

Abstract
Basonuclin (Bnc 1) is a transcription factor that has an unusual ability to interact with promoters of both RNA polymerases I and II. The action of basonuclin is mediated through three pairs of evolutionarily conserved zinc fingers, which produce three DNase I footprints on the promoters of rDNA and the basonuclin gene. Using these DNase footprints, we built a computational model for the basonuclin DNA-binding module, which was used to identify in silico potential RNA polymerase II target genes in the human and mouse promoter databases. The target genes of basonuclin show that it regulates the expression of proteins involved in chromatin structure, transcription/DNA-binding, ion-channels, adhesion/cell-cell junction, signal transduction and intracellular transport. Our results suggest that basonuclin, like MYC, may coordinate transcriptional activities among the three RNA polymerases. But basonuclin regulates a distinctive set of pathways, which differ from that regulated by MYC.
Introduction
Basonuclin (Bnc-1) is a transcription factor with highly restricted tissue distributions; it is found mainly in the basal keratinocytes of stratified epithelium (e.g., epidermal, corneal, esophageal and virginal epithelia), and the reproductive germ cells of testis and ovary. Basonuclin possesses three pairs of zinc fingers and produces three DNase I footprints on the promoters of human and mouse ribosomal RNA gene (rDNA) promoter as well as the promoter of the human basonuclin gene. Basonuclin binding sites on the rDNA are highly conserved between human and mouse, suggesting that it is functional [1-3]. And several lines of evidence also suggest that basonuclin indeed regulates rRNA transcription. However, basonuclin differs from the traditional Pol I transcription factor in that it is also present in the nucleoplasm and can interact with its own gene promoter, which suggest it may also regulate Pol II-mediated transcription [2]. This notion is supported by a recent study in the basonuclin knock-down model in mouse oocytes, in which, a large number of Pol II transcripts were perturbed [4].
Basonuclin’s potential to regulate both Pol I and Pol II transcription is rare among transcription factors. TATA binding-protein (TBP) and c-MYC are the only proteins, which have been shown to involve in the activity of all the three RNA polymerases (Pol I, II and III). TBP was isolated with the basal transcription complexes of the three polymerases and appeared to serve a basic function [5-7]. c-MYC, which plays a key role in controlling cell proliferation, growth and tumorigenesis, was shown to modulate Pol II and III transcription by interacting with Pol II gene promoters and by binding to TFIIIB, an essential transcription factor for Pol III [8, 9]. Recently, several reports showed that c-MYC (and d-MYC) also interacted with rDNA promoter and regulated rRNA transcription and processing [10, 11]. These observations, along with the previously published data, make c-MYC very unique in its ability to influence all RNA polymerases activities. Such ability is consistent with MYC’s role in promoting cell proliferation and growth, which require enhanced ribosomal biogenesis with the participation of all three RNA polymerases [12]. More importantly, it suggests a new type of transcription regulators, which coordinate the activities of the RNA polymerases. We propose that basonuclin is also such a transcription coordinator, but regulates cellular functions that differ from the MYC.
Thus, identifying basonuclin target genes transcribed by Pol II becomes a critical step in understanding basonuclin function. To this end, we take advantage of the recent development of high-throughput analysis (e.g., microarray technology and genomic databases), which is capable of examining a large number of genes in multiple genomes in silico [13] and has accelerated considerably the process of target gene identification. We searched computationally the current human and mouse promoter databases for the presence of the basonuclin binding sites. A number of screening criteria were also used to filter out the non-target genes. The candidate promoters were then verified by ChIP as well as by pathway analysis.
Materials and Methods
Computational analysis
Human (hg17) and mouse (mm5) genomic sequences were from UCSC genome database (http://genome.ucsc.edu/). DBTSS Transcription Start Site (TSS) annotation and ortholog dataset (version 5.2.0) were downloaded on June 20, 2006 from ftp://ftp.hgc.jp/pub/hgc/db/dbtss/Yamashita_NAR/ [14]. The Ensembl transcripts and human-mouse ortholog dataset were downloaded on Nov. 1, 2005 from http://www.ensembl.org/Multi/martview [15]. The basonuclin DNase I foot printing sequences were obtained from [2, 3] and Tseng, unpublished.
Consensus [16] with default parameter setting was used to define the basonuclin zinc finger-binding site. These binding sites were aligned and used to construct the computational model. The most widely used method to search for binding site is the Position Weight Matrix (PWM), which assumes independency between individual binding site positions [17]. However, this assumption of independence is not always true [18, 19] and we noted that nucleotides in the basonuclin binding site were also position dependent, for example, nucleotide ‘A’ occur at the 6th position only when T occurred at the 2nd position. We used a propensity model (Position Specific Propensity Matrix or PSPM) to capture the inter-dependency between nucleotide positions within the binding site [20]. The propensity of an oligonucleotide sequence is defined as its occurrences in the binding site sequences divided by its expected occurrence calculated from the base composition of the genome. Because basonuclin’s DNase I footprints were found between -500 and +100 (TSS being +1), this region of the promoters was extracted from the genomic sequences and masked for repetitive elements. To search for basonuclin binding site in this promoter database, we used a sliding window to find hexanucleotides with a score above the cutoff (P<0.003) (see Results). The score for each hexanucleotide (the window size) was calculated according to the PSPM model, which considers a tract of bases instead of a single base. The length of the tract was variable [20] and no greater than 5. The score of each window was the product of propensity values of the six tracts in that window, each starting at a different position.
Sequences were aligned first with LAGAN [21] and Needle in EMBOSS [22] and the machine-generated alignments were examined manually for accuracy.
Cell Culture and Chromatin Immunoprecipitation (ChIP)
HaCaT cells were grown on 100mm diameter culture dishes in DMEM supplemented with 10% fatal calf serum at 37°C and 5% CO2. For immunocytochemistry, HaCaT cells were grown on chamber slides (Nunc Inc., Naperville, IL), fixed and stained as described previously [23].
