are inhibited with 4,4′-diisothiocyanatodihyrostilbene-2, 2′-disulfonic acid (H2DIDS) and HOE694 to uncover the HCO3− permeability of the luminal membrane, which results in cytosolic acidification (see the model in Figure 5). Figure 5 shows that HCO3− permeability of WT ducts was nearly doubled by stimulation with secretin. Knock-down of CFTR had no effect on basal HCO3− permeability, but it reduced the HCO3− permeability of secretin-stimulated ducts to that of unstimulated ducts (Figure 5, WT group). Deletion of slc26a6 increased the basal luminal membrane HCO3− permeability, which was minimally increased in response to stimulation with secretin. Knock-down of CFTR markedly inhibited the basal and stimulated HCO3− permeability of slc26a6−/− ducts to 39±5 and 36±5% of stimulated, siRNA-treated WT ducts, respectively (Figure 5, slc26a6−/− group).
Figure 5.

Luminal HCO3− permeability of WT and slc26a6−/− ducts. WT (blue, red, gold, torques traces and/or columns and definition next to traces and columns) and slc26a6−/− ducts (purple, violet, green, gray traces and/or columns) were treated with scrambled or sense dicer siRNA2, incubated in HCO3−-buffered media and, as indicated, stimulated with 30 nM secretin. The ducts were then treated with 1 μM HOE694 and 0.2 mM H2DIDS and the reduction in pHi was measured. The rate of changes in pHi (ΔpH/min) was calculated and expressed as % of that of WT. The results are the mean±s.e. of at least six ducts from three WT and three slc26a6−/− mice. *P<0.01 relative to unstimulated WT, #P<0.01 relative to stimulated WT, ‡P<0.01 relative to siRNA-treated WT. (For colour figure see online version.)
The results in Figures 4 and 5 are consistent with those found by measurement of fluid secretion (Figure 2), further suggesting that fluid and HCO3− secretion are coupled and mediated by the same transporters. Moreover, these findings indicate that slc26a6 regulates the luminal membrane HCO3− permeability that also requires the activity of CFTR. Although knock-down of CFTR markedly reduces luminal HCO3− permeability in slc26a6−/− ducts, it is possible that CFTR regulates another luminal HCO3− transporter, such as another SLC26 transporter, in slc26a6−/− ducts. This interpretation is supported by the finding that the siRNA was more effective than CFTRinh-172 in inhibiting Cl−/HCO3− exchange by the slc26a6−/− ducts. It is also clear that slc26a6 inhibits the ability of CFTR to activate luminal HCO3− permeability. We note that although the luminal HCO3− permeability of slc26a6−/− ducts is as high as that of secretin-stimulated WT ducts, it does not support fluid secretion (compare Figure 2 and Figures 4 and 5). This indicates that the 1Cl−/2HCO3− exchange activity of slc26a6 (Ko et al, 2002, 2004; Xie et al, 2002) is required for stimulated ductal fluid secretion.
Slc26a6 regulates resting and stimulated CFTR activity
The results in Figures 2, 3, 4 and 5 clearly suggest that slc26a6 regulates the activity of CFTR in resting and stimulated ducts. In previous work, we showed that slc26a3 and slc26a6 activate stimulated CFTR (Ko et al, 2004). To examine the effect of slc26a6 on the activity of CFTR, we coexpressed CFTR and slc26a6 in Xenopus oocytes. Stimulation of cells expressing CFTR and CFTR+slc26a6 with 1 or 10 μM forskolin showed that slc26a6 increases the maximal current of CFTR. However, slc26a6 reduces the rate of activation of CFTR and the reduction in rate was more pronounced when the intensity of CFTR stimulation (Figure 6A and B) was lower. The reduction in the rate of CFTR activation was specific for slc26a6, as slc26a3, which also potently activates CFTR (Ko et al, 2004), increased both the rate and the extent of CFTR activity (Figure 6A, n=6).
Figure 6.

