

Abstract
The role of prolactin in human breast cancer has been controversial. However, it is now apparent that human mammary epithelial cells can synthesize prolactin endogenously, permitting autocrine/paracrine actions within the mammary gland that are independent of pituitary prolactin. To model this local mammary production of prolactin (PRL), we have generated mice that overexpress prolactin within mammary epithelial cells under the control of a hormonally nonresponsive promoter, neu-related lipocalin (NRL). In each of the two examined NRL-PRL transgenic mouse lineages, female virgin mice display mammary developmental abnormalities, mammary intraepithelial neoplasias, and invasive neoplasms. Prolactin increases proliferation in morphologically normal alveoli and ducts, as well as in lesions. The tumors are of varied histotype, but papillary adenocarcinomas and adenosquamous neoplasms predominate. Neoplasms can be separated into two populations: one is estrogen receptor alpha (ERα) positive (greater than 15% of the cells stain for ERα), and the other is ERα−(<3%). ERα expression does not correlate with tumor histotype, or proliferative or apoptotic indices. These studies provide a mouse model of hormonally dependent breast cancer, and, perhaps most strikingly, a model in which some neoplasms retain ERα, as occurs in the human disease.
Keywords: mammary cancer, prolactin, ERα, transgenic mice
Abbreviations: DCIS, ductal carcinoma in situ; E2, estradiol; ERα, estrogen receptor alpha; PRL, prolactin; MT, metallothionein; MIN, mammary intraepithelial neoplasia; NRL, neu-related lipocalin
Introduction
Mammary development is controlled by the interplay of multiple steroid and peptide hormones, together with systemic and local growth factors. Study of the physiological role of many of these factors during the estrus cycle, pregnancy, and involution following lactation has identified actions that contribute to carcinogenesis, especially for estrogens and EGF family members. Despite the recognized importance of prolactin (PRL) in mammary growth and differentiation (Horseman et al., 1997; Ormandy et al., 1997a; Brisken et al., 1999), its role in human breast cancer has been controversial (for reviews, see Goffin et al., 1999; Hankinson et al., 1999; Vonderhaar, 2000; Wennbo and Törnell, 2000; Clevenger et al., 2003). Studies in cultured mammary tumor cells have demonstrated a role for PRL in proliferation, survival, and migration, and PRL and its proteolytic products can modulate angiogenesis (for a review, see Clevenger et al., 2003). Each of these activities could contribute to neoplastic development and progression. However, human clinical studies correlating circulating PRL levels to tumor incidence and disease progression have been inconsistent. Treatment with the dopamine agonist, bromocriptine, to block transcription of pituitary PRL, had no consistent effect on tumor progression. It is now clear that circulating PRL is not the only source of this activity; multiple extrapituitary cells in primates, including normal and neoplastic human mammary epithelium, also synthesize PRL (for a review, see Ben-Jonathan et al., 1996; Vonderhaar, 2000; Clevenger et al., 2003). Furthermore, up to 98% of human mammary tumors express PRL receptor (PRLR) (Ormandy et al., 1997b; Reynolds et al., 1997; Mertani et al., 1998), and neoplastic tissue expresses higher levels of PRLR than normal adjacent tissue (Touraine et al., 1998). This potential to exert autocrine/paracrine effects within the mammary gland suggests that the role of PRL in human disease requires re-evaluation. In vivo models are necessary to explore these possibilities and dissect interactions with other neoplastic factors within a physiological context.
In contrast to the human, scant PRL is produced within the mammary epithelial cells of virgin mice (Ben-Jonathan et al., 1996; Schroeder et al., 2001). However, pituitary isografts and systemic PRL treatment increased the development of spontaneous tumors in mice, and cooperated with chemical carcinogens to induce mammary tumors in the rat (Boot et al., 1962; Welsch and Nagasawa, 1977). When PRL−/− mice were crossed with transgenic mice expressing polyoma middle-T antigen driven by the MMTV promoter (PyVT mice), pituitary transplants decreased tumor latency and increased tumor growth rate (Vomachka et al., 2000). Expression of a rat PRL transgene under the control of the widely expressed metallothionein (MT) promoter resulted in mammary tumors in virgin females, even in a line that did not have elevated circulating hormone (Wennbo et al., 1997). However, mammary gland development prior to the appearance of tumors was not described, so there remains a lack of information regarding PRL-associated preneoplastic lesions and the course of tumor progression.
Transgenic models are powerful tools to assess the role of selected growth factors and associated signaling pathways in mammary carcinogenesis. However, because most mammary-specific promoters are derived from genes normally regulated by the hormones present during pregnancy or that contain hormonal response elements (Otten et al., 1988; Amundadottir et al., 1996; Yarus et al., 1996; Qin et al., 1999; Cardiff et al., 2000), these models have not been exploited to examine the mechanisms by which hormones, including PRL, contribute to the development and/or progression of mammary cancer. We have characterized a novel promoter fragment from the neu-related lipocalin (NRL) gene (Stoesz and Gould, 1995) that selectively enriches transgene expression in mammary epithelia and is independent of PRL and estradiol (Rose-Hellekant et al., in preparation). This promoter enables us to use transgenic mice to study the interaction of factors contributing to mammary neoplasia independent of hormonal changes in transgene expression. Here we describe NRL-PRL mice that express PRL within mammary epithelia, similar to human mammary tumor cells, permitting evaluation of effects of endogenous PRL on mammary tumorigenesis. These mice develop invasive ERα+ and ERα-mammary neoplasms, providing a model to examine hormonally dependent breast cancer.
Results
Generation of NRL-PRL transgenic mice, and pattern of transgene expression
To exploit the power of murine transgenic models to explore the role of hormones in oncogene-induced mammary neoplasia, a hormone-independent, mammary-specific promoter is required. The constant level of expression of NRL during the rat estrous cycle and pregnancy (Stoesz and Gould, 1995) suggested that its promoter would be useful in this regard. As shown in Figure 1b, transcription from the proximal 3 kb promoter fragment is independent of PRL in vitro. It is also independent of estradiol (E2) in this system, and mammary TGFα expression driven by this promoter in vivo is not altered by ovariectomy or E2 supplementation (Rose-Hellekant et al., in preparation). We used this promoter to drive PRL expression from the rat PRL gene using the construct shown in Figure 1a.
Figure 1.