ChIP was performed with a kit (Upstate Cell Signaling Solutions, NY, USA) according to manufacturer’s instruction with slight modifications, and the second round ChIP was done as previously described [24, 25]. Briefly, HaCaT cells were plated at 2 X106 cells in each 100 mm dish and cultured for two days. Cells were fixed with 1% formaldehyde in PBS for 10 minutes at 37 °C to crosslink protein and DNA. The cells were then washed with iced-cold phosphate-buffered saline (PBS) twice. Cells on each dish were lysed with 300 microliters lysis buffer [1% NP-40, 0.5% sodium deoxycholate, 0.1%SDS, 5mM EDTA, 0.5 mM phenylmethanesulfonyl fluoride or PMSF, 100 nanograms/ml protease inhibitor cocktail (Roche Applied Sciences, Penzberg, Germany) in 1X PBS]. Lysates were incubated on ice for 15 minutes and sonicated on ice with an ultrasonic sonicator (Dr. Hielscher UP 100H) at amplitude=1 and duty cycle=100% in 12 one-minute pulses. The resulting DNA fragments were between 0.1 to 3.5 kb in length. The sonicated cell lysates was centrifuged at 13,000 rpm for 10 minutes at 4 °C and the supernatant was precleared with Protein A Sepharose beads (Amersham Biosciences, Piscataway, NJ) in a solution containing 0.5% BSA, 0.1% sonicated salmon sperm DNA and the Roche protein inhibitor cocktail with constant rotation at 4°C for 1 hour. The pre-cleared lysate was recovered by centrifugation at 6,000 rpm at 4 °C for 5 minutes, and 400 microliters were incubated with 2 micrograms of anti-basonuclin (Bcn1) or 2 micrograms of normal rabbit IgG as a negative control at 4 °C overnight. The immuno complexes were absorbed with 75 microliters of packed salmon sperm DNA/BSA/Protein A Sepharose beads at 4 °C for 3 hours and precipitated at 6,000 rpm at 4°C for 5 minutes. The precipitated beads were washed for 5 times with each immunoprecipitation buffers in the following order: once with the low salt buffer (0.1% SDS, 1% Triton X100, 2mM EDTA, 20 mM Tris-HCl, pH 8.1, 150 mM NaCl) for 5 minutes, once with the high salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl) for 5 minutes, once with the LiCl buffer (0.25M LiCl, 1% IGEPAL-CA630, 1% deoxycholic acid/sodium salt, 1mM EDTA, 10 mM Tris, pH 8.1) for 5 minutes, and twice with TE buffer (10 mM Tris-HCl, 1mM EDTA, pH 8.0) for 5 minutes. Immuno complexes were eluted with 100 microliters of elution buffer (1% SDS, 0.1 M NaHCO3) at room temperature for 1 hour. The eluate was diluted with 900 microliters of dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl) and used for the second round immunoprecipitation, which was identical to the first, except the immunocomplexes were eluted twice using 150 microliters of elution buffer at room temperature for one hour each time. Cross-link was reversed by incubating the precipitated chromatin in 200 mM NaCl at 65 °C overnight, and the DNA was purified by digestion with RNase (20 micrograms) and proteinase K (50 micrograms) at 55°C for two hours, followed by phenol/chloroform extraction.
Western blotting
HaCaT cell nuclear extract was prepared as previously described [24, 25]. Protein samples were electrophoresed on SDS-polyacrylamide gel (SDS-PAGE) and electro-transferred onto polyvinylidene difluoride (PVDF) membranes (Immobilon-P, Millipore, Bedford, MA), which were incubated with primary antibodies as indicated at 4°C overnight. The primary antibodies were visualized by horseradish peroxidase-conjugated secondary antibodies (1:10000 dilution) and detected with an ECL western blotting system (Amersham Biosciences, Pdscataway, NJ). Prestained molecular weight standards (New England Biolabs, MA) were used in estimating the apparent molecular weight.
Results
Building a computational model of basonuclin binding site
Basonuclin possesses three pairs of evolutionarily conserved zinc fingers, which are the only recognizable DNA-binding motifs in the protein. These zinc fingers were shown to mediate sequence specific bindings on a number of promoters of both RNA polymerase I (Pol I) and II (Pol II) [1-3]. The binding sites on rDNA promoter, as defined by DNase I foot printing, are likely functional because they are conserved between human and mouse [3]. Based on the DNase I foot printing data, we proposed a consensus sequence of basonuclin binding site (A/G/C)G(C/T)G(G/A/T)C) [3]. In order to define more precisely the binding site and to search genome-wide for potential basonuclin targets, we used computational approaches to refine the description of the binding sequence.
The initial data set comprised of 11 oligonucleotide sequences (12-18 nucleotides), which were protected by basonuclin N-terminal pair of zinc fingers in DNase-I foot printing experiments (Fig. 1A). These sequences were from three genes: human and mouse rDNA promoters (Pol I) and the human basonuclin gene promoter (Pol II). Because one pair of C2H2 type zinc fingers recognizes six nucleotides [26], we extracted the most overrepresented hexanucleotide motif from this DNase footprint data set with Consensus [16], which generates sequence matrix with a statistical significance. The most significant hexanucleotide, G(T/C)G(A/T/C)C(C/A) (Fig 1B), resembled closely to the one reported previously (A/G/C)G(T/C)G(A/T/C)C [3]. The Position Weight Matrix algorithm (PWM) [17, 20] was modified (see Methods) and employed to build a statistical model of the basonuclin binding site. A statistical distribution for the binding site model was calculated by screening 10,000 random sequences (600-nucleotide in length) from the human genome intergenic region, and a score cutoff value of 14.68, corresponding to p=0.005 was obtained. Any hexanucleotide with a score above this cutoff value was regarded as a putative basonuclin-binding site for further analysis.
Figure 1.
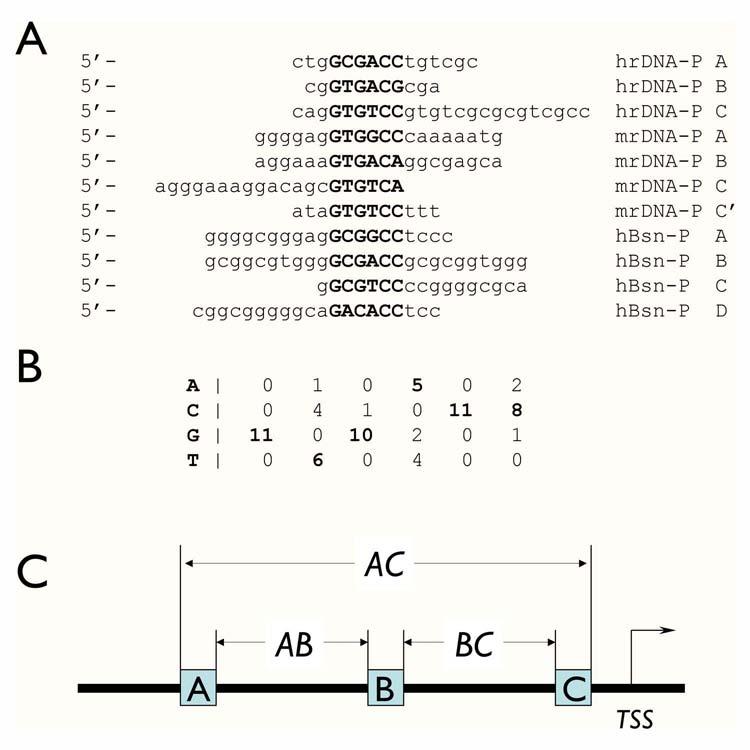
Modeling basonuclin-binding site. A) The eleven Pol I and Pol II promoter sequences, which were protected from DNase I digestion by the N-terminal zinc fingers of basonuclin, were analyzed with software program Consensus, which identifies the statistically most significant sequence matrix, which likely to be the recognition sequence of a protein. The sources of the sequence were: hrDNA-P, human rDNA promoter; mrDNA-P, mouse rDNA promoter; hBsn-P, human basonuclin gene promoter. B) The nucleotide frequency matrix of the basonuclin zinc finger-binding site derived by Consensus. The parameters associated with this matrix were: number of sequences = 11; unadjusted information = 4.79954; sample size adjusted information = 3.8627; ln(p-value) = -33.743; p-value = 2.2162E-15; ln(expected frequency) = -5.68861 expected frequency = 0.00338429. C) The basonuclin binding site module with three basonuclin binding sites constrained by their orientation and spacings (AB and BC). The other defining parameter is the distance of the binding module to the transcription start site (TSS).