Regulation of CFTR by slc26a6. (A) Oocytes expressing CFTR (dark traces), CFTR and slc26a6 (green traces) or CFTR and slc26a3 (red trace) were stimulated with 1 or 10 μM forskolin, as indicated. The summary in (B) shows the mean±s.e of six experiments. (C) WT (blue traces and columns) and slc26a6−/− (green traces and columns) parotid duct cells were used to measure CFTR current stimulated with 3 (n=7) or 10 μM (n=9) forskolin. The columns show the mean±s.e of the time to peak current. (For colour figure see online version.)
To determine the effect of slc26a6 on CFTR activity in vivo, we compared the rate of activation of CFTR by forskolin in WT and slc26a6−/− parotid duct cells that express high levels of CFTR (Ko et al, 2004). Figure 6C shows that deletion of slc26a6 markedly increased the rate of activation of CFTR when the cells were stimulated with an intermediate concentration of 3 μM forskolin. Stimulation of duct cells with 10 μM forskolin resulted in similar rates of CFTR activation in WT and slc26a6−/− cells.
Discussion
The findings in Figure 6C are the first to show regulation of native CFTR activity by any transporter and the combined results show that deletion of slc26a6 resulted in dis-regulation of CFTR in the pancreatic duct. Slc26a6 inhibits the activity of CFTR in the resting state, which likely prevents unnecessary fluid and HCO3− secretion. This is evident from the increased HCO3− permeability and fluid secretion in slc26a6−/− ducts that is eliminated by knock-down of CFTR. In addition, we showed previously that the pancreatic (Ahn et al, 2001) and salivary ducts (Luo et al, 2001; Park et al, 2002) express the HCO3− salvage mechanisms NHE3 and NBC3 that are active in the resting state and are inhibited by CFTR at the stimulated state (Ahn et al, 2001; Luo et al, 2001; Park et al, 2002). On the other hand, CFTR does not seem to regulate slc26a6 activity in the resting state because CFTRinh-172 and knock-down of CFTR did not reduce Cl−/HCO3− exchange activity of the unstimulated duct (Figure 4). Slc26a6 also regulates CFTR activity in the stimulated state by reducing the rate of CFTR activation at physiological stimulus intensity, which may also serve to prevent uncontrolled activation of CFTR.
The nature of the increased Cl−/HCO3− exchange and HCO3− permeability in slc26a6−/− cells is not known with certainty. Partial inhibition of Cl−/HCO3− exchange by CFTRinh-172 indicates that part of the increased permeability is due to dis-regulation of CFTR. However, knock down of CFTR was more effective than CFTRinh-172 in reducing Cl−/HCO3− exchange activity, suggesting that an additional CFTR-dependent, but not CFTR-mediated, mechanism was active in the slc26a6−/− ducts. This conclusion is further supported by the finding that stimulation of the slc26a6−/− duct cells clearly activated CFTR Cl− current (Figure 6C) but did not appreciably increase the luminal HCO3− permeability (Figure 5B). It is possible that activation of CFTR activates another luminal HCO3− transporter like slc26a3. In our hand, activation of slc26a3 was not due to increased expression of slc26a3.
While this manuscript was under revision, another study examined the role of slc26a6 in pancreatic HCO3− and fluid secretion and reported several findings that are significantly different from the present study (Ishiguro et al, 2006). It is possible that this resulted from the difference in the animal models. There are several pieces of data that raise questions about the physiological relevance of the model used by Ishiguro et al. The most profound difference is the change in the expression of slc26a3 in the slc26a6−/− mice model used by Ishiguro et al, which examined slc26a3 expression by RT–PCR and reported a five-fold increase in slc26a3 mRNA. RT–PCR is susceptible to overestimation artefact and Ishiguro et al seemed to extract the mRNA from the entire pancreas. Using Q-PCR and extracting the mRNA from microdissected ducts, we did not detect any appreciable change in slc26a3 mRNA in two separate HCO3−-secreting ducts, the pancreatic and SMG ducts. On the other hand, we evaluated the role of CFTR by CFTRinh-172 and downregulation with siRNA and showed that CFTR mediates a significant part of HCO3− transport in the slc26a6−/− duct. Ishiguro et al did not examine the role of CFTR in their mice. In addition, Ishiguro et al saw no stimulation of Cl−/HCO3− exchange by forskolin in WT ducts, whereas we found strong stimulation (Figure 4). It is not clear how stimulation of CFTR did not increase Cl−/HCO3− exchange activity. Moreover, the Cl−/HCO3− exchange in the WT ducts used by Ishiguro et al is very low, whereas the basal fluid secretion in these ducts is quite high. The pancreatic ducts of the WT mice obtained from our line show low spontaneous fluid secretion and significant CFTR-independent Cl−/HCO3− exchange activity (Figures 2 and 4). The increased spontaneous secretion in our ducts approaches the basal spontaneous secretion in the WT ducts used by Ishiguro et al The high basal fluid secretion by the ducts used in the Ishiguro et al study may have masked the increased spontaneous secretion in slc26a6−/− ducts. It is not clear how mice strain can be so different, but these differences may explain the different results. We believe that probing the role of CFTR in the slc26a6−/− mice and the regulation of CFTR by slc26a6 in vivo, as was done in our study, is essential to reveal the interaction between transporters and its physiological significance.
