

Abstract
Yeast flavocytochrome b2 catalyzes the oxidation of lactate to pyruvate; because of the wealth of structural and mechanistic information available, this enzyme has served as the model for the family of flavoproteins catalyzing oxidation of α-hydroxy acids. Primary deuterium and solvent isotope effects have now been used to analyze the effects of mutating the active site residue Tyr254 to phenylalanine. Both the Vmax and the V/Klactate values decrease about 40-fold in the mutant enzyme. The primary deuterium isotope effects on the Vmax and the V/Klactate values increase to 5.0, equivalent to the intrinsic isotope effect for the wild-type enzyme. In addition, both the Vmax and the V/Klactate values exhibit solvent isotope effects of 1.5. Measurement of the solvent isotope effect with deuterated lactate establishes that the primary and solvent isotope effects arise from the same chemical step, consistent with concerted cleavage of the lactate OH and CH bonds. The pH dependence of the mutant enzyme is not significantly different from that of the wild-type enzyme; this is most consistent with a requirement that the side chain of Tyr254 be uncharged for catalysis. The results support a hydride transfer mechanism for the mutant protein and, by extension, wild-type flavocytochrome b2 and the other flavoproteins catalyzing oxidation of α-hydroxy acids.
The oxidation of an α-hydroxy acid to a keto acid is catalyzed by a number of flavoenzymes, including flavocytochrome b2 (1), glycolate oxidase (2), lactate oxidase (3), lactate monooxygenase (4), mandelate dehydrogenase (5), and α-hydroxy acid oxidase (6). The three-dimensional structures of mandelate dehydrogenase (7), glycolate oxidase (8), and flavocytochrome b2 (9) show that these are homologous proteins with TIM barrel structures. The sequences of the other enzymes and the conservation of critical active site residues are consistent with all members of this family having a similar structure (10) and presumably a common catalytic mechanism for α-hydroxy acid oxidation.
Flavocytochrome b2 from Saccharomyces cerevisiae has been the most studied member of this family. This enzyme catalyzes the oxidation of L-lactate to pyruvate. Each of the four identical subunits in flavocytochrome b2 contains FMN and a heme b2 cofactor (11). The catalytic cycle begins with the transfer of a hydride equivalent from lactate to the flavin. This is followed by transfer of single electrons from the flavin to the heme and thence to the final acceptor, cytochrome c in vivo (12). The results of mechanistic studies utilizing substrate analogues, isotope effects, and site-directed mutagenesis have historically been interpreted in terms of a mechanism in which the α-proton of lactate is removed by an active site base to form a carbanion prior to oxidation of lactate and reduction of the flavin (Scheme 1, path a) (13). Analogous results with other members of this family have also been interpreted as consistent with such a carbanion intermediate (14–17). However, several authors have pointed out that the data could also be accommodated by a mechanism in which the α-hydrogen is transferred directly to the flavin as a hydride (18, 19).
Scheme 1.
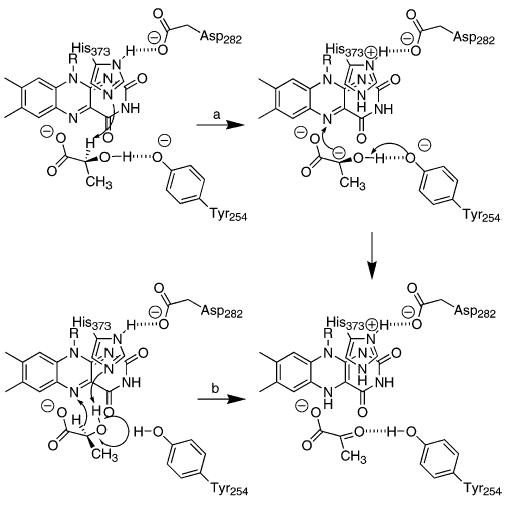
The structure of flavocytochrome b2 complexed with pyruvate has been solved at a resolution of 2.4 Å (9). When lactate is modeled into the active site in place of pyruvate, two different orientations of lactate are compatible with the structural data (20). In the first (path a in Scheme 1), the lactate α-hydrogen points toward His373; this orientation is consistent with abstraction of the substrate α-proton by His373 to form a carbanion. In the alternative orientation (path b in Scheme 1), the hydroxyl hydrogen of lactate forms a hydrogen bond with His373; this arrangement is consistent with His373 abstracting the hydroxyl proton while a hydride is transferred to the FMN. We have recently described the use of primary deuterium and solvent kinetic isotope effects to determine the relative timing of the cleavages of the lactate OH and CH bonds in flavocytochrome b2 (21). The data establish that the OH and CH bonds are not cleaved in the same transition state. This result is consistent with the predictions for formation of a carbanion intermediate and rule out concerted hydride transfer and OH bond cleavage. However, these studies also showed that there is a significant inverse solvent isotope effect on the V/K value for lactate which could mask any solvent isotope effect arising from deprotonation of the lactate hydroxyl. Thus, these data are also consistent with the mechanism of Scheme 2 in which the lactate hydroxyl is deprotonated by His373 prior to hydride transfer from lactate to the flavin.
Scheme 2.
