

Abstract
Escherichia coli cytochrome c nitrite reductase (NrfA) catalyzes the six-electron reduction of nitrite to perform an important role in the biogeochemical cycling of nitrogen. Here we describe NrfA adsorption on single-crystal Au(111) electrodes as an electrocatalytically active film in which the enzyme undergoes direct electron exchange with the electrode. The adsorbed NrfA has been imaged to molecular resolution by in situ scanning tunneling microscopy (in situ STM) under full electrochemical potential control and under conditions where the enzyme is electrocatalytically active. Details of the density and orientational distribution of NrfA molecules are disclosed. The submonolayer coverage resolved by in situ STM is readily reconciled with the failure to detect nonturnover signals in cyclic voltammetry of the NrfA films. The molecular structures show a range of lateral dimensions. These are suggestive of a distribution of orientations that could account for the otherwise anomalously low turnover number calculated for the total population of adsorbed NrfA molecules when compared with that determined for solutions of NrfA. Thus, comparison of the voltammetric signals and in situ STM images offers a direct approach to correlate electrocatalytic and molecular properties of the protein layer, a long-standing issue in protein film voltammetry.
INTRODUCTION
Enzymes adsorbed or immobilized on solid surfaces can retain their impressive catalytic specificity and selectivity to offer advantageous opportunities for exploiting and elucidating catalysis. Physical separation of catalyst from solutions of reactants, products, etc. is, for example, facilitated for catalyst recovery in bioreactors and sample analysis by biosensors (1). Appropriate choice of solid surface can also open elegant routes to control catalysis by the adsorbed enzyme, where protein film voltammetry (PFV) stands forward prominently (2–5). Here the solid is an electrode capable of direct electron exchange with a redox enzyme adsorbed as a (sub)monolayer “film”. Application of a precisely defined, but variable, potential allows control of the catalytic rate through control of the enzyme oxidation state. In turn, catalysis produces a net transfer of electrons between the electrode and substrate molecules in solution. The result is an electrical current that quantitates the rate of catalysis.
Immediate insight into the catalytic behavior of a redox enzyme is afforded by PFV in the absence of kinetic limitations from interfacial electron transfer and reactant (product) mass transport (6). New perspectives of even well-studied enzymes have emerged under such conditions by virtue of having enzyme activity defined as a function of electrochemical potential in addition to reagent concentration, temperature, and time (7–16). For example, application of an increased driving force for the catalytic reaction can boost or attenuate the steady-state catalytic rate. Presteady-state measurements can also resolve potential-dependent enzyme inhibition and activation (17–20). The next challenge is to develop mechanistic interpretations of the catalytic behavior. To do this the global activity of the film as defined by PFV must be related to descriptions at the molecular level where kinetic and thermodynamic properties of the adsorbed enzyme molecules are considered together with their surface density, orientation, and mobility.
Traditional approaches to integrate kinetic and thermodynamic enzyme properties within a catalytic mechanism can be extended to adsorbed redox enzymes (6,21). For example, interfacial electron transfer is introduced through the Butler-Volmer or Marcus formalisms, which include terms for the electrode potential and the reduction potential of the enzyme center undergoing direct electron exchange with the electrode. Calculated current-potential profiles can then be compared to those defined experimentally to test a mechanistic proposal. When applying this approach it is generally assumed that the electrocatalytically active molecules have uniform properties. This situation may be supported experimentally when the catalytic response loses magnitude over time without change in the shape or position. However, even films displaying such behavior may contain features that suggest contributions from enzyme molecules with nonuniform properties. Notably, a distribution of sluggish interfacial electron transfer rates, reflecting a range of enzyme orientations, has been suggested to account for the failure of catalytic currents to reach a constant value at high over potentials (22).
Additional molecular properties of the film may contribute to the electrocatalytic behavior even though they cannot be disclosed simply from analysis of the catalytic response. These relate to the density, long-range order, and dynamics of the adsorbed molecules. For multisubunit enzymes the integrity of intersubunit interactions after adsorption can also be questioned. Insight into some of these properties may be gained when electrocatalytically active films are studied by cyclic voltammetry in the absence of reactants (23,24). Peaks that correspond to reduction and oxidation of centers within the enzyme report on the homogeneity (or otherwise) of reduction potentials, populations of electroactive centers, and total electroactive population. However, such “nonturnover” signals are not always discernable and are usually attributed to the presence of only a small number of electroactive adsorbates in the film (7–15).
The above discussion serves to illustrate that our understanding of catalytic PFV would be greatly strengthened by direct insight into the molecular nature of a protein film. One technique that has provided such a perspective is in situ scanning tunneling microscopy (STM) (25,26). Protein films on single-crystal, atomically planar electrode surfaces submersed in aqueous buffer solution can be imaged with independent electrochemical control of the electrode, say an Au(111) electrode, and the coated STM tip (27,28). The result is a map of the electron tunneling properties of protein:electrode interface with nanometer or single-molecule resolution parallel to the Au(111) surface (29–41). Distinct regions with high conductivity define the density, ordering, and lateral dimensions of adsorbed protein molecules that can be directly related to the electrocatalytic behavior of the film.
In this report we provide new data for PFV and in situ STM imaging of Escherichia coli cytochrome c nitrite reductase (NrfA) at single crystal Au(111) electrodes. NrfA catalyzes the six-electron reduction of nitrite to ammonium, performing an important role in the biogeochemical cycling of nitrogen (42). The crystal structure has defined a decaheme-containing homodimer with asymmetric dimensions (Fig. 1) (43). At pH 7, midpoint potentials of −37 mV for Heme 2, −107 mV for Hemes 1 and 3, and −320 mV for Heme 4 and/or 5 have been defined. Nitrite binds as an axial ligand to Heme 1 and the nitrite reductase activity of NrfA has been extensively studied by catalytic PFV at pyrolytic graphite “edge” (PGE) electrodes (14,15,17,18,44,45). However, ready access to molecular resolution of these NrfA films is precluded by the unfavorable optical and topographic properties of the PGE surface. Here we present voltammetric data that show clear signals for electrocatalytic nitrite reduction by NrfA films on Au(111) electrodes. In situ STM provides imaging of adsorbed NrfA to molecular resolution under conditions where the enzyme is catalytically active. Details of the density and orientational distribution of NrfA molecules in the electrocatalytically active film are disclosed. Comparison between the voltammetric signals and the in situ STM images offers a direct approach to correlate catalytic and molecular properties of the protein film, a long-standing issue in PFV.