Our results clearly show that slc26a6 is essential for stimulated fluid and HCO3− secretion by the duct. Hence, slc26a6−/− ducts mediate mainly spontaneous secretion. A small, stimulated secretion could be elicited only by stimulation with a high concentration of forskolin. Importantly, even when the ducts were stimulated with 10 μM forskolin that rapidly and maximally activates CFTR Cl− current (Figure 6), stimulated fluid secretion by the slc26a6−/− ducts was not maximal. This indicates that slc26a6 plays an essential role and likely mediates a significant portion of fluid and HCO3− secretion by the pancreatic duct.
The present work reveals the importance of SLC26A transporters and their interaction with CFTR in pancreatic duct fluid and HCO3− secretion. It is likely that similar regulation takes place in other CFTR and SLC26A transporters expressing epithelia that secrete HCO3−, such as the salivary glands, airways, the vas deferens and the intestine. Hence, these findings have important implications for CF and epithelial diseases owing to mutations in SLC26A transporters. On the one hand, replacing the Cl− channel function of CFTR is not likely to be sufficient to correct the pathologies of CF: restoration of the CFTR-regulated and SLC26A transporter-mediated HCO3− secretion will also likely be required to alleviate the symptoms of CF. On the other hand, although the activity and fluid and HCO3− secretion by SLC26 transporters requires CFTR (Figures 2, 3, 4 and 5), if a way can be found to activate the SLC26 transporters, in particular SLC26A6, in the absence of CFTR, the lack of fluid and HCO3− secretion in CF may be corrected, in part, to alleviate the symptoms of CF related to a lack of HCO3− secretion.
Materials and methods
The CFTR and slc26a6 clones and their cRNA were the same as those used in previous works (Ko et al, 2002; Shcheynikov et al, 2004). The HEPES-buffered bath solution for sealed pancreatic ducts and parotid cells (solution A) contained (in mM) 140 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES (pH 7.4 with NaOH) and 10 glucose. Cl−-free solutions were prepared by replacement of Cl− with gluconate. HCO3−-buffered solutions were prepared by replacing 25 mM Na+-anion with 25 mM Na+-HCO3− and reducing HEPES to 2.5 mM. HCO3−-buffered solutions were gassed with 5% CO2 and 95% O2. The osmolarity of all solutions was adjusted to 310 mOsmol with the major salt. For experiments with Xenopus oocytes, the standard HEPES-buffered medium was ND96 composed of (in mM) 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 2.5 pyruvate and 5 HEPES-Na, pH 7.5 (Shcheynikov et al, 2004). The HCO3−-buffered solution contained (in mM) 71 NaCl, 25 NaHCO3, 2 KCl, 1.8 CaCl2, 1 MgCl2 and 5 HEPES-Na, pH 7.5.