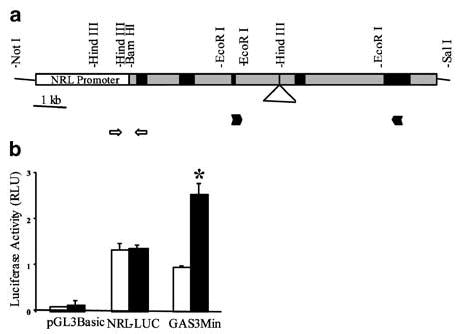
(a) Schematic diagram of the transgene containing a modified rat PRL gene with endogenous polyadenylation sequences fused to 3015 bp of the proximal NRL promoter (Rose-Hellekant et al., in preparation). NRL promoter sequences are shown as the open box; exons and introns of the PRL gene are depicted by black and gray boxes, respectively. The triangle shows the location of about 1500 bp of intron C omitted from the transgene. Locations of PCR primers used to identify transgene-positive individuals are shown by open arrows; locations of primers used for RT–PCR analyses are shown by solid arrowheads. (b) The NRL promoter is not responsive to PRL. CHO K1 cells were transiently transfected with either pGL3 Basic, NRL-Luc or GAS3MIN (positive control) in addition to CMV-β-gal (transfection control), and treated ±4nM PRL for 24 h. N=3; mean±s.e. Asterisk indicates significant difference from vehicle-treated control (P<0.001)
We identified eight NRL-PRL founder animals (four female, four male). Two founders (one female, one male: NRL-PRL 1655-8 and 1647-13, respectively) were selected for further study, based on the ability to pass the transgene, fertility, and mammary PRL expression. RT–PCR analysis demonstrated markedly elevated levels of mammary transgene mRNA in both lines (Figure 2a). Further, this expression appeared to be confined to epithelial cells; when mammary fat pads from prepubescent females were surgically ‘cleared’ of epithelium by removing the epithelial bud, no PRL mRNA was detected (Figure 2b).
Figure 2.
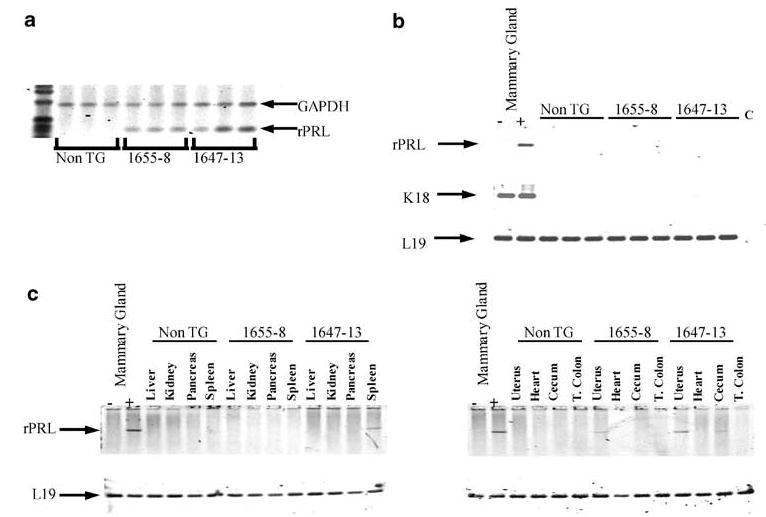
PRL transgene expression is enriched in mammary epithelial cells. (a) Total RNA isolated from mammary glands of 13-week-old mice from NRL-PRL lines 1655-8 and 1647-13, and nontransgenic (non-TG) littermates was examined for rPRL and GAPDH transcripts (positive internal control) using RT–PCR analysis as described in Materials and methods. (b) Total RNA from mammary glands that had been ‘cleared’ of epithelia from the two NRL-PRL lines and nontransgenic littermates (non-TG) was examined for rPRL, K18, and L19 transcripts. Intact mammary glands from a nontransgenic female (−) and 1655-8 littermate (+) serve as negative and positive controls, respectively. C denotes a control PCR reaction without RT. (c) Total RNA from multiple tissues of 13-week-old females (NRL-PRL 1655-8, 1647-13, nontransgenic littermates (non-TG)) were examined for rPRL transcripts. L19 transcripts serve as an internal control for RNA and loading. Intact mammary glands from a nontransgenic female (−) and 1655-8 littermate (+) serve as negative and positive controls, respectively
Analysis of transgene expression in the NRL-PRL 1655-8 lineage in eight other tissues showed undetectable expression in all nonmammary tissues except very low expression in the uterus. In NRL-PRL 1647-13, we observed low expression in the spleen and uterus, and barely detectable levels in the cecum (Figure 2c). PRL mRNA was not detectable in any tissue in nontransgenic littermates. To ascertain if transgene expression caused elevated circulating PRL, bioactive PRL in the plasma was measured using the Nb2 cell assay. PRL levels were elevated four-fold above age-matched nontransgenic controls in NRL-PRL 1655-8, but not 1647-13 (mean±s.d., N=3: non-TG: 63±20 ng/ml; NRL-PRL 1655-8: 253±28 ng/ml; NRL-PRL 1647-13: 45±13 ng/ml).
Development of mammary tumors
Mammary neoplasms, primarily adenocarcinomas, developed with age in all female founders as well as most daughters of the NRL-PRL 1647-13 and 1655-8 lineages (Figure 3, Table 1), indicating that PRL-induced neoplasia is independent of transgene integration site. Although sporadic lesions were noted in other tissues (including a cutaneous fibrosarcoma, a primary lung adenocarcinoma, and a uterine tumor), the incidence of these abnormalities was consistent with that observed in the FVB/N strain in other studies (Mahler et al., 1996; Rose-Hellekant et al., 2002), and therefore they may be unrelated to PRL transgene expression. The majority of the large mammary tumors (>80% of those in the 1655-8 line, >90% of those in the 1647-13 line) developed in the cranial glands, although mammary intraepithelial neoplasias (MINs) and other abnormalities were observed in all glands. This is similar to the distribution of WAP-TGFα as well as C3(1)/SV40 T-antigen-induced lesions in the FVB/N strain (Green et al., 2000; Rose-Hellekant et al., 2002). Several females developed more than one primary neoplasm.
Figure 3.