FIGURE 1.

The structure of E. coli NrfA. Hemes are numbered according to their order of attachment to the protein sequence. Perspectives A and C are related to B by 90° rotation around the y and x axes, respectively. The dashed curves in B indicate the region proposed to dock to the physiological redox partner NrfB. The block arrows in C indicate the proposed directionality of nitrite entry to, and ammonium egress from, the active site.
MATERIALS AND METHODS
Reagents
Cytochrome c nitrite reductase (NrfA) from E. coli was purified and quantitated as previously (43). Purified enzyme was exchanged into 50 mM Hepes, 2 mM CaCl2, pH 7.0, and stored frozen as aliquots in liquid nitrogen. Samples had a specific activity of ∼1500 μmol nitrite consumed min−1 mg−1 in 50 mM Hepes, 2 mM CaCl2, pH 7.0, using dithionite-reduced methyl viologen as the electron donor. All solutions were prepared in Millipore water (Millipore, Bedford, MA). Nitrite stock solutions (potassium nitrite, >98%; Fluka, Milwaukee, WI) were prepared daily as required (17). Phosphate buffer (2 or 5 mM, pH 7.0) was prepared by mixing K2HPO4 and KH2PO4 (Suprapur, Fluka) solutions.
Protein film voltammetry
Au(111) working electrodes were prepared by the methods of Clavilier and Hamelin (electrode diameters in the 2.5–4 mm range) and annealed for 12 h at 850°C (46,47). Electrodes were checked by cyclic voltammetry in ultrapure aqueous 0.1 M sulfuric acid on a regular basis and satisfactory comparison with reported voltammograms obtained (47). Before use the electrodes were electropolished, annealed in an oven at 860°C, further freshly annealed in a H2-flame, and quenched in H2-saturated water.
Enzyme films were prepared by covering the freshly prepared electrode surface with an ice-cold solution of 32 μM NrfA subunits. After ∼5 min, excess protein solution was removed and the electrode placed in an electrochemical cell containing a Pt wire counterelectrode and a freshly prepared reversible hydrogen reference electrode. The reference electrode was calibrated against a saturated calomel electrode (+244 mV versus standard hydrogen electrode (SHE)) after each experiment. All potentials are reported relative to SHE. The cell was flushed with a stream of pure (5N) argon and housed in a Faraday cage. Cyclic voltammetry was performed with an Autolab PGSTAT 10 instrument (EcoChemie, Utrecht, The Netherlands) using the GPES software.
Previous PFV of NrfA was performed with PGE electrodes and buffer-electrolytes containing Goods buffers such as Hepes and Mes (14,15,17). However, Goods buffers have been found to adsorb on Au(111) electrodes. Therefore, phosphate buffers were used throughout this study to minimize competition between NrfA and buffer components for adsorption on Au(111) electrodes. Control experiments at rotating PGE electrodes showed the catalytic PFV of NrfA films to be essentially independent of whether the pH 7 buffer-electrolyte contained phosphate or Goods buffer.
In situ STM imaging
The Au(111) working electrode, reference, and counter electrodes were housed in a polytetrafluoroethylene cell combined with a Teflon O-ring. Immediately before use the cell was boiled in 15% nitric acid, sonicated, and washed extensively with Millipore water. An enzyme-coated electrode, prepared as described above, was then placed in the cell. The cell was immediately filled with the desired buffer-electrolyte solution (3 mL) and potential control of the electrode initiated. The cell was mounted in the STM instrument (PicoSPM, Molecular Imaging, Tempe, AZ) inside a glass chamber that was constantly supplied with a stream of wetted, pure argon to ensure that the in situ STM was recorded under anaerobic conditions (48). The STM instrument was suspended by elastic cords in a Faraday Cage. Tips of tungsten or platinum-iridium (diameter 0.25 mm, total length ∼17 mm) coated with Apiezon wax were used. The instrument was operated in the constant current mode at 9.8 Hz.
Dimensions of features in the STM images were determined directly using SPIP software (Image Metrology, Copenhagen, Denmark). The STM instrument was calibrated against the distance (6.35 nm) between the periodic reconstruction lines on Au(111). Molecular projections perpendicular to the Au(111) surface were calibrated daily against a gold terrace (0.245 nm) while scanning a protein film. The majority of STM images of the NrfA films showed regions of high contrast with lateral dimensions >4 nm and so consistent with the crystallographically defined protein dimensions (Fig. 1). The longest lateral dimension was assigned as the molecular length. The width was measured along a normal to the longest dimension at its central point. Inspection of a considerable number of recorded images showed no immediate correlation between scan direction and the orientation of the elongated structures. Thus, the lateral dimensions can be considered as “physically” substantiated. Occasionally a region of anomalously high contrast with lateral dimensions <1 nm was observed. Such features were excluded from analysis on the basis that they were too small to correspond to protein. NrfA electrode coverage was typically determined from images of 100 × 100 nm2, molecules crossing the edge of the image were only counted if more than 50% of the molecule appeared. The presented results are representative of those from seven independently prepared electrodes and more than 100 images of each electrode.
RESULTS
PFV on Au(111) electrodes
Representative cyclic voltammograms at Au(111) electrodes freshly coated with NrfA are shown in Fig. 2. Cyclic voltammetry was restricted to potential ranges positive of −0.60 V because a strong reduction current from H2-evolution beginning at potentials negative of −0.50 V was noted. In the absence of nitrite, no signals that could be attributed to reduction, or oxidation, of centers within the enzyme were detected. In the presence of nitrite, catalytic reduction currents were apparent below −0.10 V. These currents were absent when NrfA was omitted from the experiment and are attributed to nitrite reduction catalyzed by NrfA in direct electrical communication with the electrode. The presence of an electrocatalytically active NrfA film was substantiated when the catalytic response was retained on transfer of the NrfA-coated electrodes to fresh nitrite-containing buffers.
FIGURE 2.

Representative cyclic voltammograms from NrfA films on Au(111) electrodes. Experiments were performed with the indicated level of nitrite in 2 mM potassium phosphate, pH 7, at room temperature with a scan rate of 10 mV s−1. Dashed lines indicate the first scan, solid lines the last scan, and dotted lines intermediate scans.