Basonuclin binding module
Most likely, basonuclin interacts with DNA at three individual sites simultaneously (Fig 1C). This proposition is based on the fact that basonuclin possesses three pairs of evolutionarily conserved zinc fingers ([27, 28], and Fig. 2), which are well-separated in the primary sequence; and that DNase-I foot printing experiments identified three adjacent footprints with conserved spacing in human and mouse rDNA promoters [1-3], as well as on basonuclin’s own gene promoter ([2], Tseng, unpublished). Based on these data, we built a basonuclin-binding module, which consisted of three basonuclin binding sites (the PWM model), had a defined dimension (200 nucleotides; AC in Fig. 1C) and a restricted location in relation to the transcription start site (TSS) (within -500 to +100; +1 is TSS). Within the module, the consensus binding sites were constrained by their orientation and spacing (30-65 and 70-100 nts for AB and BC, respectively. Fig. 1C).
Figure 2.

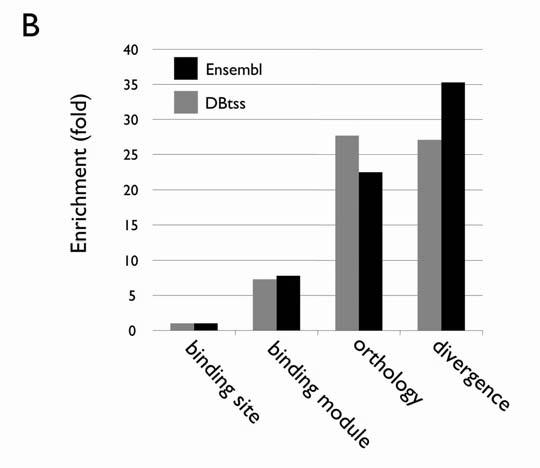
Evolutionary conservation as a powerful criterion for screening computationally derived basonuclin target candidates. (A) A comparison of basonuclin zinc fingers in some of the vertebrates. The amino acid sequences of the three pairs of basonuclin zinc fingers (ZF12, ZF34 and ZF56) from human (hBsn), chimpanzee (pBsn), mouse (mBsn), domestic dog(cBsn), cow(bBsn), fowl(gBsn) and zebrafish (dBsn) are align. The cystines and histidines are in bold and the amino acid substitutions are highlighted in yellow. (B) The enrichment of basonuclin target candidates during each step of screening. The results from screening two databases (Ensembl and DBTSS) are shown and the data are listed in Table 1.
Search for basonuclin binding module in mammalian orthologous promoters.
Because basonuclin’s DNase I footprints were found between -500 and +100 (TSS being +1), we performed a search for basonuclin-binding module in this region of the human promoter sequence in the Ensembl [15] and DBTSS [29] databases and detected the module in >2,000 promoters of human or mouse genes in each database (Table 1), representing more than 10% of the total number of promoters in the database. Because of the high degree of amino acid sequence conservation of basonuclin zinc fingers (Fig. 2A), basonuclin-binding modules should also be conserved at least in the mammalian orthologous promoters, as we showed with the rDNA promoters of human and mouse [3]. Phylogenetic footprinting in silico has been used to identify functional elements of evolutional conserved genes [30, 31]. Therefore we used evolutionary constraint to filter out the binding modules that were not conserved. According to the Ensembl and DBTSS annotations, there were a total of 27,402 and 11,900 orthologous genes between human and mouse, respectively (Table 1). The orthology constraint reduced the number of candidates in the human and mouse database from around 2,000 to 500-600, a ∼3-4 fold enrichment (Table 1, Fig. 2B). The binding modules in the human and mouse orthologs promoters were screened further by restricting the divergence of the corresponding orthologous binding sites in the module to no more than 17% (allowing one mismatch in each hexanucleotide) (Fig. 3), which was within the divergence between human and mouse orthologous cDNA sequences (∼15%) [32]. Consistent with previous reports, this restriction reduced the number of candidates to 15 (DBTSS) and 25 (Ensembl), and combined, they represented 33 unique genes, excluding that of basonuclin gene. A manual examination of the sequence alignment revealed that in six of the 33 pairs of promoters, the human and mouse basonuclin modules could not align well (Fig. 3B). These six promoters were eliminated from further analysis.
Table I.
Criteria for selecting basonuclin target promoters
| Ensembl | human | mouse | h/m orthologs |
|---|---|---|---|
| Total promoter | 25111 | 16002 | |
| Promoter with at least one binding site | 24844 | 15828 | |
| Promoter with at least one binding module | 3293 | 2227 | |
| Human and mouse orthologous promoter containing the module | 662 | 466 | |
| Binding site and sequence alignment conservation | 21 | ||
| PCR possible | 12 | ||
| Verified by ChIP | 5 (42%) | ||
| DBTSS | human | mouse | h/m orthologs |
| Total promoter | 30964 | 19023 | |
| Promoter with at least one binding site | 30478 | 18676 | |
| Promoter with at least one binding module | 3684 | 2596 | |
| Human and mouse orthologous promoter containing the module | 528 | 529 | |
| Binding site and sequence alignment conservation | 12 | ||
| PCR possible | 9 | ||
| Verified by ChIP | 6 (67%) |
Figure 3.