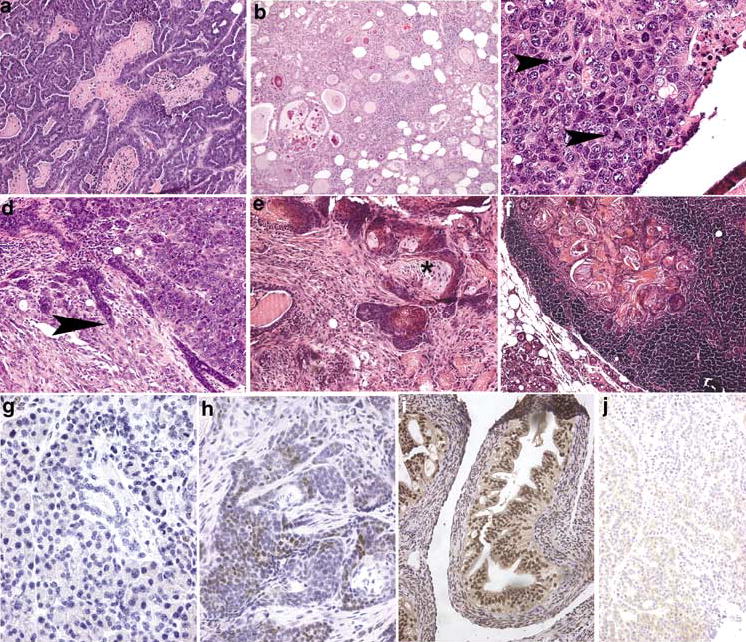
Mammary neoplasia in NRL-PRL transgenic mice. (a) Papillary adenocarcinoma, with focal necrosis. (b) Glandular adenocarcinoma, in which neoplastic epithelial cells form glands that often contain eosinophilic secretions. (c) Solid carcinoma, showing nuclear pleomorphism and multiple, often abnormal, mitotic figures (arrowheads). (d) Solid carcinoma with focal invasion into adjacent stroma (arrowhead). (e) Adenosquamous neoplasm, containing clusters of cells with central squamous differentiation (asterisk). (f) Adenosquamous neoplastic cells present in a mammary lymph node. (g) ERα− mammary neoplasm. (h) ERα+ mammary neoplasm; note the brown stain present over many nuclei. (i) ERα+ control uterus. (j) ERα+ neoplasm treated with nonimmune serum as a negative control. Original magnification: (A, B, F), ×100; (D, E, I, J), ×200; (C, G, H), ×400
Table 1.
Tumor incidence and phenotype in NRL-PRL virgin female mice
| Lineage | Tumor incidence | Mean tumor latency ±s.d.1(months) | Tumor histotypes2(number/all tumors3) |
|---|---|---|---|
| 1647-13 | 15/234 (65%) | 16.7±4.1 | Papillary (10/23; 44%)
Glandular (5/23; 22%) Solid (5/23; 22%) Adenosquamous (1/23; 4%) Fibroadenocarcinoma (1/23; 4%) Carcinosarcoma (1/23; 4%) |
| 1655-8 | 12/154 (80%) | 15.5±2.3 | Papillary (5/16; 31%)
Glandular (3/16; 19%) Solid (1/16; 6%) Adenosquamous (6/16; 37%) Fibroadenocarcinoma (1/16; 6%) |
| Nontransgenic FVB/N5 | 3/61 (5%) | 18.8±5.3 | Fibrosarcoma (1/3; 33%)
Glanular (1/3; 33%) Adenosquamous (1/3; 33%) |
Latency of tumor growth to 1.5 cm;
Many neoplasms displayed a mixed phenotype; only the dominant phenotype is noted using the classifications described (Cardiff et al., 2000). Except for the noted exceptions, all were adenocarcinomas or carcinomas;
More than one tumor was present in several individuals;
Total females for each genotype include all those in the colony ⩾19 months of age;
Both NRL-PRL lineages shared lesion histotypes (classification based on recommendations of the Annapolis Pathology Panel; Cardiff et al., 2000). Papillary adenocarcinomas represented a major class for both lines (Figure 3a). However, the NRL-PRL 1655-8 line also displayed a high fraction of adenosquamous carcinomas (Figure 3e), and the NRL-PRL 1647-13 line developed more solid carcinomas (Figure 3c, d). Some of the well-differentiated mammary masses exhibited some features of adenosis: regular cells with visible lobular organization and an enlarged central duct, and rounded edges compressing adjacent structures (Figure 3b). However, even these masses shared nuclear characteristics with the less-differentiated tumors: pleomorphism, an increased nuclear/cytoplasmic ratio, and prominent nucleoli (Figure 3c). Mitotic figures were evident frequently, consistent with the high level of BrdU labeling (Figure 4b), and occasional bizarre mitotic figures were noted, suggesting chromosomal abnormalities (arrowhead in Figure 3c). The less-differentiated neoplasms were composed of pleiomorphic cells that were organized irregularly. Most demonstrated invasion into surrounding stroma or fat pad (arrowhead in Figure 3d) and necrosis. We also observed desmoplastic stroma and areas of squamous metaplasia (asterisk in Figure 3e) as well as inflammatory cell infiltrates in some lesions. Most neoplasms displayed a mixed pattern of morphologies, consistent with the presence of clonal evolution.
Figure 4.
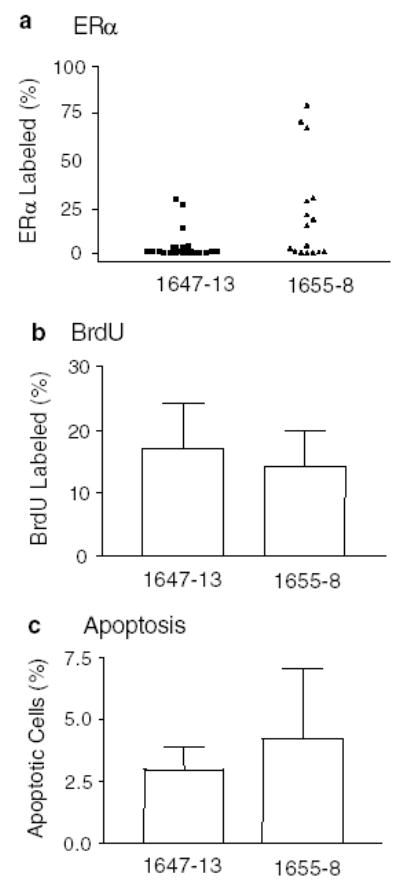
(a) Proportion of ERα+ cells in adenocarcinomas in NRL-PRL 1647-13 (▴), and NRL-PRL 1655-8 virgin females (▪). Each symbol represents a single tumor (N=23 for NRL-PRL 1647-13, and 16 for NRL-PRL 1655-8). The distribution of tumors is close to significantly different between the lines using the Mann–Whitney test (P=0.06). Proliferation (b) and apoptosis (c) in mammary tumors (N=6 tumors for NRL-PRL 1655-8; N=7 for NRL-PRL 1647-13). BrdU labeling and apoptotic indices were determined as described in Materials and methods, and expressed as means±s.d. The lineages were not significantly different
No grossly visible lung metastases were observed at the time of tumor collection. However, invasion into a local lymph noted in one 1655-8 mouse was noted, consistent with metastatic potential (Figure 3f). Fragments of a mammary neoplasm from a 1647-13 mouse transplanted into the fat pad of a nontransgenic recipient generated an invasive anaplastic tumor after 30 days.