Certain properties of the catalytic signals changed with time. Specifically, the signals decreased in magnitude and the current-potential profiles became less dependent on scan direction (Fig. 2). These changes are likely to reflect interrelated phenomena. It is reasonable to suggest that the initial and relatively rapid decrease in signal magnitude is caused by desorption of electrocatalytically active NrfA only loosely associated with the electrode. As the number of electrocatalytically active molecules on the electrode decreases there will be less competition for nitrite at the electrode surface. Any limitation on the voltammogram from substrate mass-transport, indicated by larger catalytic currents on the sweep toward more negative potentials than on the return, should then diminish as the experiments progressed (49). This was indeed the case, and over time the voltammograms tended toward a steady-state description of catalysis by the NrfA films.
Distinct steady-state signals are implied by the voltammograms in 30 and 1000 μM nitrite (Fig. 2). A dependence of the steady-state signal on enzyme substrate concentration is frequently observed in catalytic PFV (2–5). It reflects the distinct rate-limiting event of catalysis at substrate concentrations well below the Michaelis constant, KM, when compared to that for substrate concentrations greater than KM. For NrfA, 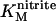 is ∼30 μM at pH 7 and the catalytic signals at Au(111) electrodes resemble those reported at rapidly rotating PGE electrodes both in their shape and the potential window for which activity is detected (14,17). It is worth noting that the catalytic response at stationary PGE electrodes is typically dominated by the consequences of nitrite depletion in the vicinity of the enzyme film. Thus, it seems reasonable to suggest that the chemically and topographically diverse PGE surface accommodates a greater density of electrocatalytically active NrfA than the much more uniform Au(111) surface.
is ∼30 μM at pH 7 and the catalytic signals at Au(111) electrodes resemble those reported at rapidly rotating PGE electrodes both in their shape and the potential window for which activity is detected (14,17). It is worth noting that the catalytic response at stationary PGE electrodes is typically dominated by the consequences of nitrite depletion in the vicinity of the enzyme film. Thus, it seems reasonable to suggest that the chemically and topographically diverse PGE surface accommodates a greater density of electrocatalytically active NrfA than the much more uniform Au(111) surface.
We have previously interpreted the steady-state catalytic PFV of NrfA in terms of a homogeneous population of electrocatalytically active molecules (5,14,15). Briefly, at nitrite concentrations 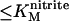 , the onset of activity occurs in a potential range that implicates reduction of Hemes 1 and 3 (Em,7 −100 mV) as a necessary step for observation of NrfA activity. At more negative potentials attenuation of the catalytic rate is proposed to result from reduction of Heme 4 and/or Heme 5 (Em,7 −320 mV) with precise mechanistic consequences that have yet to be revealed. The result is maximal steady-state catalytic current in a narrow potential window. At nitrite concentrations such as 1000 μM, that are much greater than
, the onset of activity occurs in a potential range that implicates reduction of Hemes 1 and 3 (Em,7 −100 mV) as a necessary step for observation of NrfA activity. At more negative potentials attenuation of the catalytic rate is proposed to result from reduction of Heme 4 and/or Heme 5 (Em,7 −320 mV) with precise mechanistic consequences that have yet to be revealed. The result is maximal steady-state catalytic current in a narrow potential window. At nitrite concentrations such as 1000 μM, that are much greater than 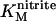 , it becomes harder to relate reduction potentials, determined in the absence of substrate, to the catalytic response. This is because events within the enzyme-substrate complex determine the catalytic rate. Under such conditions NrfA activity is no longer attenuated at low potential. In fact, in 1000 μM nitrite, NrfA activity is boosted at lower potentials. This may reflect rate-limiting electron delivery to the active site under conditions of maximal turnover. At intermediate nitrite concentrations the catalytic signal varies systematically between the limiting forms described above.
, it becomes harder to relate reduction potentials, determined in the absence of substrate, to the catalytic response. This is because events within the enzyme-substrate complex determine the catalytic rate. Under such conditions NrfA activity is no longer attenuated at low potential. In fact, in 1000 μM nitrite, NrfA activity is boosted at lower potentials. This may reflect rate-limiting electron delivery to the active site under conditions of maximal turnover. At intermediate nitrite concentrations the catalytic signal varies systematically between the limiting forms described above.
Thus, the characteristic steady-state features of NrfA catalysis at PGE electrodes are also displayed by NrfA adsorbed on Au(111) electrodes. Similar behavior has been observed for NrfA on rapidly rotating polycrystalline gold electrodes (14). However, the response at polycrystalline gold persisted for only four or five scans. By contrast, NrfA films on Au(111) electrodes displayed catalytic voltammetry at least 3 h after preparation that allowed a more critical consideration of their behavior. For example, in 1000 μM nitrite the catalytic response was shown to be essentially independent of scan rate up to 100 mV s−1. Importantly, the longevity of the NrfA film at Au(111) electrodes allowed in situ STM imaging for direct access to molecular resolution of electrocatalytically competent NrfA films as we describe below.
In situ STM on Au(111) electrodes in the absence of nitrite
Imaging of NrfA films in the absence of nitrite (tunneling current 0.15–0.30 nA) showed the presence of discrete regions of high contrast with 5–15 nm lateral dimensions (Fig. 3, A and B). This range of dimensions is in good agreement with that predicted from crystallography (Fig. 1), and these contrast regions are ascribed to NrfA molecules. That the longest STM-derived dimensions are slightly greater than defined by crystallography is a common observation that is most likely to arise from water layers around the protein or tip convolution. From the images it is clear that NrfA is adsorbed at well below monolayer coverage. Attributing each contrast to one NrfA ‘molecule’ (monomer or dimer) the coverage is 0.5 ± 0.1 picomol cm−2.
FIGURE 3.

Representative in situ STM images of NrfA adsorbed on an Au(111) electrode. Image dimensions: (A) 200 × 200 nm, (B) 100 × 100 nm, (C) 60 × 60 nm, and (D) 30 × 30 nm. Images recorded in 2 mM phosphate, pH 7, with the Au(111) electrode maintained at −0.07 V versus SHE, bias voltage −0.2 V, tunneling current 0.3 nA.