Generation of slc26a6−/− mice
The Slc26a6-targeting vector (Figure 1) was based on the plasmid pNot1-Pme-Srf (Herz et al, 1992), which contains a PolII-neobPA cassette and two copies of the herpes simplex virus thymidine-kinase (TK) gene. A short arm (consisting of a 3-kb fragment spanning exons 1–6 of the slc26a6 gene) was amplified by using isogenic 120S6/SvEvTac murine genomic DNA and the following primer pairs: 5′-CCGGCTCGAGCCAGAATTCTGATTGGTCTGAT C-3′ and 5′-CCGGCTCGAGCTAGATAACGGACAGTGGCCCA GA-3′. Sequences for the primer pairs were derived from Genbank (accession number NC 000075). An in-frame termination codon was placed at the 3′-end of the short arm and the fragment was cloned into the XhoI site of the vector. A 4-kb long arm fragment spanning from intron 9 to exon 18 was amplified with primers 5′-ATAAGAATGCGGCCGCAGCATCCCTAGCAAGT CTTCTTACACA and 5′-ATAAGAATGCGGCCGCTCACACAGTGTCCACA AAGGAGAGGGTGCT and was subcloned into the NotI site of the vector. Exons 6–9 were replaced by the Neo gene cassette. A linearized construct was introduced into murine SM-1 embryonic stem cells derived from the 129s6/SvEvTac strain by electroporation, and subsequent cloning of homologous recombinant colonies was carried out as described (Willnow and Herz, 1994). Two independent positive clones derived from the targeting construct were expanded and injected into C57BL/J blastocysts. High percentage chimeras derived from each clone were crossed with female C57BL/6J mice to generate two lines of animals carrying the disrupted slc26a6 allele. Phenotypic characterization was carried out in tissues derived from mixed C57BL/6J × 129SvEvTac animals. Slc26a6 in a congenic C57BL/6J animal was achieved by breeding for six generations before using for experiments.
Immunlocalization
The immunolocalization procedure used has been described before (Ko et al, 2004). In brief, frozen pancreatic sections were immobilized by poly-L-lysine and permeabilized with 0.5 ml of cold methanol for 10 min at −20°C. The nonspecific sites were blocked with 5% goat serum, 1% BSA and 0.1% gelatin in PBS (blocking medium). The sections were incubated with a 1:100 dilution of anti-CFTR antibodies in blocking medium overnight at 4°C. Unbound antibodies were washed and the bound antibodies were detected with goat anti-rabbit IgG tagged with FITC. Images were captured with a Zeiss 510 Laser Scanning Confocal Microscope using a 63 × 1.3 NA PlanApo objective.
Primary culture of pancreatic ducts
Age (6–10 months)- and sex-matched WT and slc26a6−/− mice were killed by cervical dislocation and the pancreas was removed and injected with 5 ml digestion buffer composed of DMEM supplemented with soy beans trypsin inhibitor (STI) 0.2 mg/ml, BSA 2 mg/ml, hyaluronidase 400 U/ml (type IV-S, Sigma) and 50 U/ml collagenase (CLSPA, Worthington). The tissue was minced into 1–2 mm pieces, gassed with 5% CO2 and 95% O2, and incubated at 37°C for 30 min and then in fresh digestion buffer for further 30 min. The digested tissue was washed with DMEM and resuspended in buffer composed of DMEM, 0.2 mg/ml STI and 30 mg/ml BSA. Intralobular ducts were microdissected from tissue fragments loosened by the digestion using sharp needles. The ducts were cultured in medium composed of DMEM supplemented with 0.1 mg/ml STI, 10% FBS and 2 mM glutamine. The ducts were cultured at 37°C in 5% CO2 for 36 or up to 48 h. During overnight culture, the ends of the ducts were sealed, leading to a slow dilatation of the ducts lumen due to the accumulation of some fluid under resting conditions. These ducts are selected as healthy, active ducts and equilibrated in HEPES-buffered media to establish the resting state so that spontaneous secretion in response to incubation in HCO3−-buffered media can be measured.