ERα expression
As PRL is able to induce ERα expression in the murine mammary gland (Muldoon, 1987), and because of the diagnostic and therapeutic importance of this receptor in clinical breast cancer, we assessed ERα expression in the neoplasms (Figure 3g–j). As shown in Figure 4a, adenocarcinomas of the NRL-PRL 1655-8 line were divided equally between ERα+ (≥15% stained cells) and ERα− tumors (≤2%). However, all but three tumors that developed in the NRL-PRL 1647-13 line were strikingly ERα− (≤3%) (Figure 3g). This difference between the two lines was not quite statistically significant (P=0.06). For both lineages, there was no correlation between ERα status and histotype, BrdU labeling, or apoptotic index.
In contrast, ERα expression in morphologically normal alveolar structures in these end-stage glands was slightly, but significantly lower in line 1655-8 females than in nontransgenic controls (Figure 5a). However, ERα expression within epithelial hyperplasias of this lineage was significantly higher than in nontransgenic controls (Figure 5c). ERα expression in ducts did not differ significantly among genotypes (Figure 5b).
Figure 5.
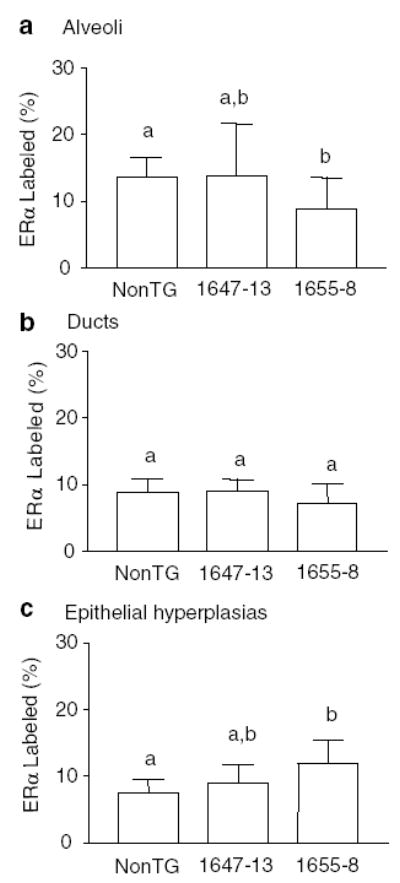
ERα+ cells in morphologically normal alveoli (a), ducts (b), and epithelial hyperplasias (c) in end-stage glands of NRL-PRL 1647-13, NRL-PRL 1655-8, or nontransgenic (non-TG) virgin females at 9 months of age. Labeling was determined as described in the Materials and methods, and data are expressed as means±s.d. Different lower case letters denote statistical differences among total structures of the different lines determined by the Kruskal–Wallis test for nonparametric data, followed by the Mann–Whitney post test (P<0.05)
Preneoplastic mammary changes
An abnormal mammary phenotype was apparent in females of both selected lines from the early stages in postpubertal development (Table 2, Figure 6). Nontransgenic glands from virgin animals displayed ductal growth throughout the fat pad, but little alveolar development, typical for young virgin adult mice of this strain (Figure 6a). By 13 weeks of age, virgin line 1655-8 females displayed increased alveolar development. By 6 and 9 months of age, abnormalities were pronounced in both lines. Ducts in many mice were enlarged, particularly in line 1647-13 (Figure 6b). Epithelial hyperplasias were present in both lines (Figure 6c), and in line 1655-8, often became very large.
Table 2.
Mammary morphology in NRL-PRL virgin female mice
| Age (months) | Line 1647-131 | Line 1655-81 |
|---|---|---|
| 3 | Focal dilated ducts (1/5) | Diffuse alveolar development (4/5)
Focal small epithelial hyperplasias (2/5) Focal dilated ducts (4/5) |
| 6 | Focal dilated ducts (3/5)
Focal irregular ductal epithelium (1/5) Focal small epithelial hyperplasias (1/5) |
Diffuse alveolar development (4/5)
Focal small epithelial hyperplasias (1/5) Focal dilated ducts (2/5) |
| 9 | Focal dilated ducts (4/5)
Increased alveolar development (3/5) Focal small epithelial hyperplasias (4/5) |
Diffuse alveolar development (3/5)
Focal large epithelial hyperplasias (2/5) Focal dilated ducts (1/5) |
| End stage | Multifocal dilated ducts with irregular surface epithelium (11/11)
Focal small epithelial hyperplasias (9/11) Squamous changes (2/11) MINs (11/11) |
Multiple large epithelial hyperplasias, many associated with ducts (13/13)
Focal fibrotic epithelial hyperplasias (6/13) Multiple dilated ducts (7/13) Squamous changes (6/13) MINs (13/13) |
Frequency of lesion type among all mice examined at that stage (compared to nontransgenic mice of the same age);
Mammary intraepithelial neoplasia
Figure 6.
