

Abstract
LOV domains are the light-sensitive portion of plant phototropins. They absorb light through a flavin cofactor, photochemically form a covalent bond between the chromophore and a cysteine residue in the protein, and proceed to mediate activation of an attached kinase domain. Although the photoreaction itself is now well-characterized experimentally and computationally, it is still unclear how the formation of the adduct leads to kinase activation. We have performed molecular dynamics simulations on the LOV1 domain of Chlamydomonas reinhardtii and the LOV2 domain of Avena sativa, both before and after the photoreaction, to answer this question. The extensive simulations, over 240 ns in duration, reveal significant differences in how the LOV1 and LOV2 domains respond to photoactivation. The simulations indicate that LOV1 activation is likely caused by a change in hydrogen bonding between protein and ligand that destabilizes a highly conserved salt bridge, whereas LOV2 activation seems to result from a change in the flexibility of a set of protein loops. Results of electrostatics calculations, principal component analysis, sequence alignments, and root mean-square deviation analysis corroborate the above findings.
INTRODUCTION
For plants to thrive in a changing light environment, they sense and respond to light. The sensory information allows them, for example, to grow toward light, regulate chloroplast positioning, and open or close their stomata. This allows plants to use as much light as possible while avoiding damage and dehydration (1). These responses are mediated by phototropins, photosensory proteins consisting of a serine-threonine kinase domain and a pair of nonidentical Light, Oxygen, or Voltage (LOV) sensitive domains (2) which contain the noncovalently bound chromophore flavin mononucleotide (FMN) (3). These domains are members of the Per/Arnt/Sim family (4). Fig. 1 gives an overview of the LOV domain, with portions important to this study highlighted.
FIGURE 1.

LOV1 domain from C. reinhardtii. Shown is the LOV1 protein in cartoon representation with the FMN chromophore and associated cysteine residue shown in licorice representation in the center. A highly conserved salt bridge between E51 and K92, and several important structural elements highlighted in this article, are labeled. The approximate location of the Jα helix (as suggested in (1)) is shown schematically as a gray rectangle.
The photocycle of the phototropin LOV domains has been well-characterized experimentally. After excitation of the chromophore into an excited singlet state, the FMN undergoes fast intersystem crossing into an excited triplet state, followed on a microsecond timescale by formation of a covalent flavin-cysteinyl adduct between a carbon atom of the flavin ring of FMN and a neighboring cysteine residue. The state with the adduct formed constitutes the active form of the LOV domain which activates an attached kinase domain for downstream signaling through an unknown mechanism; this state is only metastable since it decays via breakage of the sulfur-carbon bond on a timescale of minutes to hours, thereby returning the LOV domain to its inactive ground state (2,5,6). Throughout this article we refer to the LOV domain's ground state as the “dark” state, and to its photoactivated state as the “light” state.
Although recent spectroscopic data (7,8), in tandem with quantum mechanical studies (9,10), have yielded a wealth of information on the LOV photoreaction itself, the mechanism by which the spatially localized formation of a bond between a cysteine residue and the chromophore leads to kinase activation remains unknown. X-ray crystal structures have been determined for both the dark and light states of phototropin LOV domains from the fern Adiantum capillus-veneris and the green algae Chlamydomonas reinhardtii, but they have yielded little information on potential activation mechanisms because there is no obvious structural difference between the light and dark states in these crystal structures (4,11). This led to speculation that the changes involved in LOV domain function are actually dynamic in nature (4), arising from the mobility of secondary and tertiary structural elements. This possibility makes the LOV domain a particularly well-suited target for molecular dynamics (MD) simulations, which have previously been employed for understanding such issues as the mechanism of signal transduction in G-proteins (12) and the activation of cAMP-dependent protein kinase (13).
It has been hypothesized that activation arises as a result of the disruption of a salt bridge formed between two loops in the light state, but this remains speculation since the salt bridge is present in both light and dark state crystal structures (14). The salt bridge in question can be seen in Fig. 1; it occurs between E51 and K92 in LOV1, and between E960 and K1001 in LOV2. NMR studies on LOV2 have shown that the light and dark states may differ in the stability of a helical region, Jα (see Fig. 1), positioned next to the β-sheet in the LOV domain, but it remains unclear both how the photoreaction may cause this change and how the change may trigger kinase activation (1).
To investigate how photoadduct formation is coupled to kinase activation, we performed MD simulations on both the dark and light states of the LOV1 domain from C. reinhardtii and the LOV2 domain from Avena sativa. Our results indicate that despite the apparent structural similarities between the light and dark state crystal structures for these LOV domains, there are significant differences in the dynamic properties between these two states, which can explain how the light state triggers kinase activation. Interestingly, these dynamic changes are found to be different in the LOV1 and the LOV2 domain.
METHODS
Molecular dynamics simulations
We obtained all-atom crystal structures for the dark and light states of the LOV domains from the Protein Data Bank (15) (PDB codes 1N9L/1N9O for LOV1 (11) and 1G28/1JNU for LOV2 (4,16). These structures served as starting points for molecular dynamics (MD) simulations. For the purpose of these simulations each protein was solvated in a box of TIP3P water and its charge neutralized by adding sodium ions, giving a total system size of 48,576/48,920 atoms for the dark/light states of LOV1, and 20,903/20,918 atoms for the dark/light states of LOV2, respectively. We used a minimal number of neutralizing ions, rather than physiological strength solutions, to avoid ion-based artifacts (17,18) and to avoid making undue assumptions about the physiological environment immediately surrounding the LOV domain; we do not expect this choice to qualitatively affect our results (17). The CHARMM force-field parameters for FMN and the FMN-cysteine adduct were developed de novo; a complete description of this procedure, along with the resulting parameter set, is included in Supplementary Material.