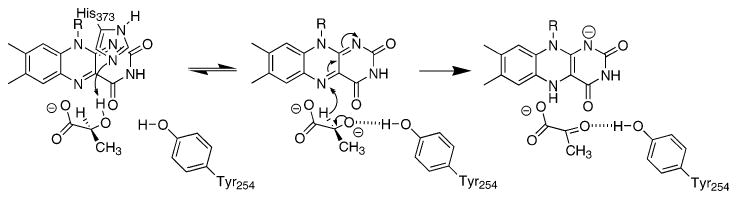
These carbanion and hydride transfer mechanisms make different predictions about the role of the active site residue Tyr254. In the carbanion mechanism of Scheme 1, Tyr254 would facilitate electron transfer from the carbanion to the flavin by deprotonating the substrate hydroxyl moiety. In the corresponding model of the lactate–enzyme complex, the phenolic oxygen of Tyr254 is 3.8 Å from the lactate alcohol proton (20). This is compatible with Tyr254 acting as a base and led Dubois et al. (20) to conclude that Tyr254 must be in the anionic form for catalysis. In a hydride transfer mechanism, Tyr254 does not act as a base but instead orients the substrate for hydride transfer; a neutral Tyr254 would be most compatible with formation of a hydrogen bond between lactate and the phenol of Tyr254. Several studies have described the effects of mutagenesis of this tyrosine to phenylalanine in an attempt to discriminate between these two roles. The crystal structures of Y254F flavocytochrome b2 and of the homologous Y129F glycolate oxidase show that the side chain of the phenylalanine is in the same position as the tyrosine side chain in the wild-type enzyme (16, 22), so this mutation is not disruptive of the active site structure. In these two mutant enzymes and in the homologous Y152F lactate monooxygenase, the rate constants for lactate oxidation are about 2 orders of magnitude smaller than the wild-type values (16, 20, 23–25). In addition, the mutant enzymes show decreases of about 2 orders of magnitude in the binding of anionic inhibitors such as sulfite and oxalate (16, 24, 25). The effects of mutating this tyrosine were typically but not exclusively (26) interpreted as supporting a carbanion mechanism, but in fact the results do not allow discrimination between carbanion and hydride transfer mechanisms.
A clearer understanding of the role of this active site tyrosine would provide significant insight into the mechanisms of the flavoproteins which oxidize α-hydroxy acids. Accordingly, we have used pH and kinetic isotope effects to further characterize Y254F flavocytochrome b2. The results of those studies and the insights they provide into the chemical mechanism of this family of flavoproteins are described herein.
EXPERIMENTAL PROCEDURES
Materials
Sodium L -[2-2H]lactate (98%) and deuterium oxide (99.9%) were purchased from Cambridge Isotope Co., Andover, MA. Lithium L-lactate and D,L-lactate were from Sigma Chemical Corp. (St. Louis, MO). Restriction endo-nucleases were from New England Biolabs Inc. Pfu DNA polymerase was obtained from Stratagene USA. Hydroxy-apatite was from Bio-Rad Laboratories (Hercules, CA). All other reagents were of the highest purity commercially available.
DNA Manipulation
The mutagenesis of Tyr254 to phenylalanine was performed following the QuikChange protocol (Stratagene). The sense primer was 5′-CAATGGTACCAACTATTTGTTAACTCTG-3′. The primers were purchased from the Gene Technology Laboratory of Texas A&M University. Plasmids were purified using kits from Qiagen, Inc. The coding region was sequenced to ensure that no unwanted mutations were incorporated during the polymerase chain reaction.
Cell Growth and Enzyme Purification
Cells were grown and enzymes were purified following previously described protocols (21).
Enzyme Assays
Initial rate assays were performed in 100 mM potassium phosphate, 1 mM EDTA, and 1 mM potassium ferricyanide, pH 7.5, 25 °C, with varied concentrations of L-lactate or L-[2-2H]lactate, by following the decrease in absorbance at 420 nm and using an ε420 value for ferricyanide of 1.04 mM−1 cm−1. Concentrations of lactate were determined by end point assays with wild-type flavocytochrome b2. For pH studies, the buffer was 50 mM Bis-Tris at pH 5.5–7.0, 150 mM HEPES at pH 7.0–8.5, and 50 mM ethanolamine at pH 8.5–10.0. All assays contained 1 mM potassium ferricyanide; this is a saturating concentration, allowing true Vmax and V/Klactate values to be obtained. Solvent isotope effects were determined in 100 mM phosphate and 1 mM EDTA, pH 7.5 or pD 8.0, with 1 mM potassium ferricyanide. D2O-containing buffers were made up using 99.9% deuterium oxide, adjusting the pD with DCl and NaOD as needed, and adding 0.4 to the value indicated by the pH meter (27). For rapid reaction analyses, heme reduction was monitored at 557 nm on an Applied Photo-physics SX.18MV stopped-flow spectrophotometer. Enzyme in 100 mM potassium phosphate and 1 mM EDTA, pH 7.5, was mixed with an equal volume of the same buffer containing varied concentrations of lactate at 25 °C.