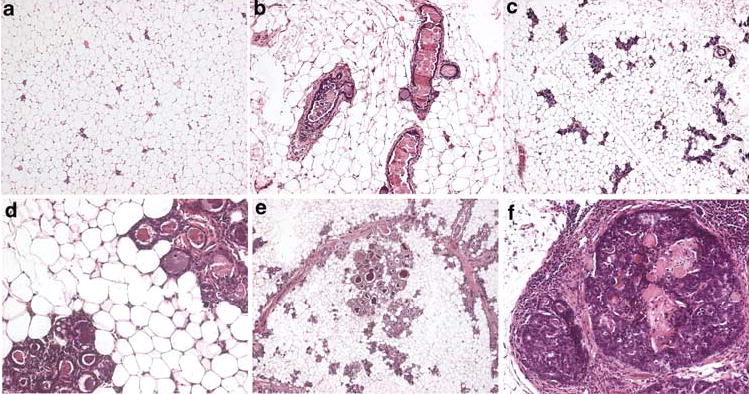
Preneoplastic lesions in mammary glands from virgin nontransgenic (non-TG), NRL-PRL line 1647-13, and NRL-PRL line 1655-8 female mice at 6 months of age. (a) Non-TG mammary gland at 6 months of age. (b) Line 1647-13 mammary gland demonstrating multiple enlarged ducts, some with irregular epithelium at 6 months of age. (c) Line 1655-8 mammary gland demonstrating diffuse alveolar development at 6 months of age. (d) Epithelial hyperplasias present in line 1655-8 mammary gland at 6 months of age. (e) Epithelial hyperplasias associated with a duct in line 1655-8 at end stage. (f) Mammary intraepithelial hyperplasia in line 1647-13 at end stage. Original magnification: (A, B, C), × 100; (D,F), × 200; (E), × 25
At ‘end stage’ (generally 14–20 months of age), both lineages exhibited marked abnormalities in the remainder of the glands, and they shared many features. All glands contained numerous foci of epithelial hyperplasia, most prominent in line 1655-8 (Figure 6d). Most foci were composed of irregular cells with atypical nuclear morphology, including large pleomorphic nuclei with prominent nucleoli. Several MINs were associated with ducts, and resembled human ductal carcinomas in situ (DCIS) (Figure 6f). Enlarged ducts with irregular epithelium were present in both lines, but were most evident in line 1647-13 (Figure 6b). Increased stroma often surrounded these structures (Figure 6e, f). Multiple foci of squamous changes were noted in both lines (Figure 6f). In addition, focal fibrotic epithelial hyperplasias were evident in some 1655-8 glands.
Since only line 1655-8 females had elevated circulating PRL, it was possible that PRL effects on ovarian hormones, particularly progesterone, might play a role in the differences in mammary pathology observed between lines. Therefore, we examined vaginal cytology for 15 days in 10 adult nontransgenic, NRL-PRL line 1647-13, and line 1655-8 females. No differences in the number of diestrus smears between groups were observed (data not shown).
Cell turnover
To examine the effects on cell turnover that may contribute to the mammary phenotype, we measured the fraction of cells in S phase using BrdU incorporation and the fraction undergoing apoptosis at two ages: 6 months, when mammary glands from transgenic females were beginning to develop morphological abnormalities, and at end stage, when tumors were collected. Although there was variation among animals of the same genotype, differences across genotypes were apparent. Transgenic PRL expression increased mitogenesis in morphologically normal alveoli and ducts in both lines (Figure 7a, c). This was evident by 6 months, and the fraction of cells in S phase was even higher, particularly in the alveoli, in the end-stage glands. Consistent with their phenotype, the NRL-PRL 1647-13 line demonstrated a higher proportion of cells in S phase in the ductal epithelium at 6 months compared to the 1655-8 line, although this difference had disappeared by end stage. Transgene expression was also associated with a slight increase in apoptosis in both alveoli and ducts (Figure 7b, d) that was statistically significant by end stage.
Figure 7.
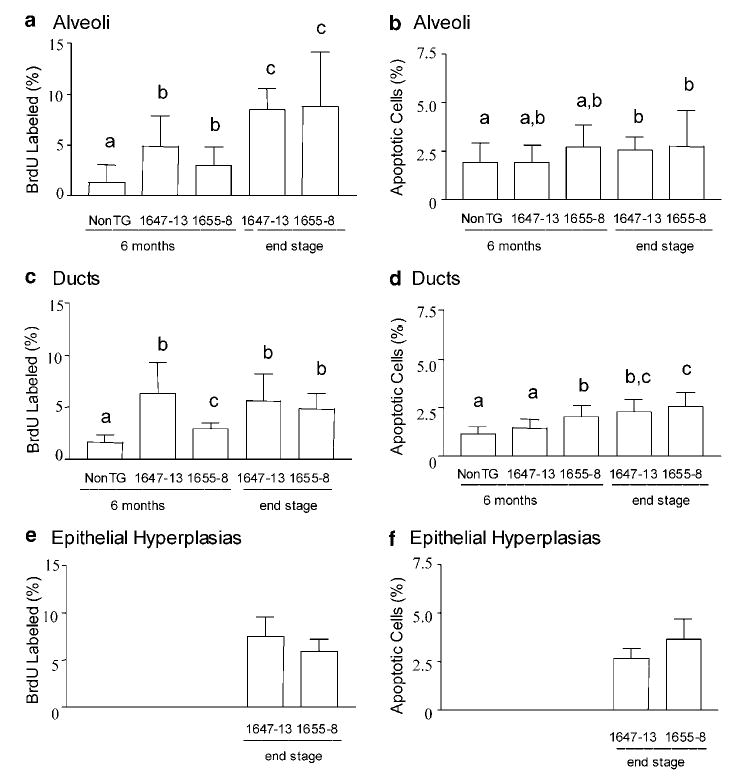
Proliferation and apoptosis in morphologically normal alveoli (a,b), ducts (c,d), and epithelial hyperplasias (e,f) in mammary glands of virgin females at 6 months of age, as well as end stage for transgenic animals (nontransgenic (nonTG), NRL-PRL 1647-13, NRL-PRL 1655-8). Epithelial hyperplasias were infrequent in glands of nontransgenic and line 1647-13 mice at 6 months of age, so only end-stage data for the transgenic lines are shown. BrdU labeling and apoptotic indices were determined from multiple structures in five animals of each genotype as described in Materials and methods, and expressed as means±s.d. Different lower case letters denote statistical differences among groups as determined by the Kruskal–Wallis test for nonparametric data, followed by the Mann– Whitney post test (P<0.05)
Discussion
We have demonstrated that mammary epithelial expression of PRL leads to the development of mammary lesions and formation of invasive ERα+ and ERα− mammary tumors in FVB/N virgin female mice. The high incidence of neoplasms in females with moderate latency that we observed in the current study is similar to the outcome in another mouse strain, C57Bl/6J × CBA-f2, carrying the MT-PRL construct (Wennbo et al., 1997). In both transgenic models, in contrast to studies using pituitary isografts, tumors were observed both in the absence as well as in the presence of elevations in circulating PRL, consistent with an ability of endogenously produced PRL to exert autocrine/paracrine actions in mammary tumor development. The general tumor histotype was similar in both transgenic models and in those obtained with the pituitary isografts in BALB/c mice (Huseby et al., 1985; Welsch and Nagasawa, 1977), indicating consistent morphological effects of PRL exposure regardless of source.