For the light and dark states of both proteins, we performed five separate 12-ns NPT ensemble simulations using the NAMD molecular dynamics package (19) at a temperature of 298 K and pressure of 1 atm, for a total of 240 ns. Our simulations used CHARMM27 (20) parameters, supplemented by our FMN force-field parameters as described above. The simulations were carried out with a 1-fs timestep, and full electrostatics calculated every two steps using the PME method (19) with a 96 × 96 × 96 Å point grid. Each simulation began from identical starting coordinates; all variations between simulations occurred due to randomized starting velocities (at 298 K) and subsequent random effects of the thermostat (19).
Sequence alignment
All sequence alignments noted were performed using CLUSTALX software (21), with Gonnet series scoring matrices and default settings. Phototropins and homologous proteins were identified via a BlastP search on the A. capillus-veneris sequence and several were selected for alignment; this set included SWISS-PROT accession numbers Q6BCT7, Q6BCT8, Q6BCU0, Q6BCU1, Q5DW42, Q8H934, Q5DW43, Q5DW44, Q8H935, Q765V9, Q9MB43, Q401Q4, and Q401Q5. Residues noted as “conserved” were perfectly conserved across this set unless otherwise specified.
Principal component analysis
To further analyze the overall motions of the LOV domain captured by our simulations, we performed principal component analysis (PCA) on the Cα atoms of the LOV domains for each of our four trajectories (22,23). PCA reveals high-amplitude concerted motions present in the analyzed trajectory, based on the eigenvectors of the covariance matrix; vectors with the largest eigenvalues correspond to the most significant collective motions. This method allows one to analyze the significant features of the trajectories and filter out random, unimportant fluctuations (13). In our analysis we include the three leading PCA modes. PCA was performed using the g_anaeig and g_covar modules of GROMACS 3.3 (24); in the case of the LOV1 dark state the N-terminal residue (G17) was omitted since this residue is missing in the light state crystal structure.
Electrostatics calculations
The electrostatic potential around the LOV domain was calculated employing the APBS software package (25), with a 0.31 Å grid spacing, protein dielectric constant of 1.0, solvent dielectric of 78.54, and mobile ions present at a concentration of 150 mM. Each electrostatic potential map presented in our study was obtained by averaging results from 50 distinct frames of our trajectories, chosen randomly from the set of frames where the distance between the E51 oxygens and K92 nitrogens was >8 Å (in the case of the LOV1 dark state) or <3.2 Å (for the LOV1 light state). This constraint was imposed to compare the full effects of formation or breakage of the E-K salt bridge and (particularly for the dark state) avoid frames where the salt-bridging partners were still in close proximity; overall, 42.5% of dark state frames and 51.6% of light state frames satisfied these criteria. In the case of the LOV2 domain, 50 frames were chosen at random without further filtering due to the absence of major differences in the E-K salt bridge of LOV2 between the light and dark states.
RESULTS AND DISCUSSION
All four simulated systems (LOV1 dark, LOV1 light, LOV2 dark, and LOV2 light) proved to be dynamically stable throughout our simulations, and in none of them were significant changes in the equilibrium structures observed over 60 ns. The Cα root mean-square deviation (RMSD) with respect to the initial state, through the simulations, was 1.41 Å/1.08 Å for the dark/light states of LOV1 and 1.12 Å/1.17 Å for the dark/light states of LOV2, respectively. Interestingly, the Cα-RMS deviation between the final frame of the light and dark state simulations showed only a 1.51 Å difference for LOV1 and 1.00 Å for LOV2, in accord with the finding from x-ray crystallography that the states are structurally very similar.
The picture changes, however, when dynamic properties are considered. Fig. 2 shows the time-averaged structures of dark and light-activated LOV1 and LOV2 domains with residues colored along a scale from blue (low mobility) to red (high mobility) according to their RMSD value. The regions of highest mobility, corresponding to residues whose RMSD is >2 SD above the mean, are highlighted in Fig. 2 with spheres centered on their Cα atoms. It is apparent that, although the mobility of most parts of the protein is similar in each case, certain regions differ greatly between the dark and light states in the extent of their motion.
FIGURE 2.

Cα-RMSD of the LOV domain resulting from our simulations. Cartoon representations are shown for each of the four protein states investigated. Colors are assigned in each case from blue (low mobility) to red (high mobility), with the color scale shown at the bottom indicating the RMSD in Å. Residues with RMSD >2 SD above the mean are highlighted with spheres.