Data Analysis
The data were analyzed using the programs Kaleidagraph (Adelbeck Software, Reading, PA), KinetAsyst (IntelliKinetics, State College, PA), or Igor (Wavemetrics, Lake Oswego, OR). Initial rate data were fit to the Michael-is–Menten equation to obtain Vmax, V/Klactate, and Klactate values. Primary deuterium and solvent isotope effects were calculated using eqs 1 and 2. Equation 1 describes separate isotope effects on the Vmax and V/Klactate values, while eq 2 yields identical isotope effects on the V and V/K values. For both, A is the concentration of the varied substrate, Fi is the fraction of heavy atom substitution in the substrate, EV is the isotope effect on the Vmax value (eq 1) or both the Vmax and the V/K values (eq 2) minus 1, and EVK is the isotope effect on the V/Klactate value (eq 1) minus 1. Data were fit to both equations; if fitting to eq 1 did not result in a decrease in the σ value for the fit, eq 2 was used. The pH profiles were obtained by fitting the Vmax and V/Klactate values obtained at each pH to eq 3; here, K1 and K2 are the dissociation constants of the ionizable groups, and C is the pH-independent value of the specific kinetic parameter. Stopped-flow traces were fit to eq 4, which describes a biphasic exponential decay; λ1 and λ2 are the first-order rate constants for each phase; A1 and A2 are the absorbances of each species at time t; and A∞ is the final absorbance. The pseudo-first-order rate constants for the rapid phase obtained at different concentrations of lactate were analyzed using eq 5; here, kred is the rate constant for flavin reduction at saturating concentrations of lactate, and Kd is the apparent dissociation constant for lactate.
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
RESULTS
Steady-State Kinetic Parameters
The steady-state kinetic parameters of Y254F flavocytochrome b2 were determined with ferricyanide as the final electron acceptor, since oxidation of the chromophores is rate limiting with cytochrome c as the electron acceptor (28). The Vmax value is 28-fold and the V/Klactate value about 50-fold smaller than the wild-type values (Table 1). The Km value for lactate for the Y254F enzyme is slightly higher. These values are consistent with previously reported data (20, 24). The changes in the steady-state kinetic parameters clearly indicate that the hydroxyl group of Tyr254 is important for enzymatic activity. For the wild-type enzyme the DVmax and D(V/Klactate) values are close to 3, consistent with CH cleavage being partially rate limiting. Removal of the hydroxyl group at position 254 makes this step more rate limiting, inasmuch as the DVmax and D(V/Klactate) values increase to an identical value of 5 (Table 1). This value is not significantly different from the intrinsic isotope effect of 5.4 ± 0.4 determined for the wild-type enzyme (21).
Table 1.
Steady-State Kinetic Parameters for Wild-Type and Y254F Flavocytochromes b2
| kinetic parametera | wild-type enzymeb | Y254F |
|---|---|---|
| Vmax, s−1 | 372 ± 12 | 13.2 ± 0.4 |
| V/Klactate, mM−1 s−1 | 2300 ± 215 | 44 ± 5.0 |
| Klactate, mM | 0.16 ± 0.02 | 0.28 ± 0.02 |
| DV | 2.9 ± 0.1 | 5.0 ± 0.3c |
| D(V/K) | 3.0 ± 0.3 | 5.0 ± 0.3 |
| D2OV | 1.38 ± 0.07 | 1.54 ± 0.07 |
| D2O(V/K) | 0.90 ± 0.04 | 1.54 ± 0.07 |
| DVD2O | ndd | 4.5 ± 0.3 |
| D(V/K)D2O | nd | 4.5 ± 0.3 |
| D2OVD | 1.1 ± 0.03 | 1.41 ± 0.05 |
| D2O(V/K)D | 0.87 ± 0.1 | 1.41 ± 0.05 |
Conditions: 100 mM potassium phosphate, 1 mM EDTA, and 1 mM ferricyanide, pH 7.5 or pD 8.0, 25 °C.
From ref 21.
For all of the isotope effects for the Y254F enzyme, the data fit better to eq 2, which describes identical isotope effects on the V and V/K values, than to eq 1, which yields different DV and D(V/K) values.
Not determined.
pH Profiles
The V/Klactate pH profile for Y254F flavocytochrome b2 is bell-shaped, consistent with two critical ionizable groups in the free enzyme (Figure 1). One group needs to be protonated for activity, and another group needs to be unprotonated. The Vmax pH rate profile for this mutant enzyme is also bell-shaped, again consistent with two ionizable groups being important for catalysis. The pKa values from both profiles for the Y254F enzyme are similar to the values for the wild-type enzyme (Table 2 and Figure 1) (21, 29).
Figure 1.
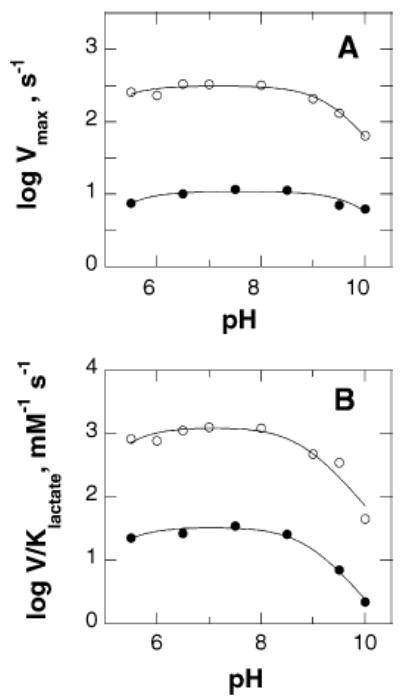
pH dependence of wild-type and Y254F flavocytochromes b2. Initial rates were determined with ferricyanide as the electron acceptor as described in Experimental Procedures. Panel A shows the Vmax profile and panel B the V/Klactate profile for the wild-type (○) and for the Y254F (•) enzymes. The lines are fits of the data to eq 3. The data for the wild-type enzyme are from ref 21.