The range of developmental abnormalities we observed prior to tumor development reflects the physiologic actions of PRL at this target. The role of PRL in normal mammogenesis has been established using genetic deletions of the PRL receptor or PRL itself (Horseman et al., 1997; Ormandy et al., 1997a). PRL plays a critical role in ductal branching at puberty, as well as lobuloalveolar development during pregnancy. The developmental abnormalities and hyperplasias associated with the ducts observed here may be a function of the pattern of PRL expression directed by the NRL promoter. However, in the pituitary isograft model, malignancies also developed in areas of intraductal hyperplasia rather than in lobuloalveolar regions (Huseby et al., 1985; Christov et al., 1996), suggesting that the ductal epithelium may be a primary target of PRL in the oncogenic process.
In our study, PRL-induced mammary tumorigenesis in the context of a virgin cycling mouse likely reflects not only the direct effects on the epithelial and stromal cells within the mammary gland, but also indirect effects such as increased responsiveness to other hormones, or increased secretion of factors from other tissues. PRLR mRNA is present in the stroma, in addition to ductal and alveolar epithelium, although the regulation of expression is different between the cell types (Bera et al., 1994; Camarillo et al., 2001; Hovey et al., 2001). Transplantation of PRLR−/− mammary epithelium into the mammary fat pad of normal recipients demonstrated that direct actions of PRL on mammary epithelium are essential for alveolar development (Brisken et al., 1999). However, the normal ductal branching and end-bud progression of the PRLR−/− epithelium in these studies prompted the hypothesis that PRL-induced stromal effects may be critical for these processes (Camarillo et al., 2001). In combination with the growing appreciation for a role for stroma in tumorigenesis (Silberstein, 2001; Imagawa et al., 2002; Rose-Hellekant et al., 2002; Wiseman and Werb, 2002), these reports suggest that stroma may mediate some PRL activities leading to the lesions observed here. The differences in mammary development and early lesions between the NRL-PRL lineages may be because of distinct integration sites resulting in a different pattern of expression, interference with another gene via insertional mutagenesis, and/or a consequence of elevated circulating PRL in the NRL-PRL 1655-8 line. The early alveolar development in the NRL-PRL 1655-8 line suggests that the latter may play a role. Endocrine PRL may support the corpus luteum, increasing progesterone, with consequences for alveolar development (Christov et al., 1996; Hovey et al., 2001; Grimm et al., 2002; Shyamala et al., 2002). Our examination of vaginal cytology did not indicate increased progesterone activity in this lineage. However, this does not rule out a period of transiently elevated progesterone earlier in life, such as that reported in response to treatment with oPRL, or pituitary isografts (Huseby et al., 1985; Hovey et al., 2001). Mammary epithelial transplantation experiments (in progress) will resolve these alternatives.
Most strikingly, transgenic mammary PRL resulted in two populations of end-stage tumors, ERα+ and ERα−. The induction of ERα+ tumors by a mammary oncogene in mice is very unusual; transgenic models for this important class of human tumors have not been available. Although increased ERα expression has been observed in early lesions induced by C3(1)/SV40 TAg expression, ERα was lost as these lesions progressed to high-grade MINs and solid tumors (Yoshidome et al., 2000). PRL has been shown to increase mammary ERα expression directly both in the entire gland in vivo, as well as tumor epithelial cells in vitro (Shafie and Brooks, 1977; Edery et al., 1985; Muldoon, 1987). However, those studies examined relatively short-term effects (<1 week), and the consequences of long-term exposure, such as that occurring in our model, may be more complex.
We observed significantly reduced ERα expression in alveoli of end-stage glands of one NRL-PRL line, despite the elevated proliferative index. This distinction between ERα expression and dividing cells also is observed in both human and murine normal glands (Clarke et al., 1997; Zeps et al., 1998; Russo et al., 1999). Interestingly, ERα expression was increased in hyperplastic structures in that same line, consistent with the observed increase in ERα+/Ki67+ cells in proliferative lesions in the human breast (Shoker et al., 1999). Consistent with the recommendations of the Annapolis Pathology Panel (Cardiff et al., 2000), we did not distinguish between alveolar and ductal hyperplasias in this study. However, murine mammary hyperplasias associated with ductal elements have been reported to express higher levels of ERα than those associated with alveoli (Medina, 2002). This pattern of ERα expression in morphologically normal structures and epithelial hyperplasias contrasted with distinct populations of ERα+ positive (>15%) and ERα− negative (<3%) tumor populations in the NRL-PRL 1655-8 line, and predominance of ERα-negative tumors in the NRL-PRL 1647-13 lineage. While our data do not directly address whether these ERα+ and ERα− tumors reflect either different populations of tumor progenitors, or distinct processes driving ERα loss as tumorigenesis proceeds, the ERα expression we observed in early MINs is consistent with the latter possibility. Interestingly, the NRL-PRL 1647-13 lineage exhibited no significant effects on ERα expression in either morphologically normal structures, or epithelial hyperplasias, compared to the nontransgenic animals. The relation between the early ERα-positive lesions and malignant tumors will be assessed by transplantation experiments. Our NRL-PRL model offers the opportunity to examine the function of ERα in PRL-induced mammary tumorigenesis as well as the role that PRL plays in the regulation of ERα expression. The relation between PRLR and ERα expression in human breast cancer is not clear from the studies reported to date (Ormandy et al., 1997b; Reynolds et al., 1997). In light of the significance of ERα expression in human breast cancer, both as a prognostic indicator and predictor of response to endocrine therapies, understanding the role that PRL may play in this process is important.