Treatment with dicer siRNA
The ducts were transfected with CFTR-specific or scrambled negative control dicer substrate RNAi duplex (siRNA) within 3 h after dissection and incubated in culture media. The dicer siRNA sequences used were: siRNA1: GUUAAGAAUCCCACCUGCUUUCAGCUU; siRNA2: GUGCAAAUUCAGAGCUUUGUGGAACAG and siRNA3: CAGAAGUGCCUGAAGGGAGUCGUACUG. The scrambled siRNA sequence was CUUCCUCUCUUUCUCUCCCUUGUGA. Isolated ducts were kept in 35 mm dishes containing 2 ml of DMEM with 10% FBS, but without antibiotics. In total, 100 pmol siRNA was diluted in 250 μl of Opti-MEM I Reduced Serum Medium and 5 μl of Lipofectamine 2000 was diluted in 250 μl of the same medium. After 5 min, the diluted siRNA and Lipofectamine 2000 were mixed and left at room temperature for 20 min before addition to the dish with the ducts. After 12 h, the medium was replaced with fresh medium without the siRNA and the ducts were used 36–48 h after the beginning of transfection.
Fluid secretion by the sealed ducts
Fluid secretion was measured by video microscopy as described before (Szalmay et al, 2001). The sealed ducts were transferred to a perfusion chamber and perfused with HEPES- and then HCO3−-buffered media containing the desired agonists. Images were captured at 2 or 5 min intervals and analyzed offline by calculating the lumen volume. Due to the variation in size between the microdissected ducts, a normalization procedure was used and the volume (V0) of the first image was set as 1. Secretion was expressed as the ratio Vt/V0, which was calculated from area ratio (At/A0) of the duct assuming a cylinder (Szalmay et al, 2001) and using equation Vt/Vo=(At/A0)3/2. Secretory rates were also calculated for the 10 min intervals of incubating the ducts in HEPES- and HCO3−-buffered media and for the first 10 min of stimulation with secretin using the average diameter and length of each group (see Ishiguro et al, 1998; Szalmay et al, 2001).
pHi measurements in the sealed ducts
The pHi of the sealed ducts was measured by microfluorometry as before (Ko et al, 2002). The sealed ducts were loaded with BCECF by incubating them for 20 min at room temperature with 2 μM of BCECF/AM. After stabilization of the fluorescence signal, the ducts were perfused with a bath solution without BCECF/AM for at least 10 min to allow completion of BCECF/AM hydrolysis and stabilization of pHi and ductal function. During the recovery, the ducts were perfused with a HEPES-buffered bath solution. BCECF fluorescence was measured at excitation wavelengths of 440 and 490 nm and collecting the light emitted at wavelengths above 530 nm. The ducts were perfused with HCO3−-buffered solutions and after stabilization of pHi, Cl−/HCO3− exchange activity was measured by alternately incubating the ducts in Cl−-free and Cl−-containing medium. HCO3− permeability of the luminal membrane was estimated by equilibrating the sealed ducts in HCO3−-buffered media. Then, resting or stimulated ducts were exposed to media containing 1 μM HOE694 and 0.2 mM H2DIDS. The rate of the ensuing acidification was used as an index of luminal membrane HCO3− permeability.
Preparation of parotid duct cells
Parotid duct cells were prepared by the enzymatic digestion procedure described before (Ko et al, 2004). In brief, the glands were removed, cleaned and rapidly minced and treated with 0.025% trypsin for 7 min at 37°C. The digestion was stopped by a wash with a medium containing 1.5 mg/ml soybean trypsin inhibitor and the tissue was further digested for 20 min with 70 U/ml collagenase CLSPA. The liberated cells were washed, suspended in solution A containing 1.5 mg/ml soybean trypsin inhibitor and kept on ice until use.
Current measurement in isolated parotid duct cells
The whole-cell configuration of the patch-clamp technique was used to measure the Cl− current as detailed before (Ko et al, 2004). The pipette solution contained (mM) 140 NMDG+-Cl−, 1 MgCl2, 2 EGTA, 5 ATP and 10 HEPES (pH 7.3 with Tris). The bath solution was Na+-free solution A. The current was recorded with an Axopatch 200A patch-clamp amplifier and digitized at 2 kHz. The membrane conductance was probed by stepping the membrane potential from a holding potential of 0 mV to membrane potentials between −80 and +60 mV at 10 mV steps for 200 ms with 500 ms intervals between steps. In some experiments, the current was recorded at a holding potential of −80 mV. Current recorded at −80 mV by both procedures was used to obtain the rate of current activation. Pipettes had resistance between 5 and 7 MΩ when filled with pipette solution and seal resistance was always more than 8 GΩ. Current recording and analysis was performed with pClamp 6.0.3 software and results were analyzed and figures were plotted with Origin 7.5 software.