Table 2.
pKa Values for Wild-Type and Y254F Flavocytochromes b2a
| enzyme | parameter | pK1 | pK2 |
|---|---|---|---|
| wild typeb | Vmax | 5.6 ± 0.1 | 9.4 ± 0.1 |
| V/Klactate | 5.8 ± 0.1 | 8.9 ± 0.1 | |
| Y254F | Vmax | 5.2 ± 0.2 | 10.0 ± 0.1 |
| V/Klactate | 5.2 ± 0.2 | 8.9 ± 0.1 |
Conditions: 100 mM potassium phosphate, 1 mM EDTA, and 1 mM ferricyanide, pH 7.5, 25 °C.
From ref 21.
Solvent Isotope Effects
With wild-type flavocytochrome b2, the D2O(V/Klactate) value is slightly inverse while the D2OVmax value is normal (Table 1) (21). For the Y254F enzyme the D2OVmax value increases and the D2O(V/Klactate) value becomes normal and equal to the D2OVmax value (Table 1). The solvent isotope effect for the wild-type enzyme was previously determined to be on a step or steps in the transfer of electrons from the reduced flavin to ferricyanide rather than on CH bond cleavage (21). To determine if the solvent and primary deuterium isotope effects were due to the same chemical step for the mutant enzyme, solvent isotope effects were measured using L-[2-2H]lactate as substrate. For the wild-type enzyme the D2OVmax value decreases substantially with the deuterated substrate, consistent with the two isotope effects arising from different steps. For the Y245F enzyme with the deuterated substrate, the D2OVmax and D(V/K) values remain identical, decreasing only slightly. This is consistent with both isotope effects arising from the same chemical step in the case of the mutant enzyme (30).
Rapid Reaction Kinetics
Direct measurement of the rate constant for FMN reduction for the Y254F enzyme by monitoring flavin reduction is not possible. The mutation decreases the rate of FMN reduction such that reduction is slower than subsequent electron transfer to the hemes (data not shown) (31). Thus, the only spectral change which is observed when the enzyme is mixed with lactate in the absence of an external electron acceptor is due to heme reduction rather than flavin reduction. With the wild-type enzyme this occurs more slowly than flavin reduction, so that the rate of heme reduction underestimates the rate of lactate oxidation (32). Still, the relative rate constants for heme reduction in the wild-type and mutant enzymes provide an alternative measure of the effect of the mutation on the reductive half-reaction. Accordingly, the rate constant for heme reduction was determined by monitoring the change in absorbance at 557 nm when the enzyme was mixed with L-lactate in a stopped-flow spectrophotometer. For both the wild-type and Y254F enzymes the reaction results in a bi-phasic increase in absorbance at 557 nm, with only the rate constant for the fast phase being substrate dependent (Figure 2). The limiting rate constant for heme reduction at saturating concentrations of lactate, the kred value, could be determined from the concentration dependence of the fast phase for each enzyme. The values for the wild-type enzyme are consistent with previously reported values (32). The kred value for the mutant enzyme is close to the Vmax value and shows a decrease of comparable magnitude to that seen in the Vmax and V/Klactate values (Table 3). This is consistent with the mutation primarily affecting the reductive half-reaction.
Figure 2.
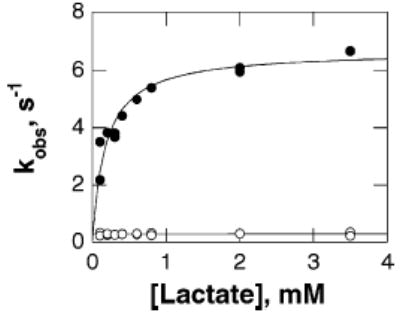
Rate of heme reduction for Y254F flavocytochrome b2 as a function of lactate concentration. The observed rate constants for the fast phase are shown in solid circles and the rate constants for the slow phase in open circles.
Table 3.
Rapid Reaction Kinetic Parameters for Heme Reduction in Wild-Type and Y254F Flavocytochromes b2a
| enzyme | kred, s−1 | Klactate, mM |
|---|---|---|
| wild type | 226 ± 11 | 0.21 ± 0.04 |
| Y254F | 6.6 ± 0.4 | 0.18 ± 0.03 |
Conditions: 100 mM potassium phosphate and 1 mM EDTA, pH 7.5, 25 °C.
DISCUSSION
The characterization of Y254F flavocytochrome b2 presented here confirms the importance of Tyr254 in the catalytic mechanism of that enzyme and provides insight into the role of this residue. More importantly, the results significantly limit the mechanistic possibilities which must be considered for this enzyme and, by extension, for all of the flavoprotein α-hydroxy acid oxidases.