In the case of LOV1, the difference between the light and dark state is particularly apparent in the Gβ-Hβ loop (residues 91–95), which is involved in the highly conserved so-called E-K salt bridge between residue E51 and residue K92. There is significant mobility in this loop in the LOV1 dark state, but not in the light state; the average RMSD of residues in the Gβ-Hβ loop of LOV1 is 2.4 Å in the dark state and 1.2 Å in the light state. Neither LOV2 simulation shows any significant motion in this loop region. The E-K salt bridge of the LOV domain has been hypothesized to break during photoactivation (14), but there is, as yet, no conclusive evidence that this actually occurs; previous molecular dynamics studies on the LOV2 domain showed no difference in this salt bridge between the dark and light states (26), in agreement with our findings.
Our simulations also show that there is another region of significant mobility in the LOV1 domain consisting of a loop formed by residues 27–32 (located on the center-right of the protein as oriented in Fig. 2), which shows significant motion in both the light and dark states. In this particular case, however, we could not find distinguishable differences in behavior between the light and dark states.
For the LOV2 domains, the primary region of high mobility in both the light and dark states is within a loop connecting two strands of the β-sheet region of the protein, the Hβ-Iβ loop (see Fig. 2). This loop has an average Cα-RMSD of 2.8 Å in the dark state and 2.7 Å in the light state. Interestingly, this loop is directly adjacent to the Jα helix, whose unfolding has been linked in NMR studies to LOV2 domain activation (1). In all four simulations the N-terminus also showed significant motion, but this is likely due to the fact that this region is relatively unconstrained due to missing adjacent portions of the protein.
Principal component analysis of LOV domain motions
To identify the most important modes of concerted motion in our trajectories, we performed PCA on the Cα atoms of all trajectories in our study. The modes corresponding to the three largest eigenvalues for each simulation (see Methods) are presented in Fig. 3; the relative magnitudes of these modes are included as Supplementary Material. In both LOV1 and LOV2, the motions corresponding to the three leading modes are concentrated in regions that also show the largest RMSD deviations (see above). The highest amplitude motions in the LOV1 dark state include both a mode of flexing in the Gβ-Hβ loop and a concerted opening/closing motion between the Gβ-Hβ loop and the loop containing E51. These modes exhibit small PCA eigenvalues in the LOV1 light state. In the case of LOV2, the most important modes involve flexing of the Hβ-Iβ loop, primarily in a direction in the plane of the main β-sheet. All four states investigated also show at least one major mode involving bending of the loop connecting the Aβ and Bβ strands (see Fig. 1); however, in no case were we able to observe a substantial difference in the conformation of this loop or contacts it makes with nearby structural regions between simulations. The PCA data corroborate the suggestion from our RMSD analysis that the most significant motions in the LOV domain are localized to a few loops.
FIGURE 3.

Porcupine plots (38) of the three largest PCA modes. For each of the four states in this study the modes are colored blue, green, and red in order of decreasing significance.
Salt-bridge formation in LOV1 and LOV2
Our findings on the mobility of the Gβ-Hβ loop prompted us to further investigate the temporal evolution of the contained E-K salt bridge over the course of our dynamics runs. We found that, contrary to previously published hypotheses on activation (14), the salt bridge is very well preserved in the LOV1 light state, but often broken in the dark state. For LOV2, we saw no significant difference in salt bridge behavior between the light and dark states. Table 1 lists the average occupancy of different states of the E-K salt bridge in our simulations. The data indicate that in LOV1, the E-K salt bridge is significantly more prevalent in the light state, being broken only 21.5% of the time as opposed to 79.7% in the dark state. In the case of LOV2, we find that there is no difference in E-K salt bridging between the dark and light states (it is formed >90% of the time in both cases), in agreement with MD data from Neiss and Saalfranck (26).
TABLE 1.
Percentage of time the E-K salt bridge is bidentate, monodentate, or broken in each of the 20 simulations performed in this study
| Bidentate | Salt-bridge status Monodentate | Broken | |
|---|---|---|---|
| LOV1 dark | |||
| Simulation 1 | 4.3% | 4.5% | 91.3% |
| Simulation 2 | 1.7% | 2.1% | 96.3% |
| Simulation 3 | 15.8% | 12.0% | 72.4% |
| Simulation 4 | 34.2% | 15.2% | 50.6% |
| Simulation 5 | 6.8% | 5.2% | 87.9% |
| Total | 12.6% | 7.8% | 79.7% |
| LOV1 light | |||
| Simulation 1 | 61.1% | 35.5% | 3.4% |
| Simulation 2 | 64.1% | 23.8% | 12.1% |
| Simulation 3 | 66.0% | 32.4% | 1.6% |
| Simulation 4 | 15.8% | 25.7% | 58.4% |
| Simulation 5 | 50.9% | 17.0% | 32.1% |
| Total | 51.6% | 26.9% | 21.5% |
| LOV2 dark | |||
| Simulation 1 | 56.6% | 19.5% | 24.0% |
| Simulation 2 | 40.5% | 58.1% | 1.4% |
| Simulation 3 | 59.1% | 40.5% | 0.3% |
| Simulation 4 | 52.7% | 22.1% | 25.2% |
| Simulation 5 | 67.0% | 32.4% | 0.5% |
| Total | 55.2% | 34.5% | 10.3% |
| LOV2 light | |||
| Simulation 1 | 73.4% | 26.4% | 0.2% |
| Simulation 2 | 37.1% | 27.1% | 35.8% |
| Simulation 3 | 72.9% | 27.0% | 0.1% |
| Simulation 4 | 72.8% | 27.2% | 0.1% |
| Simulation 5 | 61.3% | 37.3% | 1.4% |
| Total | 63.5% | 29.0% | 7.5% |
A contact is defined as formed if the lysine nitrogen-glutamate oxygen distance is <3.2 Å; the time step is labeled bidentate if both glutamate oxygens are making contact, monodentate if only one is, and broken if neither is.