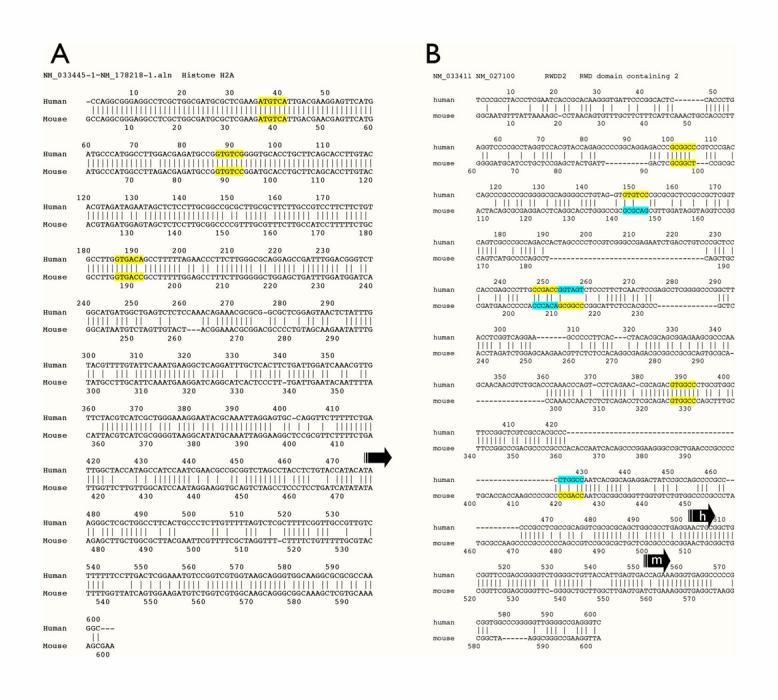
Examples of the sequence alignment of computationally derived human and mouse basonuclin target promoters. A) A good alignment, in which the promoter regions as well as the basonuclin binding sites are well aligned. Such an alignment usually indicates a higher probability of successful PCR. B) A poor alignment. Although the basonuclin binding modules are present in both human and mouse promoters, the poor alignment of the promoter sequence often led to PCR failure. Conserved binding sites are marked in yellow and not conserved sites in yellow/blue, in which yellow marked sites identified by computation. Arrows indicate the annotated transcription start sites.
Verification of basonuclin targets by ChIP
The computational model predicts basonuclin’s DNA sequence-specific interaction with the target promoters, and thus chromatin immunoprecipitation (ChIP), which detects protein-DNA interactions in vivo, was used to verify these predictions. We performed ChIP with HaCaT cells, a spontaneously established human keratinocyte line [23], which contained a high level of nuclear basonuclin (Fig. 4A). ChIPs were performed using antibodies against basonuclin [2] and rabbit immunoglobulins were used as a negative control. Western analysis showed that all detectable basonuclin was quantitatively precipitated (Fig 4B). To reduce the background, the immunoprecipitation was carried out once more and the twice-immunoprecipitated genomic DNA was analyzed by PCR. We first examined the presence of basonuclin gene promoter (Fig. 5A, BNC), because it was used to define the computational model (Fig. 1A). PCR amplified the basonuclin promoter only from basonuclin-containing precipitates, but not that obtained with the general rabbit immunoglobulins (negative control) (Fig. 5A, IgG). This result confirmed our previous in vitro results [2] and demonstrated for the first time basonuclin’s interaction with its own gene promoter in vivo. PCR with promoter specific primers (Fig. 5B) showed that 11 of the 21 promoters examined were present in basonuclin-containing ChIP precipitates. The remaining four candidate promoters could not be efficiently amplified by PCR, presumably because sequence errors in the database. Thus the average verification rate was ∼52%, which was apparently dependent on the quality of the databases (67% for the DBTSS and 42% for the Ensembl)(Table 1). In general, the target genes did not cluster or show preference regarding their chromosomal locations (Fig. 5B). These target genes spread over 14 different chromosomes, 4 of them occur in chromosome 2 and 3 in chromosome 1, which are about 1.34 and 2.62 fold than the expected based on gene distribution among the genome chromosomes. Two target genes occur in chromosomes 18, 19 and 21, which are 5.91, 1.69 and 7.77 fold high as expected.
Figure 4.
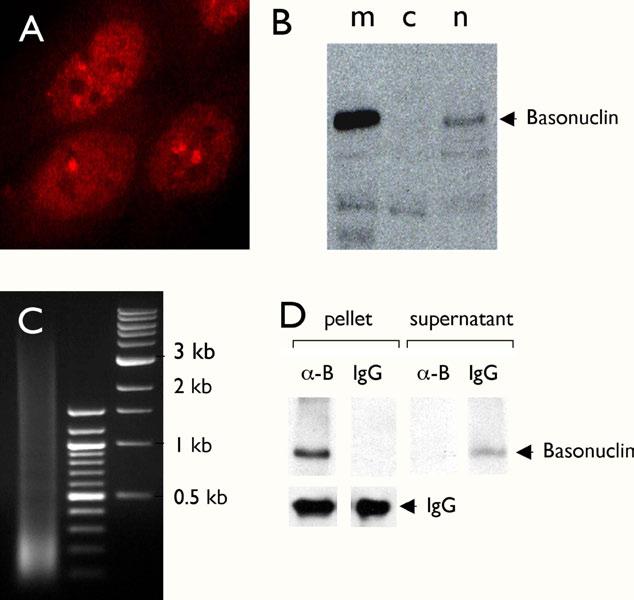
ChIP assay in HaCaT cells. Immunocytochemical staining of cultured HaCaT cells with an anti-basonuclin antibody showed that basonuclin was localized mainly in the nucleoplasm with aggregates within nucleolus and the perinucleolar region (A). Basonuclin’s nuclear localization was confirmed by Western analysis of cytoplastic (B, lane c) and nuclear (B, lane n) fractions. Lane m, exogenously expressed basonuclin in the 293 cells, serving as a molecular weight marker. For the ChIP assay, the crosslinked chromatin was sonicated to produce DNA fragments of size between 0.1 to 3.5 kb (Lane 1, C). Lanes 2 and 3 in C, molecular weight markers. Basonuclin in the ChIP lysate was quantitatively precipitated (i.e., only in the pellet, not in the supernatant) with the anti-basonuclin antibody (∼-B) but not with the control rabbit IgG (D). The lower left panels show that similar amounts of antibodies were used in the assay (IgG).
Figure 5.