The protonation state of the phenolic oxygen of Tyr254 which is required for productive catalysis has been the subject of some dispute. In the original mechanistic distinction between the roles of individual amino acid residues in carbanion and hydride transfer mechanisms (33), Tyr254 was predicted to be unprotonated in order to accept the lactate hydroxyl proton from the carbanion intermediate. There was no clear role for Tyr254 in a hydride transfer reaction. The subsequent demonstration that the Y254F enzyme had a similar Km value for lactate as the wild-type enzyme while the rate of catalysis decreased about 40-fold was taken as evidence that Tyr254 does not act as a base but instead stabilizes the transition state (20). These results and further analyses of this mutant protein led to the proposal that Tyr254 does form a hydrogen bond with lactate but this interaction is of primary importance in the transition state (24). The effect of the Y254F mutation on the pH dependence of the enzyme described here places limitations on the required protonation state of the side chain of Tyr254. The pH profiles of the mutant enzyme are similar to those of the wild-type enzyme. This eliminates Tyr254 as the residue responsible for either pKa. If Tyr254 must be unprotonated for activity as an active site base, its pKa must be well below 5; this appears unlikely in light of the change in activity in the mutant enzyme of less than 100-fold. If Tyr254 must be protonated for activity, its pKa must be above 11; this is not unreasonable for a tyrosyl residue in an enzyme active site. Thus, the most straightforward interpretation of the pH profile of the mutant enzyme is that Tyr254 must be protonated for activity, in line with the proposal of Gondry et al. (24). The requirement for a neutral side chain is also consistent with the decreased affinity for anionic inhibitors such as sulfite and oxalate seen in this mutant protein and the homologous mutants of glycolate oxidase and lactate monooxygenase (16, 25).
The effects of mutagenesis of Tyr254 on the steady-state kinetic parameters are most readily discussed in the context of the minimal kinetic mechanism of Scheme 3. Here, all chemical steps in lactate oxidation occur in the step with rate constant k2; whether this is an oversimplification will be discussed below. The subsequent steps, from pyruvate release through electron transfer to ferricyanide to regenerate the oxidized flavin, are included in the step with rate constant k3. Equations 6–11 give the relationships between the steady-state kinetic parameters and the individual rate constants of Scheme 3. The DV and D(V/K) values for the mutant enzyme are both 5.0 ± 0.3. The deuterium isotope effect on k2 for the wild-type enzyme, measured by monitoring reduction of the flavin directly, is 5.4 ± 0.4 (21). Thus, for the mutant enzyme, DV and D(V/K) are equal to Dk2. This requires that k−1 ≫ k2 (eq 8) and k3 ≫ k2 (eq 9); therefore, Vmax is equal to k2. The identity of Vmax and the rate constant for flavin reduction is confirmed by comparison of the rate constant for heme reduction with the Vmax value.
Scheme 3.
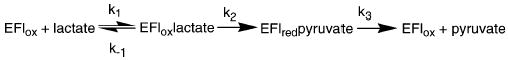
The value of k2 for the wild-type enzyme is 520 s−1 (21), so that the mutation decreases the rate of lactate oxidation 39-fold. Comparison of the V/Klactate values for the wild-type and mutant enzymes yields a comparable effect of the mutation of 52-fold. This is consistent with the mutation decreasing the transition state stabilization for lactate oxidation by 2.3 kcal/mol at 25 °C and agrees well with the conclusions previously drawn from the studies of this mutant protein (20, 24).
| (6) |
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
While the deuterium isotope effects for Y254F flavocytochrome b2 confirm previous results, it is the solvent isotope effects which are more informative regarding the effects of the mutation on chemical steps. With the wild-type enzyme, there is a significant solvent isotope effect on the Vmax value and a slightly inverse effect on the V/Klactate value (21). The solvent isotope effect on the Vmax value has previously been shown to arise from a step after pyruvate formation. The decrease in the solvent isotope effect on the Vmax value when deuterated lactate is used confirms that this solvent isotope effect arises from a different step than the primary deuterium isotope effect. For the mechanism of Scheme 3, a solvent isotope effect on the Vmax value must be due to an isotope effect on k2 or k3. With wild-type flavocytochrome, the value of k2/k3 is 1.3 ± 0.2 (21). If the solvent isotope effect were on k2, the relationship between the solvent isotope effect and the individual rate constants would be given by eq 10, so that the intrinsic solvent isotope effect on k2 would be 1.9 ± 0.4. A 5.4-fold decrease in the value of k2 when the deuterated substrate is used would increase the observed solvent isotope effect on the Vmax value to 1.7 ± 0.3. If the solvent isotope effect were instead on k3, eq 11 gives the relationship between the observed isotope effect and the rate constants, yielding a value for the intrinsic solvent isotope effect on k3 of 1.7 ± 0.2. A 5.4-fold increase in the value of k3/k2 due to the use of deuterated lactate would result in a decrease in the D2OV value to 1.1 ± 0.2. Clearly, the observed D2OV value of 1.1 when deuterated lactate is the substrate is more consistent with the latter case, confirming that for the wild-type enzyme the solvent isotope effect on the Vmax value does not arise from oxidation of lactate to pyruvate but is instead due to a subsequent step or steps.