When the Au(111) potential was maintained at a range of potentials between ∼0.3 and −0.3 V a slow decrease in NrfA coverage was noted on repeated imaging of a given electrode area (bias voltage −0.3 or −0.4 V). The decay could be approximated to a first order process with half-life of ∼60 min. Films appeared notably less stable when more negative electrode potentials were applied, and at −0.6 V the half-life for desorption was ∼10 min. When the tip was raised and moved to a fresh area of the electrode the coverage was found to be within error of that recorded initially. NrfA desorption is thus facilitated by the imaging process and accelerated by application of electrode potentials below ∼−0.3 V. Tip-induced desorption was also observed during imaging of yeast cytochrome c on Au(111) and is a general observation for noncovalently bound adsorbates (34).
Imaging at higher resolution allowed the lateral dimensions of adsorbed molecules to be determined (Fig. 3, C and D). Such an analysis was undertaken for images of the same electrode area with different Au(111) potentials (Fig. 4). With a bias voltage of −0.4 V apparent molecular lengths spanning 5–15 nm and apparent widths from 4 to 10 nm with little dependence on the Au(111) potential were observed. As a result, the molecular ‘footprints’ ranged from approximately circular to elongated oval. The smaller number of molecules recorded at lower electrode potentials reflects the enhanced desorption rates noted. There was no correlation between molecular dimensions and retention time on the surface.
FIGURE 4.

Apparent lateral dimensions of NrfA molecules adsorbed on Au(111) electrodes held at the indicated potential in the absence of nitrite. Inset: histograms representing the frequency, y axis, of a given length (black) and width (gray), x axis. Data from representative images (60 × 60 nm) recorded in 2 mM phosphate, pH 7, with bias voltage −0.4 V, tunneling current 0.15 nA.
The vertical projection of an imaged molecule reflects the tunneling current determined by electronic rather than topographic molecular structure (37). The adsorbed NrfA molecules showed apparent vertical projections of ∼0.2–0.5 nm independent of Au(111) potential from −0.25 to 0.25 V (Fig. 5). These are much smaller than the crystallographic dimensions as is mostly observed by in situ STM of redox metalloproteins (32,34,40). Although the absolute values of the projections are then less meaningful, the relative heights can still reflect differences in electronic conductivity.
FIGURE 5.

Vertical projection of molecules in 0 μM (○) and 40 μM (•) nitrite as a function of Au(111) potential. Data from representative images (60 × 60 nm) recorded in 2 mM phosphate, pH 7 with bias voltage −0.3 V and tunneling currents 0.2 nA (0 μM nitrite) and 0.5 nA (40 μM nitrite).
In situ STM on Au(111) electrodes in the presence of 40 μM nitrite
Imaging NrfA films in the presence of 40 μM nitrite revealed features with similar initial densities (0.45 ± 0.1 picomol cm−2) to those observed in the absence of nitrite. A slightly higher tunneling current, 0.5 nA, was used here for stable imaging. It was apparent that the films were less stable than those imaged under comparable conditions but lacking nitrite. Half-lives of ∼20 min were estimated from images recorded between 0.3 and −0.1 V. At lower electrode potentials the half-life decreased further and occasionally the imaged area was cleared of all adsorbed molecules over 20 min of repeated imaging. Repositioning the tip to image a fresh area of the electrode showed that NrfA desorption had been accelerated by the imaging process. It also indicated a decrease in coverage relative to that observed initially. This observation suggests that NrfA can more readily dissociate from the Au(111) surface when nitrite reduction is occurring. Quantitation of the ‘intrinsic’ rate of film loss was precluded by the difficulty of deconvolving intrinsic and imaging-induced desorption effects. However, the overall stability of the film was in good qualitative agreement with that observed with PFV where the Au(111) potential continually cycles across a range of potential.
Due to the desorption of NrfA molecules, imaging of several electrode areas was needed to assess the influence of measurement conditions on the appearance of the adsorbed molecules. The molecular dimensions were comparable to those recorded in the absence of nitrite (Fig. 6). However, the apparent heights of molecules imaged in the presence of nitrite were generally smaller and had a narrower distribution than those recorded in the absence of nitrite (Fig. 5). For example, the average apparent height was 0.39 nm with a standard deviation of 0.06 nm at 0.153 V in the absence of nitrite, whereas it was 0.26 nm with a standard deviation of 0.04 nm at 0.135 V in 40 μM nitrite. This difference is discussed below.
FIGURE 6.

Apparent lateral dimensions of NrfA molecules adsorbed on Au(111) electrodes held at the indicated potential in 40 μM nitrite. (Inset) Histograms representing the frequency, y axis, of a given length (black) and width (gray), x axis. Data from representative images (60 × 60 nm) recorded in 40 μM nitrite, 2 mM phosphate, pH 7, with bias voltage −0.4 V, tunneling current 0.5 nA.
DISCUSSION
To date, only a few studies have combined PFV with in situ scanning probe microscopies to disclose molecular structures of the adsorbed molecules. Targeted proteins include horse heart cytochrome c (29,31), horseradish peroxidase (30), Saccharomyces cerevisiae cytochrome c (34,36), Pseudomonas aeruginosa azurin (32,33,37,39,41), Pyrococcus furiosus ferredoxin (35), and Achromobacter xylosoxidans Cu-containing nitrite reductase (38). The studies presented here add to this collection with systematic observation of electrocatalytically active NrfA films imaged to molecular resolution by in situ STM. Submonolayer NrfA coverage of the Au(111) electrodes is immediately revealed and provides a starting point for consideration of the PFV.
Redox transformation of a homogeneous, noninteracting population of adsorbed molecules will give nonturnover signals with a peak current density, ip, equal to n2F2υΓ/4RT (50). Here n is the number of electrons transferred in the redox event, υ the scan rate, Γ the population of electroactive molecules, and F, R, and T have their usual meaning. For NrfA films on Au(111) electrodes Γ ≈ 0.5 picomol cm−2. With a scan rate of 10 mV s−1 as used in these experiments, an n =1 process will have ip ≈ 4 nA cm−2. Even allowing for twice the population if the molecular structures represent dimers rather than monomers, as discussed below, this is well below the level of voltammetric detection (Fig. 2). Thus, the failure to detect nonturnover signals from NrfA films is readily reconciled with the submonolayer coverage resolved by in situ STM.