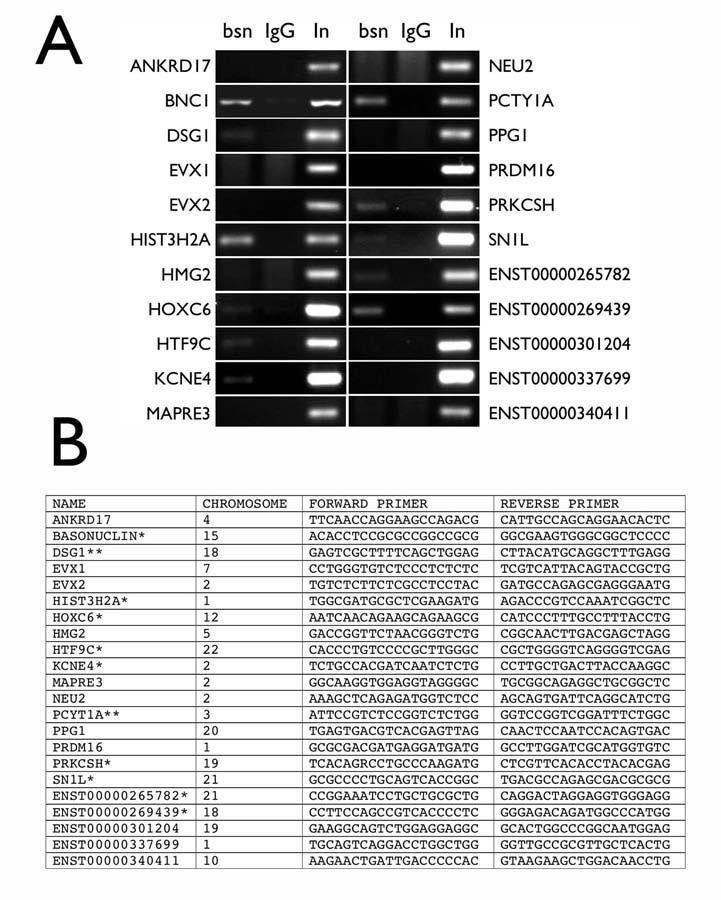
PCR verification of the basonuclin target promoters in genomic DNA precipitated by an anti-basonuclin antibody (ChIP). Electrophoretic analysis of PCR products amplified from chromatins precipitated by the anti-basonuclin antibody (bsn) and the control IgG (IgG); In, input genomic DNA normalized to be of equal amount to that of the genomic DNA precipitated with the anti-basonuclin antibody (A). The promoter identity is indicated on the left or right of the gel photo. Basonuclin gene promoter (BCN) serves as a positive control for the ChIP assay. (B) A list of the PCR primers used for the analysis shown in A. ChIP verified genes are marked with *, of which, also perturbed by basonuclin-deficiency, **. The chromosomal locations of the target genes are also listed.
Pathways perturbed by basonuclin-deficiency and the basonuclin target genes
It can be argued that basonuclin target genes should relate functionally. A manual analysis of the computation-derived candidates with known identity (18/29) showed that they clustered in six function groups: 1) chromatin structure, 2) transcription/DNA-binding, 3) adhession/intercellular junction, 4) Ion-channels, 5) signal transduction, and 6) protein transport/endoplastic reticulum (Table 2). Analysis with a web-based program GeneMerger [33] revealed similar enrichment (2-5 fold) in GO terms, which were statistically significant (see Supplementary Materials, S1), but the classification was not as precise as the manually built list. This result prompted us to compare the computation-derived basonuclin target genes with a list of genes perturbed by basonuclin-deficiency in mouse oocytes. Previously, we generated, via transgenic RNAi, a basonuclin-knock-down model for mouse oocytes, which showed that basonuclin is apparently a maternal effect gene; basonuclin deficiency in oocytes led to embryonic developmental failure [4]. In order to identify the molecular defects in the oocytes that might influence early embryogenesis, we examined, by microarray, the gene expression profile of the germinal vesicle (GV) intact oocytes with or without basonuclin. The results were published in [4] and the data deposited to the Gene Expression Omnibus Repository (Accession Number GSE4029). The GV stage oocytes were chosen because the active transcription during the proceeding growth phase comes to a halt and transcription will not resume till in the early zygotes. Thus, the mRNA composition at the GV-intact stage reflects not only the cumulative effect of Pol II transcription during oocyte growth, but also is the source of the mRNA at the beginning of oocyte maturation and thus may have important bearings on early zygotic development. This sampling time was many days after the onset of inhibition of basonuclin in oocytes, hence was not ideal for detecting the primary effect of basonuclin-deficiency and basonuclin’s direct targets [4]. It was, however, suitable for detecting the pathways perturbed by the loss of basonuclin. Indeed, only two (10%) computationally derived candidates were present among the perturbed genes identified by the microarray analysis, but the function of the computation-derived target genes matched very well with the pathways perturbed in basonuclin-deficient oocytes ([4]; Table 2).
Table 2.
Pathways analysis of basonuclin target genes (candidates)
| Pathways | Basonuclin target candidates | Perturbed genes in the similar patway in basonuclin-deficient oocytes | Fold charge |
|---|---|---|---|
| Chromatin structure | •Histone H2A | •Histone IC | -3.0 |
| •Hpall-tiny-fragments locus 9C | •DNA methyltransferse 38 | -1.5 | |
| •High mobility group 2 | •Myeloid/lymphoid or mixed-lineage leukema 3 | -1.5 | |
| •High mobility group nucleosomal binding domain 3 | +14.5 | ||
| •Nucleosome binding protein I | +2.2 | ||
| •High mobility group 20A | -1.5 | ||
| •Male-specific lethal-3 homolog I | -1.5 | ||
| •High mobility group box 2-like I | -1.6 | ||
| •RAN, RAS oncogene family | -1.9 | ||
| •High mobility group B I | -2.2 | ||
| Transcription/DNA binding* | •General transcription factor IIIC | •TATA box binding protein (Tbp)-associated factor, RNA polymerase I, A | -1.5 |
| •Homeobox C6 isoform I | •Homeo box AI | +61.0 | |
| •Homeobox A92 | •Homeo box D9 | +12.8 | |
| •Myc-associated zinc finger protein | •Paired-like homeodomain transcription factor 2 | +20.0 | |
| •Even-skipped related gene | •FBJ osteosarcoma oncogene | +12.0 | |
| •Ankyrin repeat domain protein 17 isoform b | •SRY-box containing gene 2 | +11.0 | |
| •Transcription factor AP-2, alpha | +6.7 | ||
| •Fibronectin type 3 and ankyrin repeat domains I | -1.6 | ||
| Adhesion/cell-cell junction | •Devnoglein IA | •Protocardherin I2 | +12.3 |
| •Protocardherin 18 precursor | •Protocadherin alpha I | +10.6 | |
| •Protocadherin alpha 6 | +10.6 | ||
| •Cadherin 8 | +10.3 | ||
| •Calnexin | -1.3 | ||
| Ion-channels | •Potassium voltage-gated channel, Isk- | •Gap junction membrane channel protein beta-2 | +78.0 |
| •FXYD domain-containingn ion transport regulator 6 | +47.0 | ||
| •Sodium channel, nonvoltage-gated type I alpha polypeptide | +20.7 | ||
| •Transient receptor potential cation channel subfamily M4 | +12.0 | ||
| •Calcium channel, voltage-dependent, alpha2/delat subunit 2 | +8.0 | ||
| •Gap junction membrane channel | +5.8 | ||
| •Potassium intermediate/small conductance calcium-activated channel, subfamily N, member 3 | +5.1 | ||
| •Potassium channel, subfamily K, member I | +4.9 | ||
| •Transient receptor potential cation channel, subfamily M member 6 | +4.6 | ||
| •Chloride channel 3 | +4.4 | ||
| •Potassum voltage-gated channel subfamily H(eag-related), member 2) | +4.3 | ||
| •Purinergic receptor P2X, ligand-gated ion channel 4 | +3.8 | ||
| •Calcium channel, voltage-dependent, beta 2 subunit | +3.6 | ||
| •Gp38 (Not wator channel activity) | -1.7 | ||
| •Potassium channel tetramerization domain-containing | -2.4 | ||
| •Channel-interacting PDZ domain protein | -3.0 | ||
| Signal transduction | •Protein kinase C substrate BOK-H | •Regulator of G protein signaling 7 | +24.6 |
| •Hemopoietic cell kinase | +13.8 | ||
| •Neurocumin receptor 2 •Histamine receptor H I | +11.0 | ||
| •Serino/chrooninc kinace 23 | +11.0 | ||
| •Hexokinase 2 | +11.0 | ||
| •Protein tyrosine phosphatase, receptor type, D | +9.7 | ||
| •RAP2C, member of RAS oncogene family | +7.0 | ||
| •Ectonucleoside triphosphate diphosphohydrolase 2 | +7.0 | ||
| Calcium/calmodulin-dependent protein kinase kinase | +6.5 | ||
| 2, beta | +5.5 | ||
| Mitogen activated protein kinase kinase 6 | +2.6 | ||
| SH3-binding kinase | -2.2 | ||
| Guanine nucleotide releasing factor 2 | |||
| Intracellular transport | •Microtubule-associated protein | •Phonylalkylamine Ca2+ antagonist (emopemd) binding protein | +3.5 |
| •Vesicle amine transport protein I | •Sorting nexin 15 | +3.0 | |
| •Retention in endoplasmic reticulum I | •FK506 binding protein 9 | +2.5 | |
| •Vesicle-associated membrane protein 3 | -2.3 | ||
| •RABI, member RAS oncogene family | -2.1 | ||
| •Microtubule-actin crosslinking factor I | -2.1 | ||
| •Scinderin | -2.0 | ||
| •Vascuolar protein sorting 4a | -1.9 | ||
| •Vascuolar protein sorting II | -1.6 |
1Only the target genes with a known function (name) are listed.