With the Y254F enzyme, the solvent isotope effects on the Vmax and V/Klactate values are both normal and are equal. Since the Vmax value is equal to k2, the solvent isotope effect must be on k2 also. The identities of the isotope effects on the Vmax and V/Klactate values are consistent with both isotope effects on the V/Klactate value also arising from an effect on k2. The assignment of both the solvent and primary isotope effects to a single step for the Y254F enzyme is supported by the effects of lactate deuteration on the D2OV and D2OV/K values. In contrast to the case with the wild-type enzyme, there is only a small decrease in the D2OV and D2OV/K values when deuterated lactate is used as substrate. This lack of a substantial change in the D2OV value with deuterated lactate confirms that the solvent isotope effect and the primary deuterium isotope effect arise from the same chemical step.1 The most likely explanation for these results is that cleavage of the lactate hydroxyl OH bond and cleavage of the α-CH bond are concerted in the mutant enzyme.2
It is difficult to reconcile concerted OH and CH bond cleavage with a mechanism involving a carbanion intermediate. Simultaneous removal of both the hydroxyl proton and the α-proton would generate a lactate molecule with negative charges on both the α-carbon and the adjacent oxygen; this is unlikely. In contrast, concerted OH and CH bond cleavage is fully consonant with the predictions of the hydride transfer mechanism in Scheme 1, in which the lactate hydroxyl protein is removed by His373 as the α-hydrogen is transferred to the flavin as a hydride. Thus, the data presented here support a hydride transfer mechanism for this mutant enzyme. The critical question then becomes the application of these results to the mechanism of the wild-type enzyme. With the wild-type enzyme, the OH and CH bonds are cleaved in separate steps (21), but the data do not unambiguously determine the order in which they are cleaved. The carbanion mechanism of Scheme 1 predicts that OH bond cleavage occurs after CH bond cleavage, while the hydride transfer mechanism of Scheme 2 predicts the reverse. Tyr254 has previously been proposed to be involved in stabilization of the transition state for lactate oxidation (24). This mutation decreases the affinity of the enzyme for anionic inhibitors such as sulfite and oxalate (24), and the structure of the sulfite-bound wild-type enzyme shows a hydrogen bond between the phenolic oxygen of Tyr254 and a sulfite oxygen (22). Similar decreases in the affinities for anionic inhibitors are seen when the homologous tyrosine residues in glycolate oxidase and lactate monooxygenase are mutated to phenylalanine (17, 40). The effects on inhibitor binding and the structure of the sulfite complex are consistent with the role of Tyr254 being stabilization of the lactate alkoxide via a hydrogen bond between the neutral Tyr254 phenolic oxygen and the charged lactate hydroxyl oxygen. The mechanism of Scheme 2 involves just such an interaction. The loss of this interaction would destabilize the alkoxide, making its formation more difficult and thus slower. In a hydride transfer reaction, destabilization of the alkoxide would result in a delay in its formation until the initiation of hydride transfer from the α-carbon decreased the pKa of the oxygen sufficiently for proton transfer to His373. This would make removal of the hydroxyl proton concerted with hydride transfer, as is indeed the case with the Y254F enzyme. Thus, the effects of mutation of Tyr254 can readily be reconciled with the hydride transfer mechanism of Scheme 2 for wild-type flavocytochrome b2. The present results are more difficult to reconcile with a mechanism involving formation of a carbanion. CH bond cleavage necessarily precedes OH bond cleavage in a carbanion mechanism. Loss of the base which accepts the hydroxyl proton should substantially decrease the rate of the latter step but would be expected to decrease the rate of CH bond cleavage very much less. Thus, OH bond cleavage would become more rate limiting for lactate oxidation. If this step became sufficiently slow to result in a detectable solvent isotope effect, it would then be substantially slower than CH bond cleavage. In other words, CH and OH bond cleavage would become more discrete rather than concerted. Thus, a carbanion mechanism for the wild-type enzyme should not result in concerted CH and OH bond cleavage in the Y254F enzyme.
One could propose that the mutation results in a change in the mechanism from carbanion to hydride transfer. However, the mutation only decreases the value of k2 by about 40-fold. For a decrease in the rate of carbanion formation of this magnitude to cause a change in mechanism, the wild-type enzyme must be able to catalyze the alternate mechanism, hydride transfer, at a rate within 2 orders of magnitude of the rate of the normal mechanism. Such mechanistic promiscuity would be unprecedented to our knowledge. In addition, such a mechanistic switch would seem to be unlikely to arise upon mutation of a residue other than His373, the base proposed to abstract the α-proton.
The pH dependences of the wild-type and the Y254F enzymes are also not consistent with the carbanion mechanism of Scheme 1 in which the role of Tyr254 is to abstract the hydroxyl proton. This would require that the phenolic oxygen be deprotonated; to reconcile this requirement with the pH profiles requires that the pKa value of Tyr254 be below 5. While such a depression of the pKa value cannot be ruled out, the alternate conclusion that the pKa value is shifted above 11 is less drastic.
In summary, the present results support a hydride transfer mechanism for Y254F flavocytochrome b2, with the role of Tyr254 in the wild-type enzyme being stabilization of the lactate alkoxide. The results are most readily reconciled with a stepwise hydride transfer mechanism for the wild-type enzyme. In light of the structural similarities among the several flavoproteins which catalyze oxidation of α-hydroxy acids to keto acids, the present results strongly suggest that those enzymes also utilize hydride transfer mechanisms.