Current measurement in oocytes
Electrophysiological recordings were performed with a two-electrode voltage clamp using an OC-725C Oocyte Clamp System (Warner Instrument), as detailed before (Shcheynikov et al, 2004). The microelectrodes were filled with 3 M KCl and had a resistance of 0.5–2 MΩ. Current and voltage were digitized via a Digidata 1322A A/D converter (Axon Instrument, Foster City, CA) and analyzed using a Clampex 8.1 system.
Statistical analysis
Results in all experiments are given as the mean±s.e.m. of the indicated number of experiments. Significance analysis was performed by ANOVA.
Acknowledgments
We thank Alan Verkman (University of California, San Francisco) for a gift of CFTRinh-172, and Jen-Chieh Chuang and Joyce Repa for help with the Q-PCR. This work was supported by NIH Grants DE12309, DK38938 (to SM), DK49835 (to PT) and training Grant GM-08203 (MRD), as well as CF Foundation Grant MUALLE05G0 (to SM).
References
- Ahn W, Kim KW, Lee JA, Kim JY, Choi JY, Moe OM, Milgram SL, Muallem S, Lee MG (2001) Regulatory interaction between CFTR and HCO3− salvage mechanisms in model systems and the mouse pancreatic duct. J Biol Chem 276: 17236–17243 [DOI] [PubMed] [Google Scholar]
- Choi JY, Muallem D, Kiselyvo K, Thomas PJ, Muallem S (2001) Aberrant CFTR-dependent HCO3− transport in mutations associated with cystic fibrosis. Nature 410: 94–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LL, Harline MC (1998) Dual role of CFTR in cAMP-stimulated HCO3− secretion across murine duodenum. Am J Physiol 274: G718–G726 [DOI] [PubMed] [Google Scholar]
- Cohn JA (2005) Reduced CFTR function and the pathobiology of idiopathic pancreatitis. J Clin Gastroenterol 39: S70–S77 [DOI] [PubMed] [Google Scholar]
- Cook DI, Van Lennep EW, Roberts ML, Young JA (1994) Secretion by the major salivary glands. In Textbook of Physiology of the Gastrointestinal Tract, Johnson LR (ed), pp 1061–1117. New York: Raven Press [Google Scholar]
- Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED (1997) Pendred syndrome is caused by mutation in a putative sulphate transporter gene (PDS). Nat Genet 17: 411–422 [DOI] [PubMed] [Google Scholar]
- Freel RW, Hatch M, Green M, Soleimani M (2006) Ileal oxalate absorption and urinary oxalate excretion are enhanced in Slc26a6 null mice. Am J Physiol Gastrointest Liver Physiol 290: G719–G728 [DOI] [PubMed] [Google Scholar]
- Herz J, Clouthier DE, Hammer RE (1992) LDL receptor-related prostein internalizes and degrades uPA–PAI-1 complexes and is essential for embryo implantation. Cell 71: 411–422 [DOI] [PubMed] [Google Scholar]
- Irokawa T, Krouse ME, Joo NS, Wu JW, Wine JJ (2004) A ‘virtual gland' method for quantifying epithelial fluid secretion. Am J Physiol Lung Cell Mol Physiol 287: L784–L793 [DOI] [PubMed] [Google Scholar]
- Ishiguro H, Namkung W, Yamamoto A, Wang Z, Worrell RT, Xu J, Lee MG, Soleimani M (2006) Effect of Slc26a6 deletion on apical Cl−/HCO3− exchange activity and cAMP-stimulated bicarbonate secretion in pancreatic duct. Am J Physiol Gastrointest Liver Physiol. Aug 10; (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- Ishiguro H, Naruse S, Steward MC, Kitagawa M, Ko SB, Hayakawa T, Case RM (1998) Fluid secretion in interlobular ducts isolated from guinea-pig pancreas. J Physiol 511: 407–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Asplin JR, Evan AP, Rajendran VM, Velazquez H, Nottoli TP, Binder HJ, Aronson PS (2006) Calcium oxalate urolithiasis in mice lacking anion transporter Slc26a6. Nat Genet 38: 474–478 [DOI] [PubMed] [Google Scholar]