PRL activates multiple signaling pathways in mammary epithelial cells in vitro that may contribute to the tumor development observed here, including STAT5, phosphatidylinositol 3′ kinase, MAPkinases, c-src, and others (for a review, see Clevenger et al., 2003). In MCF7 cells, these pathways lead to altered expression of several cell cycle regulators, including cyclins D1 and B, as well as p21, increasing proliferation (Brockman et al., 2002; Schroeder et al., 2002). However, although PRL has been shown to act as a survival factor in mammary tumor cells in vitro in part via bcl-2 (Beck et al., 2002), we observed increased apoptosis in morphologically normal structures as well as hyperplasias and tumors in the NRL-PRL mice, similar to the responses of murine alveoli and mammary tumors to pituitary isografts (Christov et al., 1993). Both STAT5A and AKT increase alveolar survival during pregnancy and early involution (Humphreys and Hennighausen, 1999; Hu et al., 2001; Schwertfeger et al., 2001; Ackler et al., 2002). However, other PRLR-activated mediators, such as c-myc, a powerful promoter of apoptosis as well as proliferation (Yu-Lee, 1990; Jamerson et al., 2000; Evan and Vousden, 2001), or other indirect mediators, may be responsible for our observations. Interestingly, the PRL-induced adenocarcinomas shared several characteristics with those induced by oncogenes affecting the Wnt pathway, including ductal dysmorphogenesis, focal dense stroma with inflammatory infiltrate, and squamous metaplasia (Rosner et al., 2002).
We have shown that elevated mammary PRL induces mammary epithelial neoplasias in the context of the intact, cycling virgin female. However, clinical mammary disease results from interactions of multiple oncogenic processes, many of which could be modulated by the physiologic, endogenous mammary production of PRL. Use of the NRL-PRL transgenic model will enable dissection of the actions and pathways employed by this hormone in mammary oncogenesis, as well as its interactions with ovarian hormones and other characterized oncogenes in bitransgenic models. These studies may point to clinically useful diagnostic strategies and breast cancer treatments.
Materials and methods
Materials
BrdU was obtained from Sigma Chemical Co. (St Louis, MO, USA). The anti-BrdU monoclonal (MAS-250) was obtained from Accurate Scientific, (Westbury, NY, USA). ERα-sc542 (polyclonal) was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). PRL was obtained through NHPP, NIDDK and Dr AF Parlow.
PRL responsivity of the NRL promoter
To evaluate the response of the NRL promoter to PRL, a 3015 bp fragment from the rat homolog of the human neutrophil gelatinase-associated lipocalin and mouse 24p3 (neu-related lipocalin, NRL) promoter (Stoesz and Gould, 1995, a generous gift from Dr Michael Gould) was cloned upstream from a luciferase reporter in pGL3 (NRL-luc; Promega, Madison, WI). This construct, in parallel with a control plasmid containing three copies of the STAT5 binding site from the β-casein gene upstream from the luciferase reporter (GAS3MIN, Schroeder et al., 2002), was transiently transfected into CHO K1 cells, and responsiveness to 4 nM PRL determined as described (Brockman et al., 2002).
Generation of NRL-PRL transgenic mice
The constructs pRPrl-HindIII A and pRPrl-HindIIIB, which together contain the rat PRL gene, were generous gifts from Dr Nancy Cooke (Cooke and Baxter, 1982). A 3.7 kb fragment containing the first three exons and flanking regions was PCR amplified from pRPrl-Hind IIIA (PFU, Stratagene), and was cloned into the BamHI/Hind III sites of pBluescript SK (−) (primers: F, 5′-CGCGGATCCAGTGGTTCTCTTAGGACTTCTTG-3′ and R, 5′-GCGCAAGCTTCCAATGTACTGCCTCAATATGTTG-3′). To confirm that the PCR amplification did not introduce mutations, coding regions were sequenced. An approximately 5 kb HindIII fragment from the pRPrl-Hind IIIB vector containing exons 4 and 5, the polyadenylation signal, and surrounding sequence was cloned into the HindIII site, and the orientation confirmed. Finally, the 3 kb NRL promoter fragment was cloned into the SpeI site upstream of the PRL genomic sequences, destroying that restriction site (Figure 1a). The transgene was removed from the vector by digestion with NotI and SalI, separated by agarose gel electrophoresis, and purified by electroelution. The transgene DNA was injected into the pronuclei of fertilized FVB/N eggs as described (Brinster et al., 1985). Founder mice and transgenic offspring were identified via PCR analysis of tail DNA as described (Grippo and Sandgren, 2000), using primers spanning the NRL promoter and exon 1 of the PRL gene (Figure 1a). (F, 5′-CCTCCTCATTCTCTGCTCTTC-3′; R, 5′-CCAATCACCCTTGCTCTAAACCC-3′). The NRL-PRL mice generated in these studies have been assigned the following designations: line 1647-13, TgN(Nrl-Prl)23EPS; line 1655-8, TgN(Nrl-Prl)24ES.
To measure circulating PRL levels, blood was collected retro-orbitally at 2pm from three 8-month-old females from each of the NRL-PRL 1655-8 and 1647-13 lines as well as from nontransgenic littermates under light anesthesia. The rat pre-T lymphoma cell line, Nb2, was employed to measure bioactive PRL in the plasma. These cells express elevated levels of a very high-affinity PRLR isoform, and depend on PRL for growth (Gout et al., 1980; O’Neal and Yu-Lee, 1994; Shiu et al., 1983). These characteristics make Nb2 cells a very sensitive, widely employed assay for PRL activity. Cells are maintained in Fisher’s media (Sigma), containing 10% horse serum, 10% FBS, and 0.1mM β-mercaptoethanol. For 24 h prior to the assay, cells were blocked in the cell cycle in the media mentioned above without FBS. They were then seeded at 105 cells/well in a 96 well-plate, and were incubated in the media mentioned above containing NIDDK hPRL standards (0–3.0 ng/ml) or plasma diluted 1 : 100 and 1 : 200. After 72 h, total cell number was determined by the MTS assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The amount of circulating PRL activity was calculated from the hPRL standard curve.
Animals were considered to be end stage when tumor diameter reached 1.5 cm. Mice were housed and handled in accordance with the Guide for Care and Use of Laboratory Animals in AAALAC-accredited facilities. All procedures were approved by the University of Wisconsin-Madison Animal Care and Use Committee.