Footnotes
This work was supported in part by grants to P.F.F. from the NIH (GM 58698) and from the Robert A. Welch Foundation (A-1245).
The possibility must be considered that the precision in the values of the measured isotope effects is insufficient to detect the expected change in the solvent isotope effect upon deuteration of lactate for the Y254 enzyme. If one assumes that the value of the intrinsic isotope effect on k2 for the Y254F enzyme is the same as that for the wild-type enzyme, 5.4 ± 0.4, the observed DV value for the mutant enzyme sets an upper limit on the value of k3/k2 of 5. Using this value and eq 11, the D2OV value with deuterated lactate is predicted to 1.1 ± 0.1 for the mutant enzyme. Thus, the precision in the measured kinetic parameters is sufficient to allow mechanistic discrimination.
In the case of the Y254F enzyme, there is a decrease in the measured solvent isotope with the deuterated substrate and a small but statistically insignificant decrease in the primary isotope effect in D2O. This result is most consistent with coupled motion of the alcohol proton and the lactate α-hydrogen in the transition state. A mechanism for flavocytochrome b2 involving concerted cleavage of the lactate OH and CH bonds is similar to the mechanism for glucose-6-phosphate dehydrogenase proposed by Hermes and Cleland (34). This reaction involves concerted removal of an alcohol proton by an active site base as a hydride is transferred from the carbon to NADP. The intrinsic primary deuterium isotope effect for the reaction decreases from 5.3 in H2O to 3.7 in D2O. This decrease was ascribed to coupled motion of the two hydrogens in the same transition state and tunneling. Much of the isotope effect seen upon substitution of the first hydrogen with deuterium was attributed to tunneling, since deuterium tunnels much less than hydrogen. This reduces the contribution of tunneling to the isotope effect when the second hydrogen is substituted, decreasing the magnitude of the primary deuterium isotope effect in D2O. (The solvent isotope effect with deuterated glucose 6-phosphate was not reported in this case.) In general, coupled motion and tunneling have been invoked to explain the decrease in the magnitude of an α-deuterium isotope effect when deuterium replaces the hydrogen being removed in the reaction (35–39); to our knowledge there have been no other reports of the interplay between solvent and primary isotope effects. The decrease in the primary deuterium isotope effect in D2O seen with Y254F flavocytochrome b2 is much less than that reported for glucose-6-phosphate dehydrogenase. This may reflect a smaller contribution of tunneling to the reaction catalyzed by the former. Alternatively, the extent of coupling between motion of the alcohol proton and the lactate C-2 hydrogen may be less.
References
- 1.Lederer F. In: Chemistry and Biochemistry of Flavoenzymes. Muller F, editor. II. CRC Press; Boca Raton, FL: 1991. pp. 153–242. [Google Scholar]
- 2.Schuman M, Massey V. Biochim Biophys Acta. 1971;227:500–520. doi: 10.1016/0005-2744(71)90003-9. [DOI] [PubMed] [Google Scholar]
- 3.Maeda-Yorita K, Aki K, Sagai H, Misaki H, Massey V. Biochimie. 1995;77:631–642. doi: 10.1016/0300-9084(96)88178-8. [DOI] [PubMed] [Google Scholar]
- 4.Ghisla S, Massey V. In: Chemistry and Biochemistry of Flavoenzymes. Muller F, editor. II. CRC Press; Boca Raton, FL: 1991. pp. 243–289. [Google Scholar]
- 5.Tsou AY, Ransom SC, Gerlt JA, Buechter DD, Babbitt PC, Kenyon GL. Biochemistry. 1990;29:9856–9862. doi: 10.1021/bi00494a015. [DOI] [PubMed] [Google Scholar]
- 6.Urban P, Chirat I, Lederer F. Biochemistry. 1988;27:7365–7371. doi: 10.1021/bi00419a029. [DOI] [PubMed] [Google Scholar]
- 7.Sukumar N, Xu Y, Gatti DL, Mitra B, Mathews FS. Biochemistry. 2001;40:9870–9878. doi: 10.1021/bi010938k. [DOI] [PubMed] [Google Scholar]
- 8.Lindqvist Y, Brändén CI, Mathews R, Lederer F. J Biol Chem. 1991;266:3198–3207. [PubMed] [Google Scholar]
- 9.Xia Z-x, Mathews FS. J Mol Biol. 1990;212:837–863. doi: 10.1016/0022-2836(90)90240-M. [DOI] [PubMed] [Google Scholar]