- Ko SB, Shcheynikov N, Choi JY, Luo X, Ishibashi K, Thomas PJ, Kim JY, Kim KH, Lee MG, Naruse S, Muallem S (2002) A molecular mechanism for aberrant CFTR-dependent HCO3− transport in cystic fibrosis. EMBO J 21: 5662–5672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko SB, Zeng W, Dorwart MR, Luo X, Kim KH, Millen L, Goto H, Naruse S, Soyombo A, Thomas PJ, Muallem S (2004) Gating of CFTR by STAS domain of SLC26 transporters. Nat Cell Biol 6: 343–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzelmann K, Mall M (2002) Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiol Rev 82: 245–289 [DOI] [PubMed] [Google Scholar]
- Lee JH, Choi JH, Namkung W, Hanrahan JW, Chang J, Song SY, Park SW, Kim DS, Yoon JH, Suh Y, Jang IJ, Nam JH, Kim SJ, Cho MO, Lee JE, Kim KH, Lee MG (2003) A haplotype-based molecular analysis of CFTR mutations associated with respiratory and pancreatic diseases. Hum Mol Genet 12: 2321–2332 [DOI] [PubMed] [Google Scholar]
- Lee MG, Choi JY, Luo X, Strickland E, Thomas PJ, Muallem S (1999) Cystic fibrosis transmembrane conductance regulator regulates luminal Cl−/HCO3− exchange in mouse submandibular and pancreatic ducts. J Biol Chem 274: 14670–14677 [DOI] [PubMed] [Google Scholar]
- Linsdell P, Tabcharani JA, Rommens JM, Hou YX, Chang XB, Tsui LC, Riordan JR, Hanrahan JW (1997) Permeability of wild-type and mutant cystic fibrosis transmembrane conductance regulator chloride channels to polyatomic anions. J Gen Physiol 110: 355–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XZ, Ouyang XM, Xia XJ, Zheng J, Pandya A, Li F, Du LL, Welch KO, Petit C, Smith RJ, Webb BT, Yan D, Arnos KS, Corey D, Dallos P, Nance WE, Chen ZY (2003) Prestin, a cochlear motor protein, is defective in non-syndromic hearing loss. Hum Mol Genet 12: 1155–1162 [DOI] [PubMed] [Google Scholar]
- Lohi H, Kujala M, Kerkela E, Saarialho-Kere U, Kestila M, Kere J (2000) Mapping of five new putative anion transporter genes in human and characterization of SLC26A6, a candidate gene for pancreatic anion exchanger. Genomics 70: 102–112 [DOI] [PubMed] [Google Scholar]
- Luo X, Choi JY, Ko SB, Pushkin A, Kurtz I, Ahn W, Lee MG, Muallem S (2001) HCO3− salvage mechanisms in the submandibular gland acinar and duct cells. J Biol Chem 276: 9808–9816 [DOI] [PubMed] [Google Scholar]
- Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, Verkman AS (2002) Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest 110: 1651–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makela S, Kere J, Holmberg C, Hoglund P (2002) SLC26A3 mutations in congenital chloride diarrhea. Hum Mutat 20: 425–438 [DOI] [PubMed] [Google Scholar]
- Melvin JE, Yule D, Shuttleworth T, Begenisich T (2005) Regulation of fluid and electrolyte secretion in salivary gland acinar cells. Annu Rev Physiol 67: 445–469 [DOI] [PubMed] [Google Scholar]
- Mount DB, Romero MF (2004) The SLC26 gene family of multifunctional anion exchangers. Pflugers Arch Eur J Physiol 447: 710–721 [DOI] [PubMed] [Google Scholar]