2Pathway classification is based on public electronic annotations of various sources.
Supplementary Material(s1).
Gene antology analysis for 22 putative targets
| Biological Process | |||||
|---|---|---|---|---|---|
| Name | ID | Count | Genes) | Genes) | E value |
| transcription | GO:0006350 | 6 | 20.69% | 10.37% | 0.07 |
| transcription, DNA-dependent | GO:006351 | 6 | 20.69% | 9.87% | 0.06 |
| nucleic acid metabolism | GO:0019219 | 6 | 20.69% | 10.17% | 0.07 |
| regulation of transcription | GO:0045449 | 6 | 20.69% | 10.04% | 0.06 |
| regulation of metabolism | GO:0019222 | 6 | 20.69% | 11.18% | 0.1 |
| regulation of transcription, DNA-dependent | GO:0006356 | 6 | 20.69% | 9.71% | 0.06 |
| regulation of cellular metabolism | GO:0031323 | 6 | 20.69% | 10.85% | 0.09 |
| Molecular Function | |||||
| Name | ID | Count | Genes) | Genes) | E value |
| nucleic acid binding | GO:0003676 | 8 | 27.59% | 14.60% | 0.05 |
| cation binding | GO:0043169 | 7 | 24.14% | 13.93% | 0.1 |
| DNA binding | GO:0003677 | 7 | 24.14% | 10.06% | 0.02 |
| transcription regulator activity | GO:0030528 | 5 | 17.24% | 6.22% | 0.03 |
| transcription factor activity | GO:0003700 | 5 | 17.24% | 4.49% | 8.76E-03 |
| sequence-specific DNA binding | O:0043565 | 3 | 10.34% | 2.15% | 0.02 |
| hydrclase activity, hydrolyzing D-glycosyl compounds | GO:0004553 | 2 | 6.90% | 0.43% | 6.99E-03 |
| hydrclase activity, acting on glycosyl bonds | GO:0016798 | 2 | 6.90 | 0.55% | 0.01 |
| Cellular Component | |||||
| Name | ID | Count | Genes) | Genes) | E value |
| protein complex | GO:0043234 | 6 | 20.69% | 9.78% | 0.06 |
| nucleoplasm | GO:0005654 | 3 | 10.34% | 2.90% | 0.05 |
| organelle lumen | GO:0043233 | 3 | 10.34% | 3.89% | 0.1 |
| nuclear lumen | GO:0031981 | 3 | 10.34% | 3.51% | 0.08 |
| transcription factor complex | GO:0005667 | 3 | 10.34% | 2.32% | 0.03 |
| membrane-enclosed lumen | GO:0031974 | 3 | 10.34% | 3.89% | 0.1 |
Discussion
We report here the identification of 21 basonuclin target genes (excluding the basonuclin gene) via a computational approach. Among them, 11 were verified by ChIP, of which two were perturbed by basonuclin knock-down experiment. These genes represent a minimal set of basonuclin targets, whose promoters most likely interact with basonuclin.
In our computational search, we used five criteria to screen the candidates: 1) binding site (i.e., hexanucleotide position weighed matrix), 2) binding module (i.e., three binding sites restricted by their relative orientation and position, as well as their spacing to the transcription start site, TSS), 3) orthology of promoters, 4) the evolutionary divergence of the orthologous binding sites and 5) the binding module sequence alignment within the orthologous gene promoters. Binding site criterion yielded little enrichment, which was expected because of the low information content of the binding site sequence. Screening by the binding module was more effective, which yielded 7- to 8-fold enrichment. Phylogenetics/orthology relationship is a powerful criterion for filtering out binding site/modules that are not biologically significant [34, 35]. This relationship has been used to identify unknown cis regulatory elements in a promoter [30, 31]. Our results are consistent with this notion and the enrichment by orthological relationship alone is more than 25-fold (Fig. 2B). Our results, however, suggest that phylogenetic/orthologous relationship alone may not be sufficient when the parameters of the computational model is not very stringent as is the case of our basonuclin-binding module. After applying the restriction of orthologous relationships, there were still ∼700 candidates (Table 1). By restricting the sequence divergence of the orthologous binding sites in human and mouse candidate promoters to within the divergence of the protein coding sequences (∼15%, [32]), another 35-fold enrichment was achieved. We performed the analysis on two databases and found that the DBTSS database appears to be of higher quality [36] as was reflected in the similar number of orthologs in the human and mouse datasets, the higher success rate of PCR and, importantly, the higher ChIP verification rate (67% vs 42%) (Table 1).