- 10.Diêp Lê KH, Lederer F. J Biol Chem. 1991;266:20877–20881. [PubMed] [Google Scholar]
- 11.Xia ZX, Shamala N, Bethge PH, Lim LW, Bellamy HD, Xuong NH, Lederer F, Mathews FS. Proc Natl Acad Sci USA. 1987;84:2629–2633. doi: 10.1073/pnas.84.9.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Labeyrie F, Baudras A, Lederer F. Methods Enzymol. 1978;53:238–256. doi: 10.1016/s0076-6879(78)53030-9. [DOI] [PubMed] [Google Scholar]
- 13.Lederer F. In: Flavins and Flavoproteins. Stevenson KJ, Massey V, Williams CH, editors. University of Calgary Press; Calgary, Canada: 1996. pp. 545–553. [Google Scholar]
- 14.Lehoux IE, Mitra B. Biochemistry. 1999;38:5836–5848. doi: 10.1021/bi990024m. [DOI] [PubMed] [Google Scholar]
- 15.Walsh C, Lockridge O, Massey V, Abeles R. J Biol Chem. 1973;248:7049–7054. [PubMed] [Google Scholar]
- 16.Macheroux P, Kieweg V, Massey V, Soderlind E, Stenberg K, Lindqvist Y. Eur J Biochem. 1993;213:1047–1054. doi: 10.1111/j.1432-1033.1993.tb17852.x. [DOI] [PubMed] [Google Scholar]
- 17.Müh U, Williams CH, Jr, Massey V. J Biol Chem. 1994;269:7989–7993. [PubMed] [Google Scholar]
- 18.Bright HJ, Porter DJT. In: The Enzymes. 3rd ed. Boyer P, editor. XII. Academic Press; New York: 1975. pp. 421–505. [Google Scholar]
- 19.Fitzpatrick PF. Acc Chem Res. 2001;34:299–307. doi: 10.1021/ar0000511. [DOI] [PubMed] [Google Scholar]
- 20.Dubois J, Chapman SK, Mathews FS, Reid GA, Lederer F. Biochemistry. 1990;29:6393–6400. doi: 10.1021/bi00479a008. [DOI] [PubMed] [Google Scholar]
- 21.Sobrado P, Daubner SC, Fitzpatrick PF. Biochemistry. 2001;40:994–1001. doi: 10.1021/bi002283d. [DOI] [PubMed] [Google Scholar]
- 22.Tegoni M, Cambillau C. Biochimie. 1994;76:501–514. doi: 10.1016/0300-9084(94)90174-0. [DOI] [PubMed] [Google Scholar]
- 23.Black MT, White SA, Reid GA, Chapman SK. Biochem J. 1989;258:255–259. doi: 10.1042/bj2580255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gondry M, Dubois J, Terrier M, Lederer F. Eur J Biochem. 2001;268:4918–4927. doi: 10.1046/j.0014-2956.2001.02424.x. [DOI] [PubMed] [Google Scholar]
- 25.Müh U, Williams CH, Jr, Massey V. J Biol Chem. 1994;269:7994–8000. [PubMed] [Google Scholar]
- 26.Stenberg K, Clausen T, Lindqvist Y, Macheroux P. Eur J Biochem. 1995;228:408–416. [PubMed] [Google Scholar]
- 27.Schowen KB, Schowen RL. Methods Enzymol. 1982;89:551–606. [PubMed] [Google Scholar]
- 28.Rouviere-Fourmy N, Capeillere-Blandin C, Lederer F. Biochemistry. 1994;33:798–806. doi: 10.1021/bi00169a022. [DOI] [PubMed] [Google Scholar]
- 29.Ogura Y, Ohta Y, Nakamura T. J Biochem. 1966;60:63–71. doi: 10.1093/oxfordjournals.jbchem.a128400. [DOI] [PubMed] [Google Scholar]
- 30.Cook PF, Cleland WW. Biochemistry. 1981;20:1790–1796. doi: 10.1021/bi00510a013. [DOI] [PubMed] [Google Scholar]
- 31.Gondry M, Diêp Lê KH, Manson FDC, Chapman SK, Mathews FS, Reid GA, Lederer F. Protein Sci. 1995;4:925–935. doi: 10.1002/pro.5560040512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pompon D, Iwatsubo M, Lederer F. Eur J Biochem. 1980;104:479–488. doi: 10.1111/j.1432-1033.1980.tb04450.x. [DOI] [PubMed] [Google Scholar]
- 33.Lederer F, Mathews FS. In: Flavins and Flavoproteins. Edmondson DE, McCormick DB, editors. Walter de Gruyter; Berlin: 1987. pp. 133–142. [Google Scholar]
- 34.Hermes JD, Cleland WW. J Am Chem Soc. 1984;106:7263–7264. [Google Scholar]
- 35.Kurz LC, Frieden C. J Am Chem Soc. 1980;102:4198–4203. [Google Scholar]
- 36.Cook PF, Oppenheimer NJ, Cleland WW. Biochemistry. 1981;20:1817–1825. doi: 10.1021/bi00510a016. [DOI] [PubMed] [Google Scholar]
- 37.Klinman JP. In: Enzyme mechanism form isotope effects. Cook PF, editor. CRC Press; Boca Raton, FL: 1991. pp. 127–148. [Google Scholar]
- 38.Alston WC, II, Kanska M, Murray CJ. Biochemistry. 1996;35:12873–12881. doi: 10.1021/bi960831a. [DOI] [PubMed] [Google Scholar]
- 39.Rucker J, Klinman JP. J Am Chem Soc. 1999;121:1997–2006. [Google Scholar]
- 40.Macheroux P, Mulrooney SB, Williams CH, Jr, Massey V. Biochim Biophys Acta. 1992;1132:11–16. doi: 10.1016/0167-4781(92)90046-3. [DOI] [PubMed] [Google Scholar]