- Park M, Ko SB, Choi JY, Muallem G, Thomas PJ, Pushkin A, Lee MS, Kim JY, Lee MG, Muallem S, Kurtz I (2002) The cystic fibrosis transmembrane conductance regulator interacts with and regulates the activity of the HCO3− salvage transporter human Na+-HCO3− cotransport isoform 3. J Biol Chem 277: 50503–50509 [DOI] [PubMed] [Google Scholar]
- Poulsen JH, Fischer H, Illek B, Machen TE (1994) Bicarbonate conductance and pH regulatory capability of cystic fibrosis transmembrane conductance regulator. Proc Nat Acad Sci USA 91: 5340–5344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcheynikov N, Kim KH, Kim KH, Dorwart MR, Ko SB, Goto H, Naruse S, Thomas PJ, Muallem S (2004) Dynamic control of cystic fibrosis transmembrane conductance regulator Cl−/HCO3− selectivity by external Cl−. J Biol Chem 279: 21857–21865 [DOI] [PubMed] [Google Scholar]
- Shcheynikov N, Wang Y, Park M, Ko SB, Dorwart M, Naruse S, Thomas PJ, Muallem S (2006) Coupling modes and stoichiometry of Cl−/HCO3− exchange by slc26a3 and slc26a6. J Gen Physiol 127: 511–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohma Y, Gray MA, Imai Y, Argent BE (2000) HCO3− transport in a mathematical model of the pancreatic ductal epithelium. J Membr Biol 176: 77–100 [DOI] [PubMed] [Google Scholar]
- Sokol RZ (2001) Infertility in men with cystic fibrosis. Curr Opin Pulm Med 7: 421–426 [DOI] [PubMed] [Google Scholar]
- Steward MC, Ishiguro H, Case RM (2005) Mechanisms of bicarbonate secretion in the pancreatic duct. Annu Rev Physiol 67: 377–409 [DOI] [PubMed] [Google Scholar]
- Superti-Furga A, Hastbacka J, Wilcox WR, Cohn DH, van der Harten HJ, Rossi A, Blau N, Rimoin DL, Steinmann B, Lander ES, Gitzelmann R (1996) Achondrogenesis type IB is caused by mutations in the diastrophic dysplasia sulphate transporter gene. Nat Genet 12: 100–102 [DOI] [PubMed] [Google Scholar]
- Szalmay G, Varga G, Kajiyama F, Yang XS, Lang TF, Case RM, Steward MC (2001) Bicarbonate and fluid secretion evoked by cholecystokinin, bombesin and acetylcholine in isolated guinea-pig pancreatic ducts. J Physiol 535: 795–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuo B, Riederer B, Wang Z, Colledge WH, Soleimani M, Seidler U (2006) Involvement of the anion exchanger SLC26A6 in prostaglandin E2− but not forskolin-stimulated duodenal HCO3− secretion. Gastroenterology 130: 349–358 [DOI] [PubMed] [Google Scholar]
- Wang Z, Wang T, Petrovic S, Tuo B, Riederer B, Barone S, Lorenz JN, Seidler U, Aronson PS, Soleimani M (2005) Renal and intestinal transport defects in Slc26a6-null mice. Am J Physiol Cell Physiol 288: C957–C965 [DOI] [PubMed] [Google Scholar]
- Willnow TE, Herz J (1994) Homologous recombination for gene replacement in mouse cell lines. Methods Cell Biol 43: 305–334 [DOI] [PubMed] [Google Scholar]
- Wilschanski M, Durie PR (1998) Pathology of pancreatic and intestinal disorders in cystic fibrosis. J R Soc Med 91: 40–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright AM, Gong X, Verdon B, Linsdell P, Mehta A, Riordan JR, Argent BE, Gray MA (2004) Novel regulation of cystic fibrosis transmembrane conductance regulator (CFTR) channel gating by external chloride. J Biol Chem 279: 41658–41663 [DOI] [PubMed] [Google Scholar]
- Xie Q, Welch R, Mercado A, Romero MF, Mount DB (2002) Molecular characterization of the murine SLc26a6 anion exchanger: functional comparison with SLc26a1. Am J Physiol Renal Physiol 283: F826–F838 [DOI] [PubMed] [Google Scholar]