Interestingly, the LOV1 and LOV2 sequences differ in the residues immediately adjacent to the lysine residue (K92/K1001 in LOV1/LOV2) of the E-K salt bridge: residue 93 is an aspartate in all LOV1 sequences, but residue 1002 is a serine or threonine in all LOV2 sequences examined. In our LOV1 dark state simulations, these adjacent lysine and aspartate residues formed a salt bridge (bidentate or monodentate) in 10% of all timesteps, and their charged groups were within 5 Å of each other in 20% of all timesteps. In the LOV1 light state, in contrast, the groups were never within 5 Å of each other due to the stability of the canonical E51-K92 salt bridge. The relevant segments are shown in conformations with either the E51-K92 or K92-D93 salt bridge formed in Fig. 4. It seems likely that the absence of a change of the E-K salt bridge in LOV2 is at least partly due to the absence of a negatively charged residue adjacent to the lysine; in LOV1, this negatively charged residue competes with E51 to form the salt bridge with K92, whereas in LOV2 no equivalent competitive residue exists. In both the light and dark states of LOV1, D93 also interacts with the side chain of T95 and with the main-chain nitrogens of G94 and T95 (both conserved throughout the LOV1 domains in our alignment).
FIGURE 4.

Conformation of the Gβ-Hβ loop in the LOV1 dark (left) and light (right) states. A representative timestep was chosen for each case.
As seen in Fig. 2, the loop containing K92 is much more mobile in the LOV1 dark state than in any of the other systems simulated (the average Cα-RMSD of this loop was 2.1 Å, whereas it was no higher than 1.1 Å for the other models). Furthermore, this loop takes on a slightly different conformation in the LOV1 dark state, with D93 and G94 tilted away from the rest of the protein (see Fig. 4). The LOV1 dark state conformation of this loop also appears to disfavor interaction between D93 and T95; this interaction was present in 24% of timesteps in the LOV1 dark state simulations and 97% of timesteps in the LOV1 light state simulations.
In view of the changes observed in the E-K salt bridge in LOV1, we performed electrostatic calculations using APBS to observe how this salt bridge affects the electrostatic potential surrounding the protein. The resulting potentials are shown in Fig. 5. In both the dark and light states a region of negative electrostatic potential is present on the surface opposite the β-sheet (OBS). As seen in the difference map (Fig. 5 d) between these states, the field in this region is reduced in magnitude in the LOV1 light state, due to a conformational change of E63 and D50 as well as reorientation of E51 to get into close contact with K92. Interestingly, the LOV2 domain exhibits only a region of weak negative potential (less strong than the LOV1 light state) in both the light and dark states (data not shown).
FIGURE 5.

Electrostatic potential maps of the LOV1 domain, calculated using APBS. (A) Snapshot of the LOV1 dark state oriented identically to the electrostatic surfaces. (B,C) Electrostatic potential surfaces for the LOV1 dark and light states. Isosurfaces at 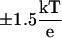 are shown in blue and red. (D) Difference of light- and dark-state potentials. A negative potential on this map indicates a region where the light-state potential is less positive than that of the dark state. Isosurfaces are shown at
are shown in blue and red. (D) Difference of light- and dark-state potentials. A negative potential on this map indicates a region where the light-state potential is less positive than that of the dark state. Isosurfaces are shown at 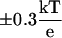 .
.
The electrostatic characteristics on the OBS surface of LOV1 and loops surrounding it suggest how the E-K salt bridge in the LOV1 light state can modulate LOV domain-kinase interaction, either by disrupting an interaction between the kinase and the negatively charged OBS surface or by enabling a strong kinase-LOV1 interaction with emerging charged regions in the loops. In the case of LOV2, aside from changes in average positioning (especially of the Hβ-Iβ loop) no significant differences were observed between the electrostatic potential of dark and light states.
Mobility of the Hβ-Iβ loop
The LOV2 domain shows significant motion of the Hβ-Iβ loop (residues 1017–1022; see Figs. 2 and 3) in both the light and dark states, with Cα-RMSDs >2.7 Å for both states, compared with <1.4 Å in LOV1. Moreover, in LOV2 this loop took on two distinct conformations in our simulations. In the more prevalent state (existing ∼71% of the time) the Hβ-Iβ loop curves toward helix Fα, whereas in the alternate state it flips away from Fα; we will call these two loop conformations HIL and HID, respectively. The net distance moved by the Cα atom of N1019 between these states is up to 10 Å (see Figs. 6 and 7). These motions are roughly perpendicular to the orientation of the putative location of the Jα helix and may be quantified by defining a function
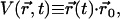 |
(1) |
where  is the relative position of the Cα atom of residue N1019 with respect to its starting position at a given time t, and
is the relative position of the Cα atom of residue N1019 with respect to its starting position at a given time t, and 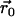 is the vector separating the extremal positions of N1019-Cα observed during the simulations. Using this measure, the HIL conformation noted above corresponds to positions where
is the vector separating the extremal positions of N1019-Cα observed during the simulations. Using this measure, the HIL conformation noted above corresponds to positions where 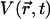 oscillates around −2.5 Å2, and the HID conformation corresponds to positions oscillating around 2.5 Å2 (see Fig. 6). Using a threshold of V = 0, the HID conformation occurs in 48.8% of timesteps in the dark state, but only in 11.9% of steps in the light state. In LOV1, only the former conformation, HIL, is significantly populated (data not shown).