Examination of transgene expression
The tissue distribution of transgene expression was evaluated in 13-week-old intact females using RT–PCR for rat PRL. To evaluate the contribution of epithelial cells to mammary expression, glands of some females were ‘cleared’ of epithelial cells by surgically excising the region of the caudal mammary fat pad surrounding the fourth nipple in 3-week-old prepubescent females (DeOme et al., 1959). The remaining fat pads were collected 9 weeks after the surgery. RT–PCR analysis was performed essentially as described (Schroeder et al., 2001). Total cellular RNA was extracted using the Qiagen RNeasy Mini Kit (Qiagen Inc., Valencia, CA, USA). First-strand complementary DNA (cDNA) was synthesized from 5 μg total RNA for all tissues except the mammary fat pads cleared of epithelial cells, from which 2 μg was employed, and 2 μl of the RT product was subjected to PCR amplification, using conditions that gave exponential product formation (K18, 25 cycles; L19, 30 cycles; rPRL, 25 cycles). Primers for the ribosomal protein, L19, and GAPDH (internal controls for RNA quality and gel loading), as well as cytokeratin 18 (K18), a marker for epithelial cells, were as described (Tseng et al., 1997). Primers for rPRL span exons 3 and 5, and discriminate rat from murine PRL: F, 5′-GGTTATTGCCAAGGCCATCAA-3′; R, 5′-CTTCATCAACTCCTTGCAGGGA-3′. Fragments were resolved on 3% agarose gels, stained with SYBR Green I, and visualized on a STORM system (Molecular Dynamics, Inc.).
Histological examination of mammary gland tissue
Mammary glands were fixed in 10% neutral buffered formalin for 18–24 h, embedded in paraffin and sectioned at 6 μm. Morphological evaluations were performed on hematoxylin and eosin-stained slides.
For evaluation of mitotic activity, mice were injected with 200 mg/kg body weight BrdU 1 h prior to the kill to label cells undergoing DNA synthesis. Positive cells were detected by immunohistochemistry, using sections of duodenum as positive controls and omission of the primary antibody as negative controls as described (Grippo and Sandgren, 2000). Apoptotic cells were identified in hematoxylin and eosin-stained sections using the standard morphological criteria of cytoplasmic shrinkage, nuclear condensation, and nuclear fragmentation. To examine ERα expression, deparaffinized slides were exposed to 0.5% H2O2 in methanol to block endogenous peroxidase activity, boiled for 15 min in 0.1 M Tris, pH 9.0, for antigen retrieval, then blocked in 0.5% milk in PBS. Slides were incubated with the primary antibody (1 : 1000), rinsed and incubated with the secondary antibody (BioGenex, San Ramon, CA, USA), rinsed and incubated with peroxidase strepavidin and 3,3′ diaminobenzidine, and counterstained with hematoxylin. An irrelevant antibody was used as a negative control, and uterine tissue was used as a positive control for ERα staining.
BrdU, apoptotic, and ERα indices were determined separately for different epithelial structures. Ducts contained a single lumen, which varied in size and shape. Alveoli were small circular structures with 1–4 small lumens each surrounded by cuboidal epithelial cells. Epithelial hyperplasias resembled alveoli, but displayed five or more lumens. For alveolar indices, a minimum of 10 separate alveoli and at least 500 cells were counted. For ducts and epithelial hyperplasias, a minimum of 10 separate structures and 1000 cells were evaluated. For tumors, 1000 cells were assessed.
Statistical analyses
Statistical analyses were performed as described using Prism v.3. 02 (GraphPad Software, Inc., San Diego, CA, USA).
Acknowledgments
We are grateful to Dr Jan Lohse for generation of transgenic mice, Kristin Wentworth and Sarah Nikolai for assistance with mouse colony management and data collection, Jennifer Gutzman for her assistance with the Nb2 cell assay, and Drs Michael Gould and Kai-Shun Chen for providing the NRL gene promoter. These studies were supported by National Institutes of Health Grants R01 CA78312 (to LAS), K01 RR00145 (to TAR-H), R01-CA64843 (to EPS), T32-AG00265 and the University of Wisconsin Center for Women’s Health and Women’s Health Research.
References
- Ackler S, Ahmad S, Tobias C, Johnson MD, Glazer RI. Oncogene. 2002;21:198–206. doi: 10.1038/sj.onc.1205052. [DOI] [PubMed] [Google Scholar]
- Amundadottir LT, Merlino G, Dickson RB. Breast Cancer Res Treat. 1996;39:119–135. doi: 10.1007/BF01806083. [DOI] [PubMed] [Google Scholar]
- Beck MT, Peirce SK, Chen WY. Oncogene. 2002;21:5047–5055. doi: 10.1038/sj.onc.1205637. [DOI] [PubMed] [Google Scholar]
- Ben-Jonathan N, Mershon JL, Allen DL, Steinmetz RW. Endocr Rev. 1996;17:639–669. doi: 10.1210/edrv-17-6-639. [DOI] [PubMed] [Google Scholar]
- Bera TK, Hwang S, Swanson SM, Guzman RC, Edery M, Nandi S. Mol Cell Biochem. 1994;132:145–149. doi: 10.1007/BF00926923. [DOI] [PubMed] [Google Scholar]
- Boot LM, Muhlbock O, Ropcke G. Gen Comp Endocrinol. 1962;2:601–603. [Google Scholar]
- Brinster RL, Chen HY, Trumbauer ME, Yagle MK, Palmiter RD. Proc Natl Acad Sci USA. 1985;82:4438–4442. doi: 10.1073/pnas.82.13.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brisken C, Kaur S, Chavarria TE, Binart N, Sutherland RL, Weinberg RA, Kelly PA, Ormandy CJ. Dev Biol. 1999;210:96–106. doi: 10.1006/dbio.1999.9271. [DOI] [PubMed] [Google Scholar]
- Brockman JL, Schroeder MD, Schuler LA. Mol Endocrinol. 2002;16:774–784. doi: 10.1210/mend.16.4.0817. [DOI] [PubMed] [Google Scholar]
- Camarillo IG, Thordarson G, Moffat JG, Van Horn KM, Binart N, Kelly PA, Talamantes F. J Endocrinol. 2001;171:85–95. doi: 10.1677/joe.0.1710085. [DOI] [PubMed] [Google Scholar]
- Cardiff RD, Anver MR, Gusterson BA, Hennighausen L, Jensen RA, Merino MJ, Rehm S, Russo J, Tavassoli FA, Wakefield LM, Ward JM, Green JE. Oncogene. 2000;19:968–988. doi: 10.1