We used two methods to verify the candidates of basonuclin targets: 1) examining the presence of basonuclin target promoter sequence in a ChIP assay; 2) comparing them with the list of genes perturbed in basonuclin-deficient mouse oocyte. Both methods are effective in validating the target genes but neither is capable of ruling out the potential target genes. ChIP assay is a more direct assessment of basonuclin-DNA interaction, which the computational model predicts. Of the 21 promoters that could be efficiently amplified by PCR, 11 (∼52%) were present in the chromatin precipitated by the anti-basonuclin antibody. The much higher verification rate of ChIP is most likely because it detects directly protein-DNA interaction. Also possible is that in the cultured HaCaT cells, more basonuclin target promoters are associated with basonuclin. Still the amount of PCR-amplified target promoter varied relative to the input, suggesting that basonuclin’s association with these promoters is not always 100%. This partial occupancy could be due to the dynamics of basonuclin-DNA interaction (e.g., a large dissociation coefficient) or the heterogeneity of the HaCaT cell population (i.e., not all cells are in the same physiological state). This variation in promoter occupancy also justifies the precaution that our ChIP results cannot be used to rule out a candidate promoter. We found two matches of the computationally derived basonuclin targets in the list of genes perturbed by inhibiting basonuclin function via RNAi in mouse oocytes. The perturbed genes were detected by microarray analysis and numbered in thousands [4]. One possible reason for the low match rate is that oocytes may not express all the basonuclin target genes. A more likely reason is that because our microarray study was designed for assessing the overall perturbation in Pol II transcription, the mRNA sampling time might not overlap optimally with the time frame of basonuclin action. Furthermore, the sampling took place at the germinal vesicle stage, which was more than 10 days after the onset of RNAi effect (i.e., at the beginning of growth phase) and hence is more suitable for investigating secondary and tertiary effects, including the compensatory reaction of the oocyte to basonuclin deficiency [4]. The fact that the pathways perturbed in oocytes matched well with the functions of the computationally derived basonuclin targets is consistent with the scenario that prior to the RNA sampling, these pathways had been disrupted by the loss of basonuclin.
The demonstration that basonuclin is associated with a group of Pol II-transcribed promoters strengthened the notion that basonuclin regulates both Pol I and Pol II transcription [2-4]. Basonuclin deficiency had opposite effects on some of the target genes, suggesting that it can function as a transcription activator as well as an inhibitor, which was not realized previously. Both desmoglein 1 (DSG 1) and phosphate cytidylyltransferase 1 gene (PCYT1A) promoters were shown to be associated with basonuclin by ChIP, but in basonuclin-deficient oocytes, desmoglein 1 mRNA level increased by 4.1 fold, whereas that of PCYT decrease by 1.6 fold. Most importantly, the identity of these potential basonuclin target genes points to the pathways, in which basonuclin plays a role. Of great interest is that basonuclin appears to associate with histone H2A (HIST3H2A) gene promoter and its occupancy is high as suggested by the ChIP assay. Furthermore, the gene promoters of HTFC9 (ChIP verified), a protein with DNA-binding and methyltransferase activity, and HMG2 (not ChIP verified), a highly abundant chromatin structure protein, also contain highly conserved basonuclin binding modules, suggesting that their expression may also be modulated by basonuclin. This notion is supported by the microarray analysis of basonuclin-deficient oocytes, which showed that the mRNA levels of histone H1C and at least five HMG proteins (Table 2) were altered by 2 to 15-fold, and may account for the chromatin de-condensation failure and fragmentation observed in the 2-cell embryos [4]. Basonuclin’s interaction with a potassium channel (KCNE4/MIR) gene promoter is also consistent with the perturbations in mRNA levels of at least 16 ion-channels from up-regulated by 78-fold to down-regulated by 3-fold (Table 2) in basonuclin-deficient oocytes, which is consistent with the dramatic oocyte cell surface change observed [4]. Combined, these observations strongly suggest that basonuclin target genes identified here are likely to be biologically meaningful. Our data also suggest the similarity in the mode of action of MYC and basonuclin. Both proteins are likely to exert their effect on Pol I and II transcription via direct interaction with the respective promoters, whereas that on Pol III via one of its transcription factors, i.e., MYC via TFIIIB [8, 9] and basonuclin, possibly via TFIIIC (GTFIIIC, Table 2). The human and mouse TFIIIC gene promoter contained well aligned basonuclin module, but repeated attempt to PCR-amplify the promoter was in vain with the input DNA (control). The Pol II target genes of MYC and basonuclin suggest that each regulates a different set of pathways ([37, 38];Table 2).
In summary, we describe a combined computational and biological approach to identify basonuclin target genes. We found that with a high quality database (DBTSS), approximately 67% of the computationally derive candidates could be verified by the ChIP assay. One of the critical criteria in filtering out non-target promoters is the evolutionary conservation, which accounted for more than 90% of the enrichment. The identity of basonuclin target genes suggests that it functions in highly diversified pathways. Basonuclin’s ability to interact with both Pol I and II promoters, and its potential to influence Pol III transcription via TFIIIC support the notion that it coordinates the activity of the RNA polymerases.
Acknowledgments
Acknowledgement: JW is a recipient of a NIH training grant (T32-HG00046). This work was supported by NIH grant AG14456 to HT.
References
- [1].Iuchi S, Green H. Basonuclin, a zinc finger protein of keratinocytes and reproductive germ cells, binds to the rRNA gene promoter. Proc Natl Acad Sci U S A. 1999;96:9628–9632. doi: 10.1073/pnas.96.17.9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tseng H, Biegel JA, Brown RS. Basonuclin is associated with the ribosomal RNA genes on human keratinocyte mitotic chromosomes. J Cell Sci. 1999;112(Pt 18):3039–3047. doi: 10.1242/jcs.112.18.3039. [DOI] [PubMed] [Google Scholar]
- [3].Tian Q, Kopf GS, Brown RS, Tseng H. Function of basonuclin in increasing transcription of the ribosomal RNA genes during mouse oogenesis. Development. 2001;128:407–416. doi: 10.1242/dev.128.3.407. [DOI] [PubMed] [Google Scholar]
- [4].Ma J, Zeng F, Schultz RM, Tseng H. Basonuclin: a novel mammalian maternal effect gene. Development. 2006;133:2053–2062. doi: 10.1242/dev.02371. [DOI] [PubMed] [Google Scholar]