oscillates around −2.5 Å2, and the HID conformation corresponds to positions oscillating around 2.5 Å2 (see Fig. 6). Using a threshold of V = 0, the HID conformation occurs in 48.8% of timesteps in the dark state, but only in 11.9% of steps in the light state. In LOV1, only the former conformation, HIL, is significantly populated (data not shown).
FIGURE 6.

Time dependence of 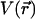 (as defined in the text) during LOV2 dark (blue) and light (orange) state simulations. Dashed lines indicate conformations HID and HIL, introduced in the text.
(as defined in the text) during LOV2 dark (blue) and light (orange) state simulations. Dashed lines indicate conformations HID and HIL, introduced in the text.
FIGURE 7.

Movement of the Hβ-Iβ loop in the dark and light states of LOV2. (A) Location of the loop in the LOV domain, with charged residues labeled. (B) Side view of the loop shown in the light and dark states at timesteps of their farthest movement; the HID conformation is shown in dark gray and the HIL conformation in light gray.
Loss of hydrogen bonding in the light state
Previous QM/MM (10) and spectroscopic studies (27) on the LOV domain indicated light-induced changes in the hydrogen-bonding network surrounding the flavin ring, involving residues N89/N998, N99/N1008, and Q120/Q1029 in LOV1/LOV2, respectively. In every crystal structure investigated, all three of these residues are involved in hydrogen bonding with the flavin ring of FMN. For example, in LOV1, N89 forms hydrogen bonds with FMN-O2 and FMN-N3, and both N99 and Q120 forms hydrogen bonds with FMN-O4 (see Fig. 8). These interactions are maintained throughout our simulations in the dark states of both LOV1 and LOV2, but undergo substantial changes in the light state in both cases. In the light states the interaction of the N99/1008 residue with FMN is either broken or exists only as a weak interaction between the side-chain nitrogen and the O4 atom of FMN. The hydrogen bond between Q120/1029 and O4 is present <35% of the time in the light state (see Fig. 8), and, in addition, a hydrogen bond occasionally forms between the side-chain oxygen of Q120 and FMN N5 (which is protonated in the light, but not the dark state); this interaction is present 54% of the time in LOV1 and 55% in LOV2. A summary of these hydrogen-bonding interactions can be found in Fig. 8. The third protein residue involved in hydrogen bonds with the flavin ring system, N89, does not appear to be affected by switching between light and dark states.
FIGURE 8.

Hydrogen bonding surrounding the flavin ring in the LOV1 dark (left) and light (right) states; in both cases a typical timestep is shown. Hydrogen-bonding distances for the displayed timestep are labeled in Å. Numbers in parenthesis underneath each residue denote the percentage of timesteps in which a given interaction was formed for that state; the value for LOV1 is given before the slash and for LOV2 after the slash.
The identity of the residues involved in the hydrogen-bonding network around FMN should be noted given the dynamic changes between the light and dark states detailed in the previous subsections: N99 and Q120 (using LOV1 numbering) are on the Hβ and Iβ strands, respectively, and thus will be mechanically coupled both to the Hβ-Iβ loop, seen to undergo a transition in LOV2, and to the loop containing the K92 residue involved in the E-K salt bridge discussed for LOV1.
A closer analysis of the changes in hydrogen-bonding pattern suggests a mechanism responsible for the broader structural transitions previously described in LOV1 and LOV2. In the case of LOV1, changes in the hydrogen bonding of N99 to FMN-O2 cause the asparagine residue to move outward from the binding pocket, occupying a position ∼0.8 Å closer to the Gβ-Hβ loop in the light state as compared to the dark state. At the same time, the adjacent side chain (F97) undergoes a 0.8 Å shift in the same direction, which in turn interacts with the backbone of residue Y90 (part of the Gβ-Hβ loop), possibly leading to the conformational change in this part of the protein as shown in Fig. 9 a. The implications of these changes will be discussed below.
FIGURE 9.

Effects of changing hydrogen-bonding patterns on LOV domain structure for LOV1 (A) and LOV2 (B); in both cases, dark-state conformations are shown in blue, and light-state in orange. All coordinates, except those of FMN, are averaged over the pertinent trajectories.
In the case of LOV2, the most significant motions associated with the changing hydrogen-bond pattern around FMN involve Q1029, which forms a hydrogen bond with FMN 84% of the time in the dark state, but only 4% in the light state; at the same time, the fraction of hydrogen bonding between the Q1029 oxygen and the side chain of S930 drops from 79.2% to 30.7%. This loss of hydrogen bonding causes Q1029 to instead occupy an alternate conformation (shown in Fig. 9 b) in which it bends further down toward the β-sheet, forming hydrogen-bonding interactions with the backbone of G1027 and V1028. At the same time, residues S930, G1027, and V1028 of the neighboring β-sheet undergo Cα-RMSD increases of 0.25 Å between the dark and light states of LOV2. The instability of this region likely contributes to the overall increase in mobility in the β-sheet of the light state of LOV2, which exhibits an increase in Cα-RMSD of 0.12 Å between dark and light state (in LOV1, the RMSD of the β-sheet drops by 0.03 Å after the photoreaction).
Comparison of LOV1 and LOV2
Experimental studies have clearly illustrated that the LOV1 and LOV2 domains play different roles in triggering phototropin activation, with the LOV2 domain responsible for the majority of kinase activation and the LOV1 domain accounting for a low level of kinase activation and some other, unknown function, possibly phototropin dimerization (28,29). Given this difference in function, it is not surprising—and indeed, it should be expected—to find different structural responses to the photoreaction in LOV1 and LOV2 (30).
The results of our simulations indicate that the most significant structural change in the LOV1 domain after photoactivation is formation of the conserved E51-K92 salt bridge, which is mostly broken in the dark state. Our finding that the state of this salt bridge does not significantly alter the overall structure of this LOV domain is consistent with experimental results on an E→Q mutant (equivalent to an E51Q mutation in C. reinhartii LOV1) (31) and spectroscopic findings that there are no major backbone rearrangements in LOV1 upon photoexcitation (30). At the very least, the change in salt bridging alters the surface characteristics and electrostatics of this region of the LOV1 domain. Obviously, such alteration can cause dimerization of phototropin or kinase regulation.
Our results on the LOV2 domain, in contrast, indicate that the E-K salt bridge does not undergo changes between dark and light states in this system. Instead, we observe a significant mobility in the Hβ-Iβ loop of the protein, with conformational switching between dark and light states. Experimental studies have demonstrated that dissociation of the Jα helix from the LOV2 domain is linked to phototropin kinase activation (1); suggestions on how the photoreaction leads to Jα dissociation, in general, have focused on changes to the β-sheet structure of the domain transduced by the hydrogen bonding of Q1029 with the FMN ligand (27,31,32). Our simulations, in contrast, do not reveal any essential changes in this β-sheet region between the light and dark states; instead, our results suggest that the dominant conformational change is in the Hβ-Iβ loop.
Possible mechanisms triggering LOV domain activation
Based on the results reported, one may speculate on how photoreaction in the LOV domains leads to kinase activation. In the case of LOV1, with its E51-K92 salt bridge broken in the dark state but formed in the light state, PCA and electrostatics calculations suggest that formation of this salt bridge reduces conformational freedom of the domain and weakens the negative potential on the domain surface. The mechanism of interaction between the LOV1 and kinase domains of phototropin is currently unknown; our results are consistent with either the presence of an interaction with a positively charged region of the kinase domain that is weakened in the light state (thus lifting repression of signaling), or with the breakage or formation of an interaction between kinase and the LOV1 Gβ-Hβ loop in response to stabilization of this region of the protein.
The changes to the hydrogen-bonding network surrounding FMN (see Loss of Hydrogen Bonding in the Light State, above) upon adduct formation may explain how light activation controls the E-K salt bridge. Formation of the photoadduct causes a loss of hydrogen bonding between residue N99 and the chromophore along with a movement of N99 toward the Gβ-Hβ loop; this movement pushes on the adjacent residue F97, which contacts the base of the Gβ-Hβ loop and causes formation of the E-K salt bridge. This mechanism is similar to one suggested by Crosson and co-workers (14).
In the case of LOV2, given that the Hβ-Iβ loop contains three negatively charged residues (two of which, D1017 and D1021, are acidic residues in all but one sequence in our alignment), the photoadduct-induced conformational change in this loop may alter electrostatic or salt-bridge interactions between the loop and some other portion of phototropin. Such interaction could involve the putative Jα helix, the dissociation of which is known to play a role in LOV2 activation (1,32).
CONCLUSIONS
This MD study suggests two distinct mechanisms for LOV domain activation of the phototropin kinase, one for LOV1 and a different one for LOV2. In either mechanism the cysteine-flavin photoadduct initiates changes in its immediate vicinity both through the motion of the cysteine residue, and through changes in the position of residues involved in hydrogen bonds with the flavin ligand. Both changes alter the dynamics of distant regions of the domains, employing for this purpose the Hβ and Iβ strands. In LOV1, the change in hydrogen bonding reorients the Gβ-Hβ loop, weakening the interaction between K92 and D93, and allowing the interloop E51-K92 salt bridge to form instead. In LOV2, the changes take hold in the Hβ-Iβ loop, causing the loop to undergo a shift in its equilibrium position that disrupts interactions with the Jα helix or portions of the protein immediately adjacent to it, leading to melting of the Jα helix (1). In hindsight, it is not surprising that the LOV1 and LOV2 domains employ different activation mechanisms given their different effects on phototropin activity (32).
In more general terms, this study established that molecular modeling methods, in particular the combination of various approaches, are in principle well suited to explain the activation of signaling proteins. Often, this activation is induced by mechanisms that do not become evident through structural analysis alone since dynamical properties and modulation of adhesion to other signaling factors is involved. Molecular modeling that includes QM/MM calculations, conventional molecular dynamics, hydrogen-bond network, normal mode, and principal component analysis, as well as advanced electrostatic calculations, is needed for a comprehensive study of signaling proteins. There are several reasons why a combination of approaches is required:
One needs to describe not only intrinsic properties of domains of signaling proteins, but also their physical properties responsible for interaction with other domains or signaling proteins.
The time domain of signaling is long compared with the nanosecond timescale covered by molecular modeling.
To detect slower changes in signaling proteins, simulations need to reach far beyond nanoseconds (here we carried out several simulations for 60 ns) or need to employ methodologies such as normal mode analysis (33) or elastic network modeling (34–36) that seem to be sensitive to long time dynamic properties of proteins.
As a result, signaling proteins pose a great challenge to molecular modeling, demanding the application of the most advanced methodologies today as well as the development of new methodologies. Meeting the challenge is definitely worthwhile scientifically because of the dramatic importance of signaling proteins in biological cells.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
All figures, except Fig. 6, were created with VMD (37).
This work was supported by grant No. MCB02-34938 from the National Science Foundation and by grant No. PHS-5-P41-RR05969 from the National Institutes of Health. The authors gladly acknowledge supercomputer time provided by the National Center for Supercomputing Applications via National Resources Allocation Committee grant No. MCA93S028. P. F. acknowledges support from the National Science Foundation Graduate Research Fellowship Program.
M. Dittrich's present address is Center for Quantitative Biological Simulation, Pittsburgh Supercomputing Center, Carnegie Mellon University, Pittsburgh, Pennsylvania.
References
- 1.Harper, S. M., L. C. Neil, and K. H. Gardner. 2003. Structural basis of a phototropin light switch. Science. 301:1541–1544. [DOI] [PubMed] [Google Scholar]
- 2.Kennis, J. T. M., S. Crosson, M. Gauden, I. H. M. van Stokkum, K. Moffat, and R. van Grondelle. 2003. Primary reactions of the LOV2 domain of phototropin, a plant blue-light photoreceptor. Biochemistry. 42:3385–3392. [DOI] [PubMed] [Google Scholar]
- 3.Christie, J., M. Salomon, K. Nozue, M. Wada, and W. Briggs. 1999. LOV (light, oxygen, or voltage) domains of the blue-light photoreceptor phototropin (NPH1): binding sites for the chromophore flavin mononucleotide. Proc. Natl. Acad. Sci. USA. 96:8779–8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crosson, S., and K. Moffat. 2002. Photoexcited structure of a plant photoreceptor domain reveals a light-driven molecular switch. Plant Cell. 14:1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swartz, T., S. Corchnoy, J. Christie, J. Lewis, I. Szundi, W. Briggs, and R. Bogomolni. 2001. The photocycle of a flavin-binding domain of the blue light photoreceptor phototropin. J. Biol. Chem. 276:36493–36500. [DOI] [PubMed] [Google Scholar]
- 6.Losi, A., T. Kottke, and P. Hegemann. 2004. Recording of blue light-induced energy and volume changes within the wild-type and mutated phot-LOV1 domain from Chlamydomonas reinhardtii. Biophys. J. 86:1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bittl, R., C. W. M. Kay, S. Weber, and P. Hegemann. 2003. Characterization of a flavin radical product in a C57M mutant of a LOV1 domain by electron paramagnetic resonance. Biochemistry. 42:8506–8512. [DOI] [PubMed] [Google Scholar]
- 8.Kay, C. W. M., E. Schleicher, A. Kuppig, H. Hofner, W. Rüdiger, M. Schleicher, M. Fischer, A. Bacher, S. Weber, and G. Richter. 2003. Blue light perception in plants. Detection and characterization of a light-induced neutral flavin radical in a C450A mutant of phototropin. J. Biol. Chem. 278:10973–10982. [DOI] [PubMed] [Google Scholar]
- 9.Neiss, C., and P. Saalfrank. 2003. Ab initio quantum chemical investigation of the first steps of the photocycle of phototropin: a model study. Photochem. Photobiol. 77:101–109. [DOI] [PubMed] [Google Scholar]
- 10.Dittrich, M., P. L. Freddolino, and K. Schulten. 2005. When light falls in LOV: a quantum mechanical/molecular mechanical study of photoexcitation in Phot-LOV1 of Chlamydomonas reinhardtii. J. Phys. Chem. B. 109:13006–13013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fedorov, R., I. Schlichting, E. Hartmann, T. Domratcheva, M. Fuhrmann, and P. Hegemann. 2003. Crystal structures and molecular mechanism of a light-induced signaling switch: the Phot-LOV1 domain from Chlamydomonas reinhardtii. Biophys. J. 84:2474–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kosztin, I., R. Bruinsma, P. O'Lague, and K. Schulten. 2002. Mechanical force generation by G-proteins. Proc. Natl. Acad. Sci. USA. 99:3575–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng, Y., Y. Zhang, and J. A. McCammon. 2006. How does activation loop phosphorylation modulate catalytic activity in the cAMP-dependent protein kinase: a theoretical study. Protein Sci. 15:672–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crosson, S., S. Rajagopal, and K. Moffat. 2003. The LOV domain family: photoresponsive signaling modules coupled to diverse output domains. Biochemistry. 42:2–10. [DOI] [PubMed] [Google Scholar]
- 15.Berman, H. M., J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, and P. E. Bourne. 2000. The protein data bank. Nucleic Acids Res. 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crosson, S., and K. Moffat. 2001. Structure of a flavin-binding plant photoreceptor domain: insights into light-mediated signal transduction. Proc. Natl. Acad. Sci. USA. 98:2995–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kastenholz, M. A., and P. H. Hunenberger. 2003. Influence of artificial periodicity and ionic strength in molecular dynamics simulations of charged biomolecules employing lattice-sum methods. J. Phys. Chem. B. 108:774–788. [Google Scholar]
- 18.Krasnovska, M. V., J. Sefcikova, K. Réblová, B. Schneider, N. G. Walter, and J. Šponer. 2006. Cations and hydration in catalytic RNA: molecular dynamics of the hepatitis δ-virus ribozyme. Biophys. J. 91:626–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phillips, J. C., R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale, and K. Schulten. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacKerell Jr., A. D., B. Brooks, C. L. Brooks, III, L. Nilsson, B. Roux, Y. Won, and M. Karplus. CHARMM: the energy function and its parameterization with an overview of the program. In The Encyclopedia of Computational Chemistry. John Wiley & Sons, Chichester, UK. 271–277.
- 21.Chenna, R., H. Sugawara, T. Koike, R. Lopez, T. J. Gibson, D. G. Higgins, and J. D. Thompson. 2003. Nucl. Acids Res. 31:3497–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ichiye, T., and M. Karplus. 1991. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins Struct. Funct. Gen. 11:205–217. [DOI] [PubMed] [Google Scholar]
- 23.Balsera, M. A., W. Wriggers, Y. Oono, and K. Schulten. 1996. Principal component analysis and long time protein dynamics. J. Phys. Chem. 100:2567–2572. [Google Scholar]
- 24.van der Spoel, D., E. Lindahl, B. Hess, G. Groenhof, A. E. Mark, and H. J. C. Berendsen. 2005. GROMACS: fast, flexible, and free. J. Comput. Chem. 26:1701–1718. [DOI] [PubMed] [Google Scholar]
- 25.Baker, N. A., D. Sept, S. Joseph, M. J. Holst, and J. A. McCammon. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 98:10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neiss, C., and P. Saalfrank. 2004. Molecular dynamics simulation of the LOV2 domain from Adiantum capillus-veneris. J. Chem. Inf. Comput. Sci. 44:1788–1793. [DOI] [PubMed] [Google Scholar]
- 27.Nozaki, D., T. Iwata, T. Ishikawa, T. Todo, S. Tokutomi, and H. Kandori. 2004. Role of Gln1029 in the photoactivation processes of the LOV2 domain in Adiantum phytochrome3. Biochemistry. 43:8373–8379. [DOI] [PubMed] [Google Scholar]
- 28.Christie, J. M., T. E. Swartz, R. A. Bogomolni, and W. R. Briggs. 2002. Phototropin LOV domains exhibit distinct roles in regulating photoreceptor function. Plant J. 32:205–219. [DOI] [PubMed] [Google Scholar]
- 29.Salomon, M., U. Lempert, and W. Rüdiger. 2004. Dimerization of the plant photoreceptor phototropin is probably mediated by the LOV1 domain. FEBS Lett. 572:8–10. [DOI] [PubMed] [Google Scholar]
- 30.Iwata, T., D. Nozaki, S. Tokutomi, and H. Kandori. 2005. Comparative investigation of the LOV1 and LOV2 domains in Adiantum phytochrome-3. Biochemistry. 44:7427–7434. [DOI] [PubMed] [Google Scholar]
- 31.Losi, A., E. Ghiraldelli, S. Jansen, and W. Gärtner. 2005. Mutational effects on protein structural changes and interdomain interactions in the blue-light sensing LOV protein YtvA. Photochem. Photobiol. 81:1145–1152. [DOI] [PubMed] [Google Scholar]
- 32.Harper, S. M., L. C. Neil, I. J. Day, P. Hore, and K. H. Gardner. 2004. Conformational changes in a photosensory LOV domain monitored by time-resolved NMR spectroscopy. J. Am. Chem. Soc. 126:3390–3391. [DOI] [PubMed] [Google Scholar]
- 33.Kitao, A., and N. Go. 1999. Investigating protein dynamics in collective coordinate space. Curr. Opin. Struct. Biol. 9:164–169. [DOI] [PubMed] [Google Scholar]
- 34.Bahar, I., A. R. Atilgan, and B. Erman. 1997. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 2:173–181. [DOI] [PubMed] [Google Scholar]
- 35.Doruker, P., A. R. Atilgan, and I. Bahar. 2000. Dynamics of proteins predicted by molecular dynamics simulations and analytical approaches: application to α-amylase inhibitor. Proteins Struct. Funct. Gen. 40:512–524. [PubMed] [Google Scholar]
- 36.Atilgan, A. R., S. R. Durell, R. L. Jernigan, M. C. Demirel, O. Keskin, and I. Bahar. 2001. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 80:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD—visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 38.Tai, K., T. Shen, U. Börjesson, M. Philippopoulos, and J. A. McCammon. 2001. Analysis of a 10-ns molecular dynamics simulation of mouse acetylcholinesterase. Biophys. J. 81:715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]