The catalytic currents recorded when nitrite is present illustrate the amplification of electron transfer achieved by NrfA catalysis. At 1000 μM nitrite the maximum catalytic rate is approached (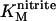 ∼ 30 μM) and several independently prepared films were studied under this condition. When the signals approached a steady-state form catalytic current densities between 4 and 15 μA cm−2 were detected at −0.4 V. Taking these currents to represent the six-electron reduction of nitrite to ammonium by 0.5 picomol of electrocatalytically active NrfA a turnover number, kcat, on the order of 30 s−1 is calculated. This is a value considerably lower than that of 770 s−1 measured using dithionite-reduced methyl viologen as the electron donor (43). Assuming that the Au(111) electrode was uniformly covered by NrfA (in situ STM imaging provided no information to contradict this) two limiting scenarios can be envisaged to account for the apparent discrepancy between solution and PFV experiments. In one scenario the adsorbed molecules exhibit identical electrocatalytic activity but with a turnover number significantly lower than that of freely diffusing NrfA. In the alternative description, the adsorbed molecules exhibit heterogeneous behavior. Here, adsorbed and diffusing NrfA have the same turnover number but only a fraction (∼5%) of the imaged structures is electrocatalytically active.
∼ 30 μM) and several independently prepared films were studied under this condition. When the signals approached a steady-state form catalytic current densities between 4 and 15 μA cm−2 were detected at −0.4 V. Taking these currents to represent the six-electron reduction of nitrite to ammonium by 0.5 picomol of electrocatalytically active NrfA a turnover number, kcat, on the order of 30 s−1 is calculated. This is a value considerably lower than that of 770 s−1 measured using dithionite-reduced methyl viologen as the electron donor (43). Assuming that the Au(111) electrode was uniformly covered by NrfA (in situ STM imaging provided no information to contradict this) two limiting scenarios can be envisaged to account for the apparent discrepancy between solution and PFV experiments. In one scenario the adsorbed molecules exhibit identical electrocatalytic activity but with a turnover number significantly lower than that of freely diffusing NrfA. In the alternative description, the adsorbed molecules exhibit heterogeneous behavior. Here, adsorbed and diffusing NrfA have the same turnover number but only a fraction (∼5%) of the imaged structures is electrocatalytically active.
To take this discussion further it is necessary to consider the properties of individual molecules. Here the in situ STM images combine with the intrinsic asymmetric dimensions of the NrfA molecule to provide further valuable information. The molecular structures show a range of lateral dimensions that lie outside the uncertainty inherent to such measurements, suggesting heterogeneity in the properties of the adsorbed molecules. Dimensions derived from in situ STM imaging are inextricably linked to the electronic properties of the adsorbed molecules. However, previous studies have found broad agreement between STM-derived lateral dimensions and those defined crystallographically (32,34,35). The slightly larger dimensions commonly recorded by the former is attributed to the participation of structured water layers in image contrast or tip convolution. In this context the lateral NrfA dimensions disclosed here suggest molecules projecting distinct ‘footprints’ onto the Au(111) surface (Fig. 7). Thus, NrfA may adsorb with a distribution of orientations and possibly as both monomer and dimer forms.
FIGURE 7.

Superposition of the crystallographically defined dimensions of NrfA monomers (white) and dimers (gray) on the lateral dimensions derived from in situ STM as in Fig. 4 with the Au(111) electrode at 1 mV versus SHE.
The structure of NrfA, like that of many multicentered metalloenzymes, suggests regions tailored to facilitate specific steps in catalysis. For example, channels that are proposed to facilitate substrate/product exchange between the NrfA active site and bulk solution have been identified in addition to ‘hot spots’ for productive intermolecular electron exchange (Fig. 1). As a consequence the electrocatalytic behavior of adsorbed NrfA is expected to be orientation dependent. If the majority of NrfA molecules are adsorbed to restrict access to the channels that allow substrate entry to, and product egress from, the active site or, in a manner that fails to support facile interfacial electron transfer as required for catalysis, the apparently low catalytic activity of the film is explained.
The work presented here and the discussions above serve to illustrate the complexity that may be inherent to an electrocatalytically active protein film. The in situ STM data point to a low enzyme surface coverage that accords with the electrochemical data but is also caused in part by the imaging process and the close tip contact. The molecular structures show a distribution of lateral dimensions suggestive of a range of orientations that may include a distribution of dimer and monomer enzyme forms. However, resolution of electrocatalytic activity at the single molecule level will be required to definitively account for the behavior that is observed.
A number of recent studies of transition metal complexes (51), organic redox molecules (52,53), and the small redox metalloprotein, Pseudomonas aeruginosa azurin (41), supported by theoretical frames (54), have opened routes to in situ STM not only as a high-resolution imaging tool but also as a single-molecule configuration with device-like properties. Such observations could hold other prospects for addressing electronic properties of redox metalloenzymes toward the single-molecule level. The overpotential dependence of the electrocatalytic current reflects, for example, the interfacial electron exchange in the active and resting enzyme states, also inherent in the in situ STM tunneling current. The different electronic structures of the enzyme and enzyme-substrate complex would also in principle give different in situ STM signals. The present data show a smaller apparent height for the enzyme-substrate complex than for the substrate-free enzyme. A slightly higher tunneling current was used for stable imaging in the presence of substrate, i.e., 0.5 nA vs. 0.15–0.3 nA. However, if the tunneling current follows the expected exponential distance dependence, such a current change appears too small to induce the observed change in apparent height. In addition, the presence of nitrite appeared to weaken the enzyme binding to the electrode as evidenced by the decreased half-life for desorption. Disregarding different degrees of tip interference with the enzyme and enzyme-substrate complex, then taken at face value, the in situ STM data suggest that the enzyme-substrate complex has a more compact structure and is more loosely surface bound with a lower conductivity than the free enzyme.
The inferences of the small apparent height changes, i.e., changes in electronic conductivity, on either Au(111) potential variation or nitrite binding require further substantiation. However, they serve to illuminate a general issue in redox metalloprotein mapping by in situ STM. Close relations between voltammetric and in situ STM patterns can be expected for small redox molecules such as transition metal complexes (51,54). When the redox center constitutes only a small part of the target molecule such as in metalloenzymes, a multitude of other off-resonance electron tunneling channels become increasingly competitive. These are visible in STM and in situ STM but not in PFV where electron trapping at the redox center is essential. To this adds the nonuniform adsorption patterns of NrfA on the bare Au(111)-surface, which would further erode the subtle tunneling spectroscopic features for uniform adsorption. Approaches to favorable and uniform adsorption would be based on systematic studies of NrfA on modified Au(111) electrode surfaces with monolayers of linker molecules with functional groups suitable for gentle linking of the enzyme in uniform orientations. This would also ascertain efficient electronic coupling to the electrode surface such as shown recently (41).
CONCLUSIONS
Reported studies of the electrocatalytic behavior of redox metalloenzymes on well-defined single-crystal electrode surfaces are few in number. As shown by reports on small electron transfer proteins, such approaches, however, hold prospects for introducing novel technology such as in situ STM to address metalloprotein behavior at a resolution approaching that of the single molecule (29–41). This would further address outstanding issues in PFV, particularly the protein coverage of the electrode and the number of electrochemically and electrocatalytically active protein molecules in the protein surface monolayer.
This study has illuminated some of these issues. The target metalloenzyme, NrfA, is electrocatalytically active on unmodified single-crystal Au(111) electrode surfaces but no voltammetric signals were detected in the absence of nitrite substrate. The electrocatalytic signals decreased in magnitude over time most likely due to weak NrfA adsorption. In situ STM under electrochemical potential control has disclosed a low density of adsorbed molecules that readily explains the failure to detect voltammetric signals in the absence of substrate. Correlating catalytic currents with the number of adsorbed molecules produces a turnover number much lower that that observed in solution studies. A reason for this is suggested by the wide distribution of lateral molecular-size structures, which reflects a distribution of metalloenzyme adsorption modes, only some of which may be electrocatalytically active. Thus, the mutual support between the macroscopic PFV and the single-molecule in situ STM approach is strikingly illuminated in this way. Together with previous studies of electron transfer proteins (27,32–36,39,40) and redox metalloenzymes (30,38), the study suggests ways of approaching both stable electrocatalytic voltammetry and parallel structural and functional mapping toward single-molecule resolution.
Acknowledgments
We are grateful to Christine Moore for purification of NrfA and to David Richardson, Tom Clarke, Bénédicte Burlat, Myles Cheesman, and Andrew Hemmings for helpful discussion.
This work was funded by the Engineering and Physical Sciences Research Council (Doctoral Training Account award to J.D.G.), the Biotechnology and Biological Sciences Research Council (grants 83/B17233 and 83/B18695), and a Joint Infrastructure Fund award for Biophysical Chemistry at the University of East Anglia (062178). Financial support from the Danish Research Council for Technology and production Sciences (26-00-0034) to J.U. is acknowledged.
References
- 1.Tischer, W., and V. Kasche. 1999. Immobilized enzymes: crystals or carriers? Trends Biotechnol. 17:326–335. [DOI] [PubMed] [Google Scholar]
- 2.Léger, C., S. J. Elliott, K. R. Hoke, L. J. C. Jeuken, A. K. Jones, and F. A. Armstrong. 2003. Enzyme electrokinetics: using protein film voltammetry to investigate redox enzymes and their mechanisms. Biochemistry. 42:8653–8662. [DOI] [PubMed] [Google Scholar]
- 3.Vincent, K. A., and F. A. Armstrong. 2005. Investigating metalloenzyme reactions using electrochemical sweeps and steps: fine control and measurements with reactants ranging from ions to gases. Inorg. Chem. 44:798–809. [DOI] [PubMed] [Google Scholar]
- 4.Armstrong, F. A. 2005. Recent developments in dynamic electrochemical studies of adsorbed enzymes and their active sites. Curr. Opin. Chem. Biol. 9:110–117. [DOI] [PubMed] [Google Scholar]
- 5.Butt, J. N. 2003. Fresh perspectives on nitrogen-cycle enzymes from protein film voltammetry. Recent Res. Devel. Biochem. 4:159–180. [Google Scholar]
- 6.Heering, H. A., J. Hirst, and F. A. Armstrong. 1998. Interpreting the catalytic voltammetry of electroactive enzymes adsorbed on electrodes. J. Phys. Chem. B. 102:6889–6902. [Google Scholar]
- 7.Elliott, S. J., K. R. Hoke, K. Heffron, M. Palak, R. A. Rothery, J. H. Weiner, and F. A. Armstrong. 2004. Voltammetric studies of the catalytic mechanism of the respiratory nitrate reductase from Escherichia coli: how nitrate reduction and inhibition depend on the oxidation state of the active site. Biochemistry. 43:799–807. [DOI] [PubMed] [Google Scholar]
- 8.Anderson, L. J., D. J. Richardson, and J. N. Butt. 2001. Catalytic protein film voltammetry from a respiratory nitrate reductase provides evidence for complex electrochemical modulation of enzyme activity. Biochemistry. 40:11294–11307. [DOI] [PubMed] [Google Scholar]
- 9.Frangioni, B., P. Arnoux, M. Sabaty, D. Pignol, P. Bertrand, B. Guigliarelli, and C. Léger. 2004. In Rhodobacter sphaeroides respiratory nitrate reductase, the kinetics of substrate binding favors intramolecular electron transfer. J. Am. Chem. Soc. 126:1328–1329. [DOI] [PubMed] [Google Scholar]
- 10.Heffron, K., C. Léger, R. A. Rothery, J. H. Weiner, and F. A. Armstrong. 2001. Determination of an optimal potential window for catalysis by E. coli dimethyl sulfoxide reductase and hypothesis on the role of Mo(V) in the reaction pathway. Biochemistry. 40:3117–3126.11258926 [Google Scholar]
- 11.Hirst, J., B. A. C. Ackrell, and F. A. Armstrong. 1997. Global observation of hydrogen/deuterium isotope effects on bidirectional catalytic electron transport in an enzyme: direct measurement by protein-film voltammetry. J. Am. Chem. Soc. 119:7434–7439. [Google Scholar]
- 12.Hudson, J. M., K. Heffron, V. Kotlyar, Y. Sher, E. Maklashina, G. Cecchini, and F. A. Armstrong. 2005. Electron transfer and catalytic control by the iron-sulfur clusters in a respiratory enzyme, E. coli fumarate reductase. J. Am. Chem. Soc. 127:6977–6989. [DOI] [PubMed] [Google Scholar]
- 13.Butt, J. N., J. Thornton, D. J. Richardson, and P. S. Dobbin. 2000. Voltammetry of a flavocytochrome c3: the lowest potential heme modulates fumarate reduction rates. Biophys. J. 78:1001–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Angove, H. C., J. A. Cole, D. J. Richardson, and J. N. Butt. 2002. Protein film voltammetry reveals distinctive fingerprints of nitrite and hydroxylamine reduction by a cytochrome c nitrite reductase. J. Biol. Chem. 277:23374–23381. [DOI] [PubMed] [Google Scholar]
- 15.Gwyer, J. D., D. J. Richardson, and J. N. Butt. 2005. Diode or tunnel-diode characteristics? Resolving the catalytic consequences of proton coupled electron transfer in a multi-centered oxidoreductase. J. Am. Chem. Soc. 127:14964–14965. [DOI] [PubMed] [Google Scholar]
- 16.Bradley, A. L., S. E. Chobot, D. M. Arciero, A. B. Hooper, and S. J. Elliott. 2004. A distinctive electrocatalytic response from the cytochrome c peroxidase of Nitrosomonas europaea. J. Biol. Chem. 279:13297–13300. [DOI] [PubMed] [Google Scholar]
- 17.Gwyer, J. D., D. J. Richardson, and J. N. Butt. 2004. Resolving complexity in the interactions of redox enzymes and their inhibitors: contrasting mechanisms for the inhibition of a cytochrome c nitrite reductase revealed by protein film voltammetry. Biochemistry. 43:15086–15094. [DOI] [PubMed] [Google Scholar]
- 18.Gwyer, J. D., H. C. Angove, D. J. Richardson, and J. N. Butt. 2004. Redox-triggered events in cytochrome c nitrite reductase. Bioelectrochemistry. 63:43–47. [DOI] [PubMed] [Google Scholar]
- 19.Léger, C., S. Dementin, P. Bertrand, M. Rousset, and B. Guigliarelli. 2004. Inhibition and aerobic inactivation kinetics of Desulfovibrio fructosovorans NiFe-hydrogenase studied by protein film voltammetry. J. Am. Chem. Soc. 126:12162–12172. [DOI] [PubMed] [Google Scholar]
- 20.Léger, C., A. K. Jones, W. Roseboom, S. P. J. Albracht, and F. A. Armstrong. 2002. Enzyme electrokinetics: hydrogen evolution and oxidation by Allochromatium vinosum NiFe-hydrogenase. Biochemistry. 41:15736–15746. [DOI] [PubMed] [Google Scholar]
- 21.Reda, T., and J. Hirst. 2006. Interpreting the catalytic voltammetry of an adsorbed enzyme by considering substrate mass transfer, enzyme turnover, and interfacial electron transport. J. Phys. Chem. B. 110:1394–1404. [DOI] [PubMed] [Google Scholar]
- 22.Léger, C., A. K. Jones, S. P. J. Albracht, and F. A. Armstrong. 2002. Effect of a dispersion of interfacial electron transfer rates on steady state catalytic electron transport in NiFe-hydrogenase and other enzymes. J. Phys. Chem. B. 106:13058–13063. [Google Scholar]
- 23.Aguey-Zinsou, K. F., P. V. Bernhardt, and S. Leimkuhler. 2003. Protein film voltammetry of Rhodobacter capsulatus xanthine dehydrogenase. J. Am. Chem. Soc. 125:15352–15358. [DOI] [PubMed] [Google Scholar]
- 24.Turner, K. L., M. K. Doherty, H. A. Heering, F. A. Armstrong, G. A. Reid, and S. K. Chapman. 1999. Redox properties of flavocytochrome c3 from Shewanella frigidimarina NCIMD400. Biochemistry. 38:3302–3309. [DOI] [PubMed] [Google Scholar]
- 25.Gewirth, A. A., and H. Siegenthaler, editors. 1995. Nanoscale Probes of the Solid/Liquid Interface. Kluwer, Dordrecht, The Netherlands.
- 26.Danilov, A. 1995. Scanning tunnelling and atomic force microscopy in the electrochemistry of surfaces. Russ. Chem. Rev. 64:767–781. [Google Scholar]
- 27.Zhang, J., Q. Chi, A. M. Kuznetsov, A. G. Hansen, H. Wackerbarth, H. E. M. Christensen, J. E. T. Andersen, and J. Ulstrup. 2002. Electronic properties of functional biomolecules at metal/aqueous solution interfaces. J. Phys. Chem. B. 106:1131–1152. [Google Scholar]
- 28.Zhang, J. D., Q. J. Chi, T. Albrecht, A. M. Kuznetsov, M. Grubb, A. G. Hansen, H. Wackerbarth, A. C. Welinder, and J. Ulstrup. 2005. Electrochemistry and bioelectrochemistry towards the single-molecule level: theoretical notions and systems. Electrochim. Acta. 50:3143–3159. [Google Scholar]
- 29.Andersen, J. E. T., P. Moller, M. V. Pedersen, and J. Ulstrup. 1995. Cytochrome c dynamics at gold and glassy-carbon surfaces monitored by in situ scanning tunnel microscopy. Surf. Sci. 325:193–205. [Google Scholar]
- 30.Zhang, J., Q. Chi, S. Dong, and E. Wang. 1996. In situ electrochemical scanning tunnelling microscopy investigation of structure for horseradish peroxidase and its electrocatalytic property. Bioelectrochem. Bioenerg. 39:267–274. [Google Scholar]
- 31.Andersen, J. E. T., K. G. Olesen, A. I. Danilov, C. E. Foverskov, P. Moller, and J. Ulstrup. 1997. Covalently immobilised cytochrome c imaged by in situ scanning tunnelling microscopy. Bioelectrochem. Bioenerg. 44:57–63. [Google Scholar]
- 32.Chi, Q. J., J. D. Zhang, J. U. Nielsen, E. P. Friis, I. Chorkendorff, G. W. Canters, J. E. T. Andersen, and J. Ulstrup. 2000. Molecular monolayers and interfacial electron transfer of Pseudomonas aeruginosa azurin on Au(111). J. Am. Chem. Soc. 122:4047–4055. [Google Scholar]
- 33.Chi, Q. J., J. D. Zhang, J. E. T. Andersen, and J. Ulstrup. 2001. Ordered assembly and controlled electron transfer of the blue copper protein azurin at gold (111) single-crystal substrates. J. Phys. Chem. B. 105:4669–4679. [Google Scholar]
- 34.Hansen, A. G., A. Boisen, J. U. Nielsen, H. Wackerbarth, I. Chorkendorff, J. E. T. Andersen, J. D. Zhang, and J. Ulstrup. 2003. Adsorption and interfacial electron transfer of Saccharomyces cerevisiae yeast cytochrome c monolayers on Au(111) electrodes. Langmuir. 19:3419–3427. [Google Scholar]
- 35.Zhang, J. D., H. E. M. Christensen, B. L. Ooi, and J. Ulstrup. 2004. In situ STM imaging and direct electrochemistry of Pyrococcus furiosus ferredoxin assembled on thiolate-modified Au(111) surfaces. Langmuir. 20:10200–10207. [DOI] [PubMed] [Google Scholar]
- 36.Bonanni, B., D. Alliata, A. R. Bizzarri, and S. Cannistratto. 2003. Topological and electron-transfer properties of yeast cytochrome c adsorbed on bare gold electrodes. ChemPhysChem. 4:1183–1188. [DOI] [PubMed] [Google Scholar]
- 37.Zhang, J. D., A. M. Kuznetsov, and J. Ulstrup. 2003. In situ scanning tunnelling microscopy of redox molecules. Coherent electron transfer at large bias voltages. J. Electroanal. Chem. 541:133–146. [Google Scholar]
- 38.Zhang, J. D., A. C. Welinder, A. G. Hansen, H. E. M. Christensen, and J. Ulstrup. 2003. Catalytic monolayer voltammetry and in situ scanning tunneling microscopy of copper nitrite reductase on cysteamine-modified Au(111) electrodes. J. Phys. Chem. B. 107:12480–12484. [Google Scholar]
- 39.Alessandrini, A., M. Salerno, S. Fabbroni, and P. Facci. 2005. Single-metalloprotein wet biotransistor. App. Phys. Lett. 86:133902. [Google Scholar]
- 40.Friis, E. P., J. E. T. Andersen, Y. I. Kharkats, A. M. Kuznetsov, R. J. Nichols, J. D. Zhang, and J. Ulstrup. 1999. An approach to long-range electron transfer mechanisms in metalloproteins: in situ scanning tunneling microscopy with submolecular resolution. Proc. Natl. Acad. Sci. USA. 96:1379–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chi, Q. J., O. Farver, and J. Ulstrup. 2005. Long-range protein electron transfer observed at the single-molecule level: in situ mapping of redox-gated tunneling resonance. Proc. Natl. Acad. Sci. USA. 102:16203–16208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simon, J. 2002. Enzymology and bioenergetics of respiratory nitrite ammonification. FEMS Microbiol. Rev. 26:285–309. [DOI] [PubMed] [Google Scholar]
- 43.Bamford, V. A., H. C. Angove, H. E. Seward, A. J. Thomson, J. A. Cole, J. N. Butt, A. M. Hemmings, and D. J. Richardson. 2002. Structure and spectroscopy of the periplasmic cytochrome c nitrite reductase from Escherichia coli. Biochemistry. 41:2921–2931. [DOI] [PubMed] [Google Scholar]
- 44.Gwyer, J. D., D. J. Richardson, and J. N. Butt. 2006. Inhibiting Escherichia coli cytochrome c nitrite reductase: voltammetry reveals an enzyme equipped for action despite the chemical challenges it may face in vivo. Biochem. Soc. Trans. 34:133–135. [DOI] [PubMed] [Google Scholar]
- 45.Burlat, B., J. D. Gwyer, S. Poock, T. Clarke, J. A. Cole, A. M. Hemmings, M. R. Cheesman, J. N. Butt, and D. J. Richardson. 2005. Cytochrome c nitrite reductase: from structural to physicochemical analysis. Biochem. Soc. Trans. 33:137–140. [DOI] [PubMed] [Google Scholar]
- 46.Clavilier, J., R. Faure, G. Guinet, and R. Durand. 1980. Preparation of mono-crystalline Pt microelectrodes and electrochemical study of the plane surfaces cut in the direction of the (111) and (110) planes. J. Electroanal. Chem. 107:205–209. [Google Scholar]
- 47.Hamelin, A. 1996. Cyclic voltammetry at gold single-crystal surfaces.1. Behaviour at low-index faces. J. Electroanal. Chem. 407:1–11. [Google Scholar]
- 48.Zhang, J., and J. Ulstrup. 2006. Oxygen-free in situ scanning tunnelling microscope. J. Electroanal. Chem. In press.
- 49.Armstrong, F. A., A. M. Bond, H. A. O. Hill, I. S. M. Psalti, and C. G. Zoski. 1989. A microscopic model of electron-transfer at electroactive sites of molecular dimensions for reduction of cytochrome c at basal-plane and edge-plane graphite-electrodes. J. Phys. Chem. 93:6485–6493. [Google Scholar]
- 50.Bard, A. J., and L. R. Faulkner. 2001. Electrochemical Methods: Fundamentals and Applications. John Wiley & Sons, New York.
- 51.Albrecht, T., A. Guckian, J. Ulstrup, and J. G. Vos. 2005. Transistor-like behavior of transition metal complexes. Nano Lett. 5:1451–1455. [DOI] [PubMed] [Google Scholar]
- 52.Haiss, W., H. van Zalinge, S. J. Higgins, D. Bethell, H. Höbenreich, D. J. Schiffrin, and R. J. Nichols. 2003. Redox state dependence of single molecule conductivity. J. Am. Chem. Soc. 125:15294–15295. [DOI] [PubMed] [Google Scholar]
- 53.Li, Z., B. Han, G. Meszaros, I. Pobelov, T. Wandlowski, A. Blaszcyk, and M. Mayor. 2006. Two-dimensional assembly and local redox-activity of molecular hybrid structures in an electrochemical environment. Faraday Discuss. 131:121–143. [DOI] [PubMed] [Google Scholar]
- 54.Kuznetsov, A. M., and J. Ulstrup. 2000. Mechanisms of in situ scanning tunnelling microscopy of organized redox molecular assemblies. J. Phys. Chem. A. 104:11531–11540. Errata: J. Phys. Chem. A. 105:7494. [Google Scholar]