- [5].Comai L, Tanese N, Tjian R. The TATA-binding protein and associated factors are integral components of the RNA polymerase I transcription factor, SL1. Cell. 1992;68:965–976. doi: 10.1016/0092-8674(92)90039-f. [DOI] [PubMed] [Google Scholar]
- [6].Comai L, Zomerdijk JC, Beckmann H, Zhou S, Admon A, Tjian R. Reconstitution of transcription factor SL1: exclusive binding of TBP by SL1 or TFIID subunits. Science. 1994;266:1966–1972. doi: 10.1126/science.7801123. [DOI] [PubMed] [Google Scholar]
- [7].Henry RW, Sadowski CL, Kobayashi R, Hernandez N. A TBP-TAF complex required for transcription of human snRNA genes by RNA polymerase II and III. Nature. 1995;374:653–656. doi: 10.1038/374653a0. [DOI] [PubMed] [Google Scholar]
- [8].Felton-Edkins ZA, Kenneth NS, Brown TR, Daly NL, Gomez- Roman N, Grandori C, Eisenman RN, White RJ. Direct regulation of RNA polymerase III transcription by RB, p53 and c-Myc. Cell Cycle. 2003;2:181–184. [PubMed] [Google Scholar]
- [9].Gomez-Roman N, Grandori C, Eisenman RN, White RJ. Direct activation of RNA polymerase III transcription by c-Myc. Nature. 2003;421:290–294. doi: 10.1038/nature01327. [DOI] [PubMed] [Google Scholar]
- [10].Arabi A, Wu S, Ridderstrale K, Bierhoff H, Shiue C, Fatyol K, Fahlen S, Hydbring P, Soderberg O, Grummt I, Larsson LG, Wright AP. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat Cell Biol. 2005;7:303–310. doi: 10.1038/ncb1225. [DOI] [PubMed] [Google Scholar]
- [11].Grandori C, Gomez-Roman N, Felton-Edkins ZA, Ngouenet C, Galloway DA, Eisenman RN, White RJ. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat Cell Biol. 2005;7:311–318. doi: 10.1038/ncb1224. [DOI] [PubMed] [Google Scholar]
- [12].Oskarsson T, Trumpp A. The Myc trilogy: lord of RNA polymerases. Nat Cell Biol. 2005;7:215–217. doi: 10.1038/ncb0305-215. [DOI] [PubMed] [Google Scholar]
- [13].Stormo GD. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- [14].Yamashita R, Suzuki Y, Wakaguri H, Tsuritani K, Nakai K, Sugano S. DBTSS: DataBase of Human Transcription Start Sites, progress report 2006. Nucleic Acids Res. 2006;34:D86–89. doi: 10.1093/nar/gkj129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kasprzyk A, Keefe D, Smedley D, London D, Spooner W, Melsopp C, Hammond M, Rocca-Serra P, Cox T, Birney E. EnsMart: a generic system for fast and flexible access to biological data. Genome Res. 2004;14:160–169. doi: 10.1101/gr.1645104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hertz GZ, Hartzell GW, 3rd, Stormo GD. Identification of consensus patterns in unaligned DNA sequences known to be functionally related. Comput Appl Biosci. 1990;6:81–92. doi: 10.1093/bioinformatics/6.2.81. [DOI] [PubMed] [Google Scholar]
- [17].Stormo GD, Schneider TD, Gold L, Ehrenfeucht A. Use of the ‘Perceptron’ algorithm to distinguish translational initiation sites in E. coli. Nucleic Acids Res. 1982;10:2997–3011. doi: 10.1093/nar/10.9.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Man TK, Stormo GD. Non-independence of Mnt repressor-operator interaction determined by a new quantitative multiple fluorescence relative affinity (QuMFRA) assay. Nucleic Acids Res. 2001;29:2471–2478. doi: 10.1093/nar/29.12.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bulyk ML, Johnson PL, Church GM. Nucleotides of transcription factor binding sites exert interdependent effects on the binding affinities of transcription factors. Nucleic Acids Res. 2002;30:1255–1261. doi: 10.1093/nar/30.5.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang J, Hannenhalli S. A mammalian promoter model links cis elements to genetic networks. BBRC. doi: 10.1016/j.bbrc.2006.06.062. (in press) [DOI] [PubMed] [Google Scholar]
- [21].Wang J, Hannenhalli S. Generalizations of Markov model to characterize biological sequences. BMC Bioinformatics. 2005;6:219. doi: 10.1186/1471-2105-6-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Brudno M, Do CB, Cooper GM, Kim MF, Davydov E, Green ED, Sidow A, Batzoglou S. LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003;13:721–731. doi: 10.1101/gr.926603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- [24].Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dignam JD, Martin PL, Shastry BS, Roeder RG. Eukaryotic gene transcription with purified components. Methods Enzymol. 1983;101:582–598. doi: 10.1016/0076-6879(83)01039-3. [DOI] [PubMed] [Google Scholar]
- [27].Pavletich NP, Pabo CO. Zinc finger-DNA recognition: Crystal structure of a Zif268-DNA complex at 2.1 Å. Science. 1991;252:809–817. doi: 10.1126/science.2028256. [DOI] [PubMed] [Google Scholar]
- [28].Tseng H, Green H. Basonuclin: a keratinocyte protein with multiple paired zinc fingers. Proc Natl Acad Sci U S A. 1992;89:10311–10315. doi: 10.1073/pnas.89.21.10311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Matsuzaki K, Iuchi S, Green H. Conservation of human and mouse basonuclins as a guide to important features of the protein. Gene. 1997;195:87–92. doi: 10.1016/s0378-1119(97)00176-5. [DOI] [PubMed] [Google Scholar]
- [29].Suzuki Y, Yamashita R, Sugano S, Nakai K. DBTSS, DataBase of Transcriptional Start Sites: progress report 2004. Nucleic Acids Res. 2004;32:D78–81. doi: 10.1093/nar/gkh076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Blanchette M, Tompa M. Discovery of regulatory elements by a computational method for phylogenetic footprinting. Genome Res. 2002;12:739–748. doi: 10.1101/gr.6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chin CS, Chuang JH, Li H. Genome-wide regulatory complexity in yeast promoters: separation of functionally conserved and neutral sequence. Genome Res. 2005;15:205–213. doi: 10.1101/gr.3243305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Makalowski W, Zhang J, Boguski MS. Comparative analysis of 1196 orthologous mouse and human full-length mRNA and protein sequences. Genome Res. 1996;6:846–857. doi: 10.1101/gr.6.9.846. [DOI] [PubMed] [Google Scholar]
- [33].Castillo-Davis CI, Hartl DL. GeneMerge--post-genomic analysis, data mining, and hypothesis testing. Bioinformatics. 2003;19:891–892. doi: 10.1093/bioinformatics/btg114. [DOI] [PubMed] [Google Scholar]
- [34].Castillo-Davis CI. The evolution of noncoding DNA: how much junk, how much func? Trends Genet. 2005;21:533–536. doi: 10.1016/j.tig.2005.08.001. [DOI] [PubMed] [Google Scholar]
- [35].Pavesi G, Mereghetti P, Mauri G, Pesole G. Weeder Web: discovery of transcription factor binding sites in a set of sequences from co-regulated genes. Nucleic Acids Res. 2004;32:W199–203. doi: 10.1093/nar/gkh465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Aerts S, Thijs G, Dabrowski M, Moreau Y, De Moor B. Comprehensive analysis of the base composition around the transcription start site in Metazoa. BMC Genomics. 2004;5:34. doi: 10.1186/1471-2164-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Grandori C, Eisenman RN. Myc target genes. Trends Biochem Sci. 1997;22:177–181. doi: 10.1016/s0968-0004(97)01025-6. [DOI] [PubMed] [Google Scholar]
- [38].Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, Golub TR. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci U S A. 2000;97:3260–3265. doi: 10.1073/pnas.97.7.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]