

Abstract
The membrane dye FM 1-43 has frequently been used to quantify exocytosis in neurons. In epithelia, intense lateral intracellular space staining and fluctuations in baseline labeling produced inconsistent results. Membrane retrieved in the presence of FM 1-43 retains the dye, however, and cells that undergo compensatory endocytosis during and following evoked exocytosis contain punctate, fluorescent particles after washout of external stain. As an alternative measure of trafficking, we quantified the fluorescent puncta retained after dye washout and tested our method on both coverslip-grown cell clusters and filter-grown intact monolayers. Images for analysis were acquired using serial sectioning with either epifluorescence or confocal microscopy. Tests with an intestinal goblet cell line that exhibits basal and ATP-stimulated granule trafficking confirmed that 1), the algorithm identified the same number of internalized particles with either epifluorescence or confocal microscopy acquired images; 2), low density clusters exhibited significantly more internalized particles per cell than either filter-grown monolayers or high density clusters; 3), ATP stimulation significantly increased the number of internalized particles in all preparations; and 4), the number of particles internalized was comparable to capacitance measurements of exocytosis. This method provides a single technique for quantifying membrane trafficking in both monolayers and unpolarized cells.
INTRODUCTION
Membrane trafficking in epithelia is known or suspected to regulate the number and lifetime of several ion channels in the plasma membrane (1–3), as well as the secretion of large proteins such as mucin that play an important role in maintaining and protecting the epithelial surface (4). Intensive effort is underway to identify the regulatory pathways controlling membrane trafficking in epithelia. Single cell electrophysiology and fluorescence techniques exist for the measurement of vesicle fusion, retrieval, and product release in real time (5,6), but their application to epithelial cells is often questioned: do single cells represent an adequate model of the intact epithelial monolayer?
The epithelium provides a barrier between external (luminal) and plasma compartments, as well as vectorial transport of salt, water, nutrients, and waste products between these compartments. Development of the polarized epithelial monolayer progresses through cell adhesion, proliferation of cells to form clusters exhibiting spatial contact, and formation of tight junctions between cells. At each step of this process, a complex interaction of proteins and cell signaling occurs, and some of these interactions are also known to regulate trafficking and secretion (7–9). For example, integrins active during cell adhesion and spreading mediate activation of phospholipase C, which in turn produces diacylglycerol and inositol triphosphate, leading to an elevation of intracellular calcium and activation of protein kinase C (8). The activation of second messengers as a function of cell polarity suggests that differences in protein or ion secretion might be expected in a particular epithelial cell line at different stages of monolayer development.
One ion channel shown to exhibit functional changes based on polarization is the epithelial sodium channel (ENaC). In the mouse cortical collecting duct, ENaC currents were not present in single cells but appeared once these same cells had grown to confluency and exhibited active vectorial transport (10). Our measurements of mucin and chloride secretion in an HT29 subclone also identified different secretory responses in single cells (11,12) versus monolayers (13). However, the separate measurement techniques used to record current and capacitance changes in single cells versus monolayers may also contribute to different results. Whole cell patch clamping with measurement of capacitance records net exocytosis in the single cell, but impedance analysis is used to track exocytosis in the intact monolayer. The monolayer configuration limits access to the intracellular environment, averages the response of all cells in the monolayer, and offers a relatively low time resolution (14). The lack of a uniformly applicable assay has potentially limited study of the impact cell differentiation status has on protein and ion secretion; much of the information known about monolayer differentiation is derived from studies exploring the development of malignancies (15).
Research in the neural and neuroendocrine fields has used the fluorescent membrane dye FM 1-43 to assess rates of exocytosis and endocytosis using microscopy (5). The basic concept is straightforward (16): FM 1-43 in solution is minimally fluorescent; upon binding membranes, it becomes intensely fluorescent. Plasma membranes exposed to solution containing FM 1-43 will fluoresce, and since the dye does not cross the lipid bilayer, increases in fluorescence after preliminary labeling are attributed to increases in membrane surface area due to granule or vesicle fusion. Granules or vesicles will retain the staining during endocytosis in the continued presence of extracellular dye, and therefore the fluorescence signal reflects both fused and endocytosed membrane. Plasma membrane staining can be removed by perfusing with dye-free solution, and this technique has been used to prestain vesicles to monitor recycling events.
We examined the application of FM 1-43 to assess membrane trafficking in the Cl.16E subclone of the human colonic cancer cell line HT29, which forms confluent monolayers with at least 50% of cells exhibiting goblet cell properties including large numbers of mucin granules in the apical pole (17). Several techniques have demonstrated that purinergic agonists stimulate exocytosis in this line: electron micrographs of control and ATP-stimulated monolayers, which demonstrate compound exocytosis (18); capacitance measurements in isolated cells (11,12) and impedance analysis of intact monolayers (13), which demonstrate that ATP stimulates a transient increase in capacitance; and detection of secreted, radiolabeled mucins. We report here our refinement to the FM 1-43 technique that allows quantifying membrane trafficking in single cells, cell clusters of variable density, and filter-grown intact monolayers, as well as our preliminary observations on the behavior of cells in these different configurations.
MATERIALS AND METHODS
Cell culture
HT29-Cl.16E cells were propagated in Falcon culture flasks (25 cm2) in a humidified atmosphere of 95% air, 5% CO2 at 37°C. The cells were fed daily with a standard media consisting of Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 4 mM L-glutamine. Cultures were periodically tested for mycoplasma using a commercial kit (Mycotect, Invitrogen, Carlsbad, CA) and found free of contamination. The passage numbers for the reported experiments were between 23 and 45. Polarized cell monolayers were grown on Vitrogen-coated, Transwell Clear (size 6.5 mm) filters. Cl.16E cells were seeded at a density of 4–6 × 105/filter, became visually confluent after 7 days, and were used between days 9 and 14. Single cells and cell clusters were grown on No. 1, 25-mm glass coverslips and typically seeded using 1/10 the concentration used for filters. Coverslips were used for microscopy studies either 2–3 days or 7–9 days after plating.
DMEM, FBS, and L-glutamine were purchased from Invitrogen. Vitrogen was purchased from Collagen (Palo Alto, CA), and Transwell filters from Millipore (Bedford, MA). The fluorescent dyes FM 1-43, FM 1-43FX, and DRAQ5 were purchased from Invitrogen-Molecular Probes (Eugene, OR). All other chemicals were purchased from Sigma (St. Louis, MO).
FM 1-43 labeling
Image acquisition and analysis were performed after dye exposure and washout, thus two labeling techniques were possible: perfused labeling performed on the microscope stage and static labeling performed in an incubator. Since endocytosis is a dynamic process with an expectation that endocytosed particles will eventually be recycled or degraded, serial sectioning needed to be performed as quickly as possible within a finite interval after dye washout on live cells, or preparations needed to be fixed for later imaging. This time constraint led to perfused labeling on the microscope as the primary choice for live cell preparations. FM 1-43 is quite labile and our initial experiments fixing FM 1-43 labeled cells were unsuccessful; however, a fixable variant of FM 1-43 is available and was used when fixation was to be performed.
A second constraint when choosing a labeling method was the ease with which high resolution serial sectioning could be performed. High numerical aperture (NA) objectives limit the depth of focus and are therefore desirable for sectioning, but they usually have a small working distance (separation between the objective and specimen). Cells grown on No. 1 thickness coverslips met the limits of high NA objectives, but cells grown on permeable filters did not, due to the space required for perfusion. Because the filter membrane can be removed from the holder and mounted on a slide after fixation to allow high resolution serial sectioning, static labeling was preferred with monolayers.
Perfused labeling
All experiments were performed at 37°C. Coverslips were mounted in a PDMI-2 open perfusion microincubation chamber with a TC-202 temperature controller (Harvard Apparatus, Holliston, MA), which was then mounted on the stage of an inverted microscope (Diaphot 300; Nikon, Tokyo, Japan) equipped for epifluorescence. The 300-μl chamber was perfused at 1 ml/min. Perfusion was momentarily stopped for solution changes; delay from perfusion reservoir to chamber was 60 s.
Each experiment consisted of 7-min perfusion with 4 μM FM 1-43 to label cells and assess basal trafficking, followed by 5-min perfusion with 4 μM FM 1-43 ± 200 μM ATP to stimulate mucin secretion. Finally, extracellular FM 1-43 labeling was removed by perfusing with bath solution for 8 min. Phosphate-buffered bath solution (PBS) was used for all experiments (in mM, 145 NaCl, 0.4 KH2PO4, 1.6 K2HPO4, 1.0 MgCl2, 1.5 CaCl2, and 5 glucose at pH 7.3). During perfusion, images were acquired every 20 s at a focal plane of 5 μm (above the coverslip). The exposure time per image was 0.16 s. Images collected during this phase were used for qualitative assessments of labeling and washout. Immediately after the 8-min washout, serial sectioning was performed as described below.
Static labeling
Static labeling experiments were performed on either coverslips or Transwell filters and used the fixable variant of FM 1-43. All solutions were maintained at 37°C unless noted. Cell culture media were removed, and cells rinsed twice with PBS. After rinsing, 10 μM FM 1-43FX in PBS was added and cells returned to the incubator for 7 min. This solution was then exchanged with PBS + 10 μM FM 1-43FX ± ATP and incubated for 5 min. Cells were then rinsed three times with PBS and returned to the incubator for 4 min. This destaining step was repeated once to give 8 min total in dye-free bath. After the final incubation, the warm bath was immediately replaced with ice-cold PBS to stop endocytosis and prepare cells for fixation. During the steps above, coverslips received 1-ml bath ± additives whereas filters, incubated in Transwell holders, received 800-μl bath to the basolateral compartment and 100-μl bath ± additives to the apical compartment.
Within minutes of the final incubation, the cells were incubated in 4% paraformaldyhyde at 4°C for 20 min. Cells were rinsed twice with cold PBS then incubated in a nuclear staining cocktail (5 μM Hoechst + 5 μM DRAQ5) at 4°C for 40 min. Both stains were used to allow imaging with epifluorescence or laser light sources. After a final rinse, coverslips and excised filters were mounted on glass slides for later microscopy.
Microscopy
Serial sectioning with epifluorescence microscopy
The coverslip was illuminated using a 75-W xenon lamp and 485-nm excitation filter through a 40×, NA 1.3 objective. A multiple wavelength emission filter was used to capture FM 1-43 fluorescence at 530 nm and above 590 nm; for fixed coverslips, acquisition of the Hoechst nuclear stain was also performed with excitation at 380 nm and emission at 460 nm. The signal was recorded using a C4742-95 charge-coupled device (CCD) camera (Hamamatsu, Hamamatsu, Japan). The coverslip was identified using transmitted light and set as the reference plane (Z = 0 μm). A typical field of view (640 × 512 pixels) contained several individual cells and/or cell clusters.
Serial sectioning was performed by stepping the focal plane in 1-μm steps from below the coverslip to 25 μm above the coverslip with an image acquisition at each step. The exposure time per image was 0.16 s, and the stack was acquired in succession without delay. All images collected during this phase were stored in one file (stacked tagged image file (TIF) format) to facilitate automated data analysis. Shutter, excitation filter, and z axis position were under software control (SimplePCI, Compix, Sewickley, PA) through a LUDL (LEP, Hawthorne, NY) controller.
Serial sectioning with confocal microscopy
Confocal sectioning was performed on a Leica (Wetzlar, Germany) TCS-SL upright microscope. Laser power settings never exceeded 30% of maximum, and pinhole size was set at optimal (as determined by Leica software) for the 64×, 1.4 NA objective used. To capture FM 1-43 and DRAQ5 fluorescence, laser illumination and emission filters were alternated between green (480 nm) and far red (680 nm+) settings, respectively. Serial sections were taken every 0.45 μm through the cells starting just above the level of the apical membrane to the point where the support became visible. All images collected during this phase were stored in one file (stacked TIF format) to facilitate automated data analysis. Source and z axis position were under Leica software control.
Dimensional limits
The theoretical resolution limits in the lateral (x-y) and axial (z) planes are defined from basic optical principles as the radius of the first dark ring in the Airy diffraction image (rAiry) and the distance from the center of the three-dimensional diffraction pattern to the first axial minimum (zmin), respectively, where the diffraction pattern results from an infinitely small point source (19):
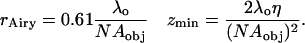 |
For the epifluorescence microscope used, the primary emission wavelength (λo) is 530 nm, the refractive index (η) for an oil objective is 1.515, and the NA of the objective is 1.3, giving the theoretical resolutions as 0.25 μm laterally and 0.95 μm axially.
The actual lateral resolution is also a function of the magnifying power of the objective and the binning of individual elements (pixels) of the CCD camera. The transmitted light resolution measured using a calibrated scale with the 40× objective and no binning (1280 × 1024 pixel image) was 0.163 μm; with 2 × 2 binning (640 × 512 pixel image) the actual lateral resolution is 0.325 μm.
The excitation light of an epifluorescence microscope produces significant intensity above and below the plane in focus, reducing the contrast of focused objects and thus degrading the axial resolution (19). The manner in which light emanating from a source outside of the focal plane impinges upon a focused image can be modeled by measuring the point spread function (PSF) of the experimental setup. This was accomplished by collecting images of a point source, modeled by a 175-nm fluorescent bead (PS-Speck, Molecular Probes), at 1-μm intervals across the z axis range and plotting the intensity changes in the point as a function of the z axis position (20). The apparent resolution, or depth of field (δ), can be estimated as (19)
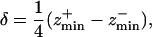 |
where these zmin values represent the axial minima to either side of the central maximum. The PSF for the epifluorescence experimental setup measured an apparent depth of field of 3.25 μm.
Image analysis
Each image file produced from serial sectioning was processed to provide quantification of internalized membrane. For low density cell preparations, each cell or cell cluster was processed separately: a region of interest (ROI) was drawn around the perimeter of the individual cell or cell cluster to identify the outermost boundary, using the focal plane exhibiting the largest diameter or surface area, respectively (typically 4–6 μm above the coverslip). For high density cell preparations, a rectangular ROI was used encompassing 100–200 cells. Each image in the stack was then subjected to the following steps to determine the location and size of all punctate objects: 1), A new (gradient) image was generated by filtering the original image with a Laplacian (gradient) operator (below) to identify significant gradients in intensity. 2), The gradient image was smoothed to average out noise speckling, using a 3 × 3 median filter (21). 3), A binary mask was generated by tagging pixels in the smoothed, gradient image that exceeded a fixed threshold. 4), The tagged pixels in the mask were examined to ensure they did not mark residual junction staining, using an operator to identify tags whose length was at least four times their width. 5), The binary mask was then used as a template to identify and analyze objects in the original, unprocessed image. These analysis steps were performed using intrinsic functions available in our imaging software (SimplePCI) and are graphically demonstrated for a single plane (see Fig. 3).
FIGURE 3.

Image-processing steps to identify internalized FM 1-43 labeled particles. (A) An ROI (in red) is manually drawn around the cluster perimeter; only particles inside the ROI will be considered. (B) A Laplacian filter and smoothing filter are applied to the image to highlight intensity gradients (Materials and Methods), generating a new image. (C) A binary map (in green) is generated by tagging pixels in the gradient image with values above a fixed threshold. (D) Tagged pixels in the map are marked (blue) then removed if they are overly elongated with respect to their width (to eliminate nonpunctate tags). (E) Pixels in the original image that lie under the map (green) are analyzed for frequency and area.
Detection algorithm
Each image plane in a stack collected after FM 1-43 washout exhibited punctate fluorescence from objects within the focal plane, more diffuse punctate fluorescence from near focal plane objects, and general diffuse fluorescence from residual plasma membrane staining and objects well outside of the focal plane. The simplest method to detect punctate fluorescence would be to tag all pixels exceeding a fixed threshold of fluorescence after determining what constituted the desired particle intensity. In practice, significant variability in the desired particle intensity occurs at different heights in the cell and between experiments. This variability results because diffuse fluorescence from outside the focal plane impinges on the fluorescence of objects in the focal plane with varying magnitudes of intensity and due to experimental variability such as small fluctuations in the concentration of FM 1-43. To detect punctate objects unambiguously, we applied a Laplacian operator to identify significant, abrupt transitions in pixel-to-pixel intensity (21). The Laplacian operator generates a new image (B) that approximates the gradients in intensity I of the original image (A):
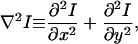 |
through convolution with a Laplacian kernel (L). The Laplacian kernel is a 3 × 3 matrix with values:
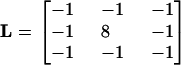 |
In our implementation, original image A is represented by a 640 × 512 matrix whose elements store the intensity of each pixel using a 16-bit grayscale. The convolution is performed through a simple sum for each neighborhood defined by the 3 × 3 kernel:
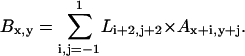 |
A scaling factor of 1/2 × 216 is added to each element to avoid negative values and results in a medium gray pixel in the absence of a gradient. The edge values (B1,y; B640,y; Bx,1; Bx,512) are discarded. The symmetry of the Laplacian kernel provides for a greater response to puncta over boundary lines (21), an optimal response for this application.
A Laplacian filter is an intrinsic function in many image-processing software packages and can be utilized if it conforms to the kernel and operation defined above, simplifying implementation of the detection algorithm. The image-processing steps described here were programmed as a macro in our software and performed automatically but allowed user intervention at key decision points. The macro produced a spreadsheet containing size, intensity, and area information for every identified punctate object throughout each cell's volume. Processing time for a typical stack of images was <15 min; without user intervention pauses, a stack of 25 images can be processed in <1 min.
Normalizing puncta to cell area
After image processing, the total number of puncta, their average size in pixels, their distribution throughout the analyzed volume, and surface area of the ROI used to collect the information were known. Spatial calibration was performed for each combination of microscope, objective, and image binning used as follows: a precision micrometer slide (Leica) displaying a 2-mm scale bar divided into 0.01-mm units was illuminated and captured for display using SimplePCI image acquisition software. A line was drawn on the image spanning 10 units (100 μm), and the line length in pixels used to calculate a spatial conversion factor in pixels/μm. These conversion factors were used to convert pixel data to spatial equivalents.
Quantifying the results on a per-cell basis required accounting for the total number of cells in the ROI used to count puncta. For small cell clusters, the total number of cells was easily counted, and during perfused labeling an image acquired before dye washout facilitated the process due to dye permeation between cells (Fig. 2 B). Cell counting was tedious for large or dense cell clusters, but fixed cells counterstained with a nuclear stain simplified the task to counting the better defined nuclei in an ROI. Results can be presented as number of puncta at specific cell heights (sections) or summed for all sections to provide total number of puncta per cell. Since the average and total area of puncta are also known, results may also be presented in terms of labeled area.
FIGURE 2.

A low density cell cluster during perfusion with FM 1-43 + ATP. The corresponding MFI for each image was shown in Fig. 1. Focal plane is 5 μm above the coverslip. (A) Preliminary labeling of the cells with FM 1-43 after 7 min in dye. Bar represents 10 μm. (B) After 4-min exposure to ATP, the LIS are intensely stained. Some internalization of fluorescence is evident on the periphery of the cell cluster (arrowhead), whereas the intense staining of the LIS between cells (arrow) makes it difficult to discern internalization. (C) After washout of extracellular FM 1-43, internalized dye is evident as puncta within each cell. The degree of internalization does not appear different between peripheral (arrowhead) and LIS adjacent (arrow) cells. The intensity range of C was expanded to reveal the low fluorescence detail.
Electrophysiology methods for measuring exocytosis use capacitance as a measure of surface area, with increases in capacitance attributable to the surface area of exocytosed granules and vesicles. It was desirable to determine whether the puncta internalized in the presence of FM 1-43 were comparable to capacitance measurements of exocytosis. If the area of each punctate object is taken as the two-dimensional cross section of a spherical object, then the corresponding surface area is calculated as 4× the cross section, based on the relationship between the surface area of a circle (πr2) and the surface area of a sphere (4πr2). Thus an equivalent capacitance per cell attributable to exocytosis was calculated using the measured pixel to pixel dimensions and the standard conversion factor of 1 μF/cm2 (see Tables 1–3 below).
TABLE 1.
Membrane internalized in low density coverslipped cells
| 2–3-day-old cells on coverslips | No. particles per cell | Average particle diameter (nm) | Calculated capacitance (pF/cell) | Effective cross section area per cell (μm2) |
|---|---|---|---|---|
| Basal (n = 17) | 316 ± 31 | 570 ± 10 | 3.3 ± 0.4 | 277 ± 21 |
| ATP (n = 21) | 443 ± 28* | 580 ± 10 | 4.7 ± 0.3* | 222 ± 7* |
p < 0.05.
TABLE 3.
Membrane internalized in high density coverslipped cells
| 7–11-day-old cells on coverslips | No. particles per cell | Average particle diameter (nm) | Calculated capacitance (pF/cell) | Effective cross section area per cell (μm2) |
|---|---|---|---|---|
| Basal (n = 3) | 8 ± 3 | 490 ± 10† | 0.06 ± 0.02 | 118 ± 11† |
| ATP (n = 6) | 30 ± 8* | 560 ± 20* | 0.33 ± 0.10* | 123 ± 7† |
p < 0.05, basal versus ATP.
p < 0.05, high density coverslip versus filter.
TABLE 2.
Membrane internalized in filter-grown monolayers
| Confluent monolayers on filters | No. particles per cell | Average particle diameter (nm) | Calculated capacitance (pF/cell) | Effective cross section area per cell (μm2) |
|---|---|---|---|---|
| Basal (n = 6) | 4 ± 1 | 560 ± 10 | 0.04 ± 0.01 | 44.2 ± 5.1 |
| Epifluorescence | ||||
| ATP (n = 3) | 15 ± 2* | 560 ± 20 | 0.15 ± 0.03* | 62.8 ± 5.8* |
| Epifluorescence | ||||
| Basal (n = 4) | 4 ± 1 | 410 ± 10† | 0.02 ± 0.01* | 47.2 ± 1.7 |
| Confocal | ||||
| ATP (n = 4) | 16 ± 4* | 390 ± 10*† | 0.08 ± 0.02* | 65.2 ± 6.0* |
| Confocal |
p < 0.05, basal versus ATP.
p < 0.05, epifluorescence versus confocal.
Statistics
Tabulated data are shown as means ± SE of the mean of n experiments, where each experiment represents an ROI containing ≥100 cells for filters and high density coverslips or an average of five cells for low density coverslips. Statistical comparisons were carried out using the Student's t-test, with p < 0.05 considered significant.
RESULTS
Measurement of exocytosis
The fluorescent dye FM 1-43 rapidly and reversibly labels the extracellular membranes of cells to which it is exposed. The traditional use of FM 1-43, to quantify exocytosis in neurons, equates an agonist-induced increase in mean fluorescent intensity (MFI) after a finite labeling period to the labeling of new membrane inserted during exocytosis (5). Labeled membrane retains its fluorescence during endocytosis, providing a cumulative measure of exocytosis as the sum of MFI from internalized and newly inserted surface membrane. We tested this traditional technique on HT29-Cl.16E cells, using a stimulation protocol previously demonstrated to result in a 15% increase in the membrane capacitance of single cells (11).
Several differences between the 16E cells and reported results in neurons were immediately evident. First, as FM 1-43 was perfused over cells (Fig. 1), the rapid increase in MFI expected during preliminary labeling of membranes persisted for at least 2 min in all cases (n = 56) and did not reach a steady state over the course of 12 min (control cases, n = 13). This preliminary labeling period was six times longer than the volume exchange rate of the microscope chamber (20 s; Materials and Methods). The 2-min labeling period markedly exceeded the 5 s typically reported in neurons (22), and the failure to reach a constant MFI in the absence of stimulation indicated that significant basal membrane turnover was continuously occurring. Second, during stimulation, intense staining of the lateral intracellular spaces (LIS) between cells occurred (Fig. 2). Third, washout of the dye after stimulation clearly revealed significant endocytosis, evident as a pattern of fluorescent puncta, but there was no evidence that significantly more internalization had occurred in the near LIS region compared to other near membrane regions of cells. Additional anomalies are demonstrated in the Supplementary Material.
FIGURE 1.

Changes in MFI do not reliably reflect total exocytosis. The average MFI of 12 cells from separate clusters of 1–10 cells each was measured as dye was perfused onto the coverslip then washed off. The two experiments shown (±200 μM ATP) were recorded from consecutive coverslips on the same day, with age in culture, focal plane above the coverslip, and concentration of FM 1-43 identical for each. Even with these balanced conditions, the difference in response ± ATP is not statistically significant. Data are presented as mean ± SE of the 12 cells. Lettered arrows correspond to images in Fig. 2.
Quantifying endocytosed membrane
The discrete puncta evident in cells after FM 1-43 washout (Fig. 2 C) suggested an alternate technique for measuring trafficking in epithelia: quantify the membrane internalized during the stimulation protocol. Capacitance measurements in HT29-Cl.16E cells had shown that ATP-stimulated capacitance increases were transient: cell capacitance returned to baseline levels within minutes after stimulation (11,13). This suggested that cells retrieved membrane equivalent to the new membrane inserted during granule exocytosis, in agreement with stimulated secretion responses in neural and neuroendocrine cells (23). To provide a quantitative assessment of endocytosis, measurement of the size of all the fluorescent, punctate objects per cell was required. An automated technique that would ignore residual plasma membrane labeling, analyze the entire cell volume, and if possible identify puncta in images collected with epifluorescence microscopy was desirable.
Two steps were required to implement this approach: collect images representative of the cell volume, then process the images to identify endocytosed puncta. The first step, serial sectioning, was accomplished by capturing images at discrete, equally spaced focal planes starting at the coverslip and moving up through the cell height, and produced a stack of images representing the cell's volume. The spacing between focal planes (Δz) is typically a compromise between the theoretical and actual z axis resolution of the microscope (Materials and Methods): out-of-focus fluorescence degrades the epifluorescence image, and all images suffer as the NA of the objective decreases. For epifluorescence experiments a Δz = 1 μm was used with an objective NA ≥ 1.3, whereas a Δz = 0.45 μm was typically used with confocal microscopy.
We generated an algorithm using standard image-processing tools to perform the second step. To identify fluorescent puncta in each image, a map was created identifying the location and size of all the punctate objects found. With epifluorescence microscopy, out-of-focus objects and nonspecific labeling create background fluorescence that is hazy with small graduations in intensity (Fig. 3 A). Conversely, when puncta are in the focal plane, the graduations in intensity are sharp due to the clear focus. We used a Laplacian filter, which produces a new image whose grayscale values reflect the intensity gradients in the original image, to identify focused puncta (Fig. 3 B). A smoothing filter was applied to the Laplacian image to average out single pixel gradients (noise speckle, (21)) in the dark regions of the image.
A threshold detection operation was applied to the gradient image to generate the map of punctate objects (Fig. 3 C). In the absence of a gradient due to a bright puncta, the corresponding pixel in the gradient image is medium gray (Fig. 3 B); a positive gradient results in a lighter gray pixel or larger value. The threshold for detecting a pixel gradient and assigning it to the map was 5% above the medium gray level for epifluorescence images and 2% for confocal images. The map itself is a simple binary image: one color (green) if the pixel below it represented a punctate object, another (black) if it did not. After the binary map was generated from the gradient image, it was checked for unusual tags: if any contiguous group of green pixels was four times longer than wide, cell-cell junction labeling was suspected and the pixels were deselected (Fig. 3 D). The final binary map, overlaid on the original image, was used to identify which pixels to analyze as internalized puncta (Fig. 3 E).
Cell density affects quantity of membrane internalized
We tested this algorithm on data collected from cells grown on glass coverslips as well as monolayers grown on permeable filters and compared FM 1-43 labeling techniques and confocal versus epifluorescence microscopy for serial sectioning. Cells grown on coverslips and cultured for 2–3 days (low density, five cells/cluster on average) exhibited significant basal internalization, 316 ± 31 particles/cell (n = 17; Table 1). With 200 μM ATP included in the FM 1-43 solution for 5 min before washout, the number of internalized particles/cell significantly increased to 443 ± 28 (n = 21). These values correspond to equivalent capacitances of 3.3 and 4.7 pF/cell, respectively (Table 1).
Whole cell patch clamping has demonstrated that ATP stimulates a 15% increase in membrane capacitance in HT29-Cl.16E (11,12), with the distribution of stimulated changes in capacitance (Fig. 4) a reflection of the range in cell size. Interestingly, no change in membrane capacitance is recorded under basal conditions, presumably because cell surface area is maintained at steady state. In comparing the FM 1-43 results with patch clamping, we considered two possibilities: 1) ATP-stimulated secretion occurs on top of basal secretion, or 2) ATP stimulation overrides basal secretion. For the first case, the capacitance increase attributed to ATP would be 1.4 pF/cell (E1 line in Fig. 4; ΔCm = 4.7 pF/cell − 3.3 pF/cell). For the second case, with ATP present for only half of the FM 1-43 exposure period (time course in Fig. 1), we needed to subtract the basal internalization that occurred before ATP addition. Assuming the amount of basal internalization that occurred before ATP addition was half the value measured for basal turnover alone, the capacitance increase attributed to ATP would be 3.1 pF/cell (E2 line in Fig. 4; ΔCm = 4.7 pF/cell − (1/2) × 3.3 pF/cell). For either case, the FM 1-43 internalization measurement was comparable to patch clamping (Fig. 4) and also revealed that significant trafficking occurred in the absence of stimuli even though it did not result in a significant change in net membrane area.
FIGURE 4.

Equivalent capacitance of FM 1-43 labeled membrane internalized during ATP stimulation falls within the range of ATP-stimulated capacitance increases measured using whole cell patch clamp. The distribution of ATP-stimulated capacitance increases (bars) reflects the size distribution of cells (10–30 pF); recorded increase is 15 ± 4% (n = 27). The mean increase using patch clamp (dashed line M) was 2.9 pF. The FM 1-43 method measured significant basal internalization not recorded by the patch clamp technique, and the stimulated increase resulting from ATP was calculated for two different cases: ATP secretion occurs in addition to basal secretion (dotted line E1, 1.4 pF), and ATP secretion overrides basal secretion (dotted line E2, 3.1 pF).
Impedance analysis has been used to monitor capacitance in intact monolayers, and we have estimated that ATP-stimulated capacitance increases in HT29-Cl.16E correspond to a range of 40–170 granules per cell (13). Using the fixable variant of FM 1-43, we tested intact monolayers in response to ATP. Using epifluorescence microscopy to perform serial sectioning, we found the monolayers exhibited much less basal internalization (4 ± 1 particles/cell, n = 6; Table 2) than low density coverslipped cells and were responsive to ATP (15 ± 2 particles/cell, n = 3). Since the monolayers were fixed, we also performed serial sectioning on the same preparations using confocal microscopy. The confocal technique eliminates much of the out-of-focus haze evident in epifluorescence images, clearly rendering the internalized puncta (Fig. 5 C) and making it relatively easy to identify them. Application of the gradient process to confocal images resulted in significant noise speckling, and the smoothing operation was mandatory (Fig. 5, A and B). We used our algorithm to count puncta in the confocal sections (Table 2) and found the same numbers of particles internalized per cell as identified in the epifluorescence-acquired sections, confirming our algorithm was accurately identifying puncta in the presence of out-of-focus haze. The confocal sections were acquired with higher magnification and twice the z axis resolution but did not detect additional particles, indicating that the sampling dimensions used with epifluorescence acquisition were adequate.
FIGURE 5.

Confocal serial section of fixed, FM 1-43 labeled confluent monolayer. The filter surface is uneven, and cells in the dark zone to the right are below the image plane. (A) Substantial noise speckling is evident in the gradient image. (B) Two passes with a smoothing filter eliminates noise speckling. (C) Punctate particles are clearly visible in the raw, unprocessed image.
We next asked if the significant difference in internalized puncta between the low density, coverslip-grown cells and the filter-grown monolayers reflected the intact barrier between apical and basolateral membranes in the monolayers by testing high density cell clusters grown on coverslips. In these preparations the individual clusters, cultured for at least 7 days, contained at least 100 cells. Using the perfused labeling method, dye migration between cells was evident (Fig. 6), indicating that these high density clusters did not have an intact barrier at the cluster edges. However, the number of internalized puncta was similar to the values found in the monolayers: 8 ± 3 particles/cell (n = 3) under basal conditions, increasing to 30 ± 8 particles/cell in ATP-stimulated clusters (Table 3). Both the high density coverslipped cells and the intact monolayers had average cell cross sectional areas less than half the value noted in the low density coverslipped cells (Tables 1–3).
FIGURE 6.

Dye permeation into LIS occurs over time in high density, coverslipped cell clusters. (A) FM 1-43 initially labels the peripheral cells of the cluster, indicating the basolateral membrane is accessible from the bath. (B) After 2 min, migration of dye has stained almost all the LIS. (C) Four minutes into preliminary dye perfusion, all the LIS are stained. The brightly stained puncta in the lower center of the cluster is debris.
The high density cell preparations were cultured for at least 7 days, suggesting that time in culture could be a factor in reducing trafficking rates. We plated coverslips with a higher density of cells to induce greater variability in cluster size during a 2-day culture period to address this issue. As shown in Fig. 7 A for basal uptake, the number of objects per cell dropped exponentially with increasing cluster size in cells cultured for the shorter period. Combining the basal uptake data of Tables 1 and 3 in a single plot (Fig. 7 B) and fitting it to an exponential relation resulted in the same decay rate, strongly suggesting that cell density rather than age in culture was the determining factor in trafficking rates.
FIGURE 7.

The magnitude of basal internalization decreases exponentially with cell cluster density independent of cell culture age. Data are fit to a single exponential decay of the form 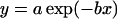 . (A) Cells plated at higher density exhibit a wider distribution of cells per cluster after 2 days in culture and an exponential decay in particles/cell with increasing density. Fit parameters for the solid line are a = 352, b = 0.037; static labeling was used to stain cells. (B) Data from low density coverslips cultured for 2–3 days (Table 1) and high density coverslips cultured for 7+ days (Table 3) were combined in one set and fit, with resulting fit parameters for the solid (gray) line of a = 363, b = 0.034. The fit curve from panel A is shown as a dashed line. Coverslips were stained using perfused labeling; the low density data distribution between fixed (A) and perfused (B) labeling is the same.
. (A) Cells plated at higher density exhibit a wider distribution of cells per cluster after 2 days in culture and an exponential decay in particles/cell with increasing density. Fit parameters for the solid line are a = 352, b = 0.037; static labeling was used to stain cells. (B) Data from low density coverslips cultured for 2–3 days (Table 1) and high density coverslips cultured for 7+ days (Table 3) were combined in one set and fit, with resulting fit parameters for the solid (gray) line of a = 363, b = 0.034. The fit curve from panel A is shown as a dashed line. Coverslips were stained using perfused labeling; the low density data distribution between fixed (A) and perfused (B) labeling is the same.
DISCUSSION
Our initial attempts to use FM 1-43 as a marker of exocytosis in HT29-Cl.16E resulted in several inconsistencies, including an unusually long preliminary labeling period, failure to reach a consistent plateau (Fig. 1), and an ATP-induced intense staining of the LIS that disappeared during continuous, dye-free perfusion and thus was not well correlated with trafficking events (Fig. 2). Additional ambiguities are demonstrated in the Supplementary Material. Brumback et al. (24), who extended the use of FM 1-43 to monitor neuropeptide granule dynamics, reported several caveats that may account for these effects. There are no simple solutions for correcting these problems, however, and they preclude using the MFI of FM 1-43 as a dynamic marker of exocytosis in these epithelial cells.
After washout of the external plasma membrane FM 1-43 staining, we observed significant internalization of dye in the form of puncta distributed throughout the volume of the cell (Fig. 2 C). It is well known that membrane endocytosed in the presence of FM 1-43 will retain its fluorescent label (16,22), and washout of extracellular plasma membrane stain after a round of stimulation has been used to prelabel vesicles to study recycling (16). Since the problems observed with FM 1-43 were related to the extracellular labeling, and prior capacitance measurements demonstrated that stimulated membrane increases were transient, we hypothesized that the puncta evident after dye washout represented endocytosed granules that had undergone exocytosis in the presence of FM 1-43 and could provide a measure of membrane exocytosed and recaptured during stimulation. As discussed above, this concept was supported by the complete reversal of evoked increases in membrane capacitance during continuous agonist exposure and was also in agreement with the coupling of exocytosis and endocytosis in neurons and neuroendocrine cells (23). To test this, we developed a technique to quantify the internalized puncta.
Quantifying internalized puncta
Our goal was to develop a simple algorithm that could be implemented with basic microscopy tools. Acquisition of the images used for analysis requires a microscope capable of performing serial sectioning with high resolution. Most laboratory microscopes equipped for epifluorescence measurements will suffice provided they have software control of z axis position and movement and are equipped with a high NA objective (Materials and Methods). Image analysis was performed using intrinsic functions available in the standard software package used for acquisition and analysis with our microscope and were easily incorporated into a macro for automated analysis. This allowed a typical experiment to be analyzed in <15 min with moderate user interaction.
The challenging task for image analysis was to identify sharply focused puncta present over diffuse fluorescence of varying magnitudes. We used a Laplacian (gradient) operation to help identify sharp transitions in fluorescence while ignoring gradual intensity transitions, a technique also insensitive to baseline fluorescence magnitudes (Materials and Methods; Figs. 3 B and 5 B). The symmetry of the Laplacian kernel gives greater weight to point changes over line changes, making it optimal for identifying puncta while showing decreased sensitivity to the more linear residual staining of LIS (21). The Laplacian operation can produce noise speckling in the image from minor, pixel-to-pixel gradients, however, requiring a smoothing filter to average these transitions. Noise speckling was particularly severe in confocal images (Fig. 5, A and B), where the absence of diffuse fluorescence exposes minor pixel-to-pixel variability, and two passes with the smoothing filter were used. Finally, conversion of lateral (x-y) data from pixels to meters required calibration of each objective used as described in Materials and Methods, and axial (z) dimensions were calibrated based on stepper motor performance.
Verification of measurements
There were two considerations to validate the data collected: did the image-processing algorithm accurately count internalized puncta, and were the number of puncta counted representative of the membrane trafficking expected during ATP stimulation. To address the first question, we compared data collected with both confocal and epifluorescence microscopy. As shown in Table 2, both serial sectioning techniques produced images that resulted in the same number of internalized particles under either basal or ATP-stimulated conditions. The confocal sections were acquired at half the z-step (0.45 μm) as the epifluorescence sections (1 μm), indicating that the larger spacing did not miss particles. In addition, the focal depth of the epifluorescence acquisition was measured as 3.25 μm, which raised the concern that puncta a micron above or below the acquisition plane might have enough intensity gradient to be tagged in more than one plane. Again, the similar values acquired with confocal sectioning, which closely approximates the theoretical axial resolution, eliminated this concern and confirmed the threshold values used for pixel tagging. The diameter of puncta is reduced in the confocal analysis compared to epifluorescence (Table 2), which probably reflects some loss due to the two smoothing operations used on confocal sections but may also reflect the improvement in lateral resolution achieved with the higher NA objective and 1 × 1 binning used for confocal acquisition (0.325 μm vs. 0.23 μm).
To assess whether the number of internalized puncta represented a measure of the granules involved in stimulated exocytosis, we compared the results against capacitance measurements. The image analysis identifies the two-dimensional area of internalized puncta, whereas capacitance measures their three-dimensional contribution to membrane increases. We assigned an equivalent capacitance to identified puncta by assuming they represented the cross section of spheres and calculated the spherical surface area from the puncta diameter. In the low density cells (Table 1), the equivalent capacitance internalized is of the same order reported for ATP-stimulated capacitance changes due to exocytosis (11,12). Since it is not known whether basal mucin secretion continues during stimulated secretion (4), we compared equivalent capacitance changes for both cases (Fig. 4). Although both cases produce changes in the range observed with patch clamping, the case for ATP-dominated secretion produced a change nearly identical to the mean value reported with patch clamping.
In the high density monolayers, impedance analysis had measured an ATP-stimulated 1-μF increase in capacitance per cm2 of filter area within 3 min (13). Recovery back to baseline capacitance was much slower than observed in single cells: 5 min after ATP addition, monolayer capacitance had dropped 12% from its peak value, or 0.12 μF per cm2 of filter area. The monolayers in this study were exposed to ATP + FM 1-43 for 5 min before washout, and granules endocytosed after washout are not labeled. We expected the FM 1-43 analysis to identify internalized membrane equivalent to 0.12 μF per cm2 of filter area. From Table 2, the effective cross section of a cell on a filter was 63 μm2. This predicts an ATP-stimulated change in equivalent capacitance per cell of 0.076 pF (0.12 μF/cm2 × 63 μm2/cell), the same magnitude predicted from FM 1-43 (Table 2). Thus FM 1-43 labeled, internalized puncta represent a reasonable measure of membrane trafficked in both low density cells and intact monolayers.
Differences in puncta internalized
It was clear from earlier electrophysiology studies in HT29-Cl.16E that monolayers had significant trafficking differences from single cells. Single cells exhibited a larger capacitance increase (per cell) and a significantly faster response and recovery to ATP stimulation than monolayers did (11,13). Because the monolayer response reflected the average of a million cells, it was not clear whether the slower timing and smaller capacitance change in response to ATP was due to a staggered response among cells behaving as observed during patch clamping, or whether the response time of individual cells was significantly slower with a smaller magnitude of secretion. Our preliminary results support a smaller and slower response in individual cells as their neighbors increase in number. The role of density on response is not yet clear: cell-cell contact and gap junction communication may play a role, as well as agonist access and available surface area for granule docking and fusion. Clearly as the cells grow denser and potentially respond to contact inhibition, the effective membrane presented to the apical bath would appear to shrink considerably (Tables 1–3). A surprising finding was that the monolayer behavior was adequately duplicated by high density cell clusters grown on glass coverslips (Table 3), suggesting that a barrier between apical and basolateral membranes was not the determining factor for the observed difference in response. This finding suggests that high density cells can achieve a similar differentiation status with respect to mucin secretion as cells polarized on filters (Fig. 7).
These differences between cell configurations raise questions concerning the validity of extending an observation in single cells to the polarized epithelium but may also be exploited in advancing our knowledge of disease states. For example, the relatively high rates of basal secretion in the single cell and low density clusters are not present in the high density cells (Tables 1–3), raising the question of whether basal secretion is a significant factor in this epithelium. Interestingly, recent studies in subclones of the HT29 family have correlated their mucin secretory properties with invasiveness and drug resistance, identifying them as models of human colon carcinoma (25,26). Disruption of cell polarity is receiving attention as a promoter of malignancy (9,15), and the polarity disruption that precedes cell plating for whole cell patch clamping and propagation may temporarily induce the same state observed in these carcinoma models. In addition, it is well known that significant increases in mucin secretion occur during the inflammatory response to infection (27) and chronic obstructive lung diseases (28), and thus the single cell model may provide clues about the in vivo epithelium's response to infection. The ability to use the same functional assay across different epithelial cell configurations is an asset in studying the regulation of these poorly understood phenomena and the manner in which they adapt during differentiation.
Choice of staining methods
The ability to perform FM 1-43 staining offline presented several advantages, such as allowing high resolution imaging of filter-grown monolayers. A benefit to perfused staining was that the cells were continuously washed during the destaining step. Our experiments suggested that coverslipped cells, which did not inhibit the flow of dye into LIS (Fig. 6), benefited from continuous perfusion during the washout phase to reverse the dye permeation into the LIS. If dye exposure can be limited to the apical face of the membrane, as is the case with filter-grown monolayers, then the problem of LIS dye permeation no longer exists.
CONCLUSIONS AND GENERAL APPLICABILITY OF THE METHOD
We have provided a detailed technique for quantifying membrane internalized during endocytosis in epithelia at variable stages of monolayer development and differentiation, and we have demonstrated the accuracy of the analysis and validity of the method as a predictor of granules involved in stimulated exocytosis and retrieval in HT29-Cl.16E epithelia. These cells exhibit tight coupling between exocytosis and endocytosis and internalized particle diameters in the 500-nm range, comparable to earlier EM studies of granules in this cell line (18). To use the method as a predictor of exocytosis requires that cells exhibit compensatory endocytosis; however, as shown for neurons and neuroendocrine cells (23), these two processes are most likely coordinated in all secretory cells to regulate cell membrane area. The primary question is most likely not if cells retrieve membrane inserted through exocytosis but when, and timing is an easily adjusted parameter in this assay.
Beyond measurements of stimulated exocytosis, evidence suggests many cells undergo rapid recycling and internalization of membrane (29). The primary concern with measurement of internalized membrane then becomes size limitations: the minimum particle size that can be accurately detected is a function of NA, ∼225 nm in diameter with optimal components. This is a limitation of light microscopy in general, and clearly the area of a 100-nm vesicle will not be accurately reported by this method. If the analysis identifies numerous puncta of a single pixel in area, the user should suspect that these puncta could be smaller than the microscope resolution and take appropriate precautions. Vesicles are expected to fuse with endosomes of the sorting and recycling paths, however, and recent reports have suggested that these compartments are within the size limitations of light microscopy (30,31) at 200–750 nm, amenable to investigation with this FM 1-43 method.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
Algorithms are available for download from the Department of Cell Biology and Physiology, University of Pittsburgh at http://www.pitt.edu/∼cbertra/.
We thank Dr. Michael Butterworth for assistance with confocal microscopy, Bill Burnip for helpful discussions on SimplePCI imaging software, and Matthew Green, Maitrayee Sahu, and Sharawnda Smith for excellent technical assistance.
Resources providing partial support for this work included Cystic Fibrosis Foundation grants BERTRA00F0 (to C.A.B.) and HOPFER99P0 (to U.H.), and National Institutes of Health grants DK07678 (to U.H.), DK54774 (to R.J.B.), DK68196 (to R.A.F), and DK72506 (to R.A.F.).
R. J. Bridges's present address is Dept. of Physiology and Biophysics, Rosalind Franklin University of Medicine and Science, North Chicago, IL.
References
- 1.Bertrand, C. A., and R. A. Frizzell. 2003. The role of regulated CFTR trafficking in epithelial secretion. Am. J. Physiol. 285:C1–C8. [DOI] [PubMed] [Google Scholar]
- 2.Jentsch, T. J., T. Maritzen, and A. A. Zdebik. 2005. Chloride channel diseases resulting from impaired transepithelial transport or vesicular function. J. Clin. Invest. 115:2039–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stein, M.-P., A. Wandinger-Ness, and T. Roitbak. 2002. Altered trafficking and epithelial cell polarity in disease. Trends Cell Biol. 12:374–381. [DOI] [PubMed] [Google Scholar]
- 4.Forstner, G. 1995. Signal transduction, packaging and secretion of mucins. Annu. Rev. Physiol. 57:585–605. [DOI] [PubMed] [Google Scholar]
- 5.Angleson, J. K., and W. J. Betz. 1997. Monitoring secretion in real time: capacitance, amperometry and fluorescence compared. Trends Neurosci. 20:281–287. [DOI] [PubMed] [Google Scholar]
- 6.Hille, B., J. Billiard, D. F. Babcock, T. Nguyen, and D.-S. Koh. 1999. Stimulation of exocytosis without a calcium signal. J. Physiol. (Lond.). 520:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braga, V. M. M., and A. S. Yap. 2005. The challenges of abundance: epithelial junctions and small GTPase signalling. Curr. Opin. Cell Biol. 17:466–474. [DOI] [PubMed] [Google Scholar]
- 8.Gilcrease, M. Z. Integrin signaling in epithelial cells. Cancer Lett. In press. [DOI] [PubMed]
- 9.D'Souza-Schorey, C. 2005. Disassembling adherens junctions: breaking up is hard to do. Trends Cell Biol. 15:19–26. [DOI] [PubMed] [Google Scholar]
- 10.Korbmacher, C., A. S. Segal, G. Fejes-Toth, G. Giebisch, and E. L. Boulpaep. 1993. Whole-cell currents in single and confluent M-1 mouse cortical collecting duct cells. J. Gen. Physiol. 102:761–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bertrand, C. A., H. Danahay, C. T. Poll, C. Laboisse, U. Hopfer, and R. J. Bridges. 2004. Niflumic acid inhibits ATP-stimulated exocytosis in a mucin-secreting epithelial cell line. Am. J. Physiol. 286:C247–C255. [DOI] [PubMed] [Google Scholar]
- 12.Guo, X. W., D. Merlin, C. Laboisse, and U. Hopfer. 1997. Purinergic agonists, but not cAMP, stimulate coupled granule fusion and Cl-conductance in HT29–C1.16E. Am. J. Physiol. 273:C804–C809. [DOI] [PubMed] [Google Scholar]
- 13.Bertrand, C. A., C. L. Laboisse, and U. Hopfer. 1999. Purinergic and cholinergic agonists induce exocytosis from the same granule pool in HT29-Cl.16E monolayers. Am. J. Physiol. 276:C907–C914. [DOI] [PubMed] [Google Scholar]
- 14.Bertrand, C. A., D. M. Durand, G. M. Saidel, C. Laboisse, and U. Hopfer. 1998. System for dynamic measurements of membrane capacitance in intact epithelial monolayers. Biophys. J. 75:2743–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bilder, D. 2004. Epithelial polarity and proliferation control: links from the Drosophila neoplastic tumor suppressors. Genes Dev. 18:1909–1925. [DOI] [PubMed] [Google Scholar]
- 16.Cochilla, A. J., J. K. Angleson, and W. J. Betz. 1999. Monitoring secretory membrane with FM 1-43 fluorescence. Annu. Rev. Neurosci. 22:1–10. [DOI] [PubMed] [Google Scholar]
- 17.Augeron, C., and C. L. Laboisse. 1984. Emergence of permanently differentiated cell clones in a human colonic cancer cell line in culture after treatment with sodium butyrate. Cancer Res. 44:3961–3969. [PubMed] [Google Scholar]
- 18.Merlin, D., C. Augeron, X.-Y. Tien, X. Guo, C. L. Laboisse, and U. Hopfer. 1994. ATP-stimulated electrolyte and mucin secretion in the human intestinal goblet cell line HT29-Cl.16E. J. Membr. Biol. 137:137–149. [DOI] [PubMed] [Google Scholar]
- 19.Inoué, S. 1995. Foundations of confocal scanned imaging in light microscopy. In Handbook of Biological Confocal Microscopy. J. B. Pawley, editor. Plenum Press, New York. 1–17.
- 20.Shaw, P. J. 1995. Comparison of wide-field/deconvolution and confocal microscopy for 3D imaging. In Handbook of Biological Confocal Microscopy. J. B. Pawley, editor. Plenum Press, New York. 373–387.
- 21.Russ, J. C. 1999. The Image Processing Handbook. CRC Press, Boca Raton, FL.
- 22.Smith, C. B., and W. J. Betz. 1996. Simultaneous independent measurement of endocytosis and exocytosis. Nature. 380:531–534. [DOI] [PubMed] [Google Scholar]
- 23.Gundelfinger, E. D., M. M. Kessels, and B. Qualmann. 2003. Temporal and spatial coordination of exocytosis and endocytosis. Nat. Rev. Mol. Cell Biol. 4:127–139. [DOI] [PubMed] [Google Scholar]
- 24.Brumback, A. C., J. L. Lieber, J. K. Angleson, and W. J. Betz. 2004. Using FM1-43 to study neuropeptide granule dynamics and exocytosis. Methods. 33:287–294. [DOI] [PubMed] [Google Scholar]
- 25.Leteurtre, E., V. Gouyer, K. Rousseau, O. Moreau, A. Barbat, D. Swallow, G. Huet, and T. Lesuffleur. 2004. Differential mucin expression in colon carcinoma HT-29 clones with variable resistance to 5-fluorouracil and methotrexate. Biol. Cell. 96:145–151. [DOI] [PubMed] [Google Scholar]
- 26.Truant, S., E. Bruyneel, V. Gouyer, O. D. Wever, F.-R. Pruvot, M. Mareel, and G. Huet. 2003. Requirement of both mucins and proteoglycans in cell-cell dissociation and invasiveness of colon carcinoma HT-29 cells. Int. J. Cancer. 104:683–694. [DOI] [PubMed] [Google Scholar]
- 27.Jobin, C., and R. B. Sartor. 2000. The IkB/NF-kB system: a key determinant of mucosal inflammation and protection. Am. J. Physiol. 278:C451–C462. [DOI] [PubMed] [Google Scholar]
- 28.Perez-Vilar, J., J. K. Sheehan, and S. H. Randell. 2003. Making more MUCS. Am. J. Respir. Cell Mol. Biol. 28:267–270. [DOI] [PubMed] [Google Scholar]
- 29.Hao, M., and F. R. Maxfield. 2000. Characterization of rapid membrane internalization and recycling. J. Biol. Chem. 275:15279–15286. [DOI] [PubMed] [Google Scholar]
- 30.Ganley, I. G., K. Carroll, L. Bittova, and S. Pfeffer. 2004. Rab9 GTPase regulates late endosome size and requires effector interaction for its stability. Mol. Biol. Cell. 15:5420–5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haas, A. K., E. Fuchs, R. Kopajtich, and F. A. Barr. 2005. A GTPase-activating protein controls Rab5 function in endocytic trafficking. Nat. Cell Biol. 7:887–893. [DOI] [PubMed] [Google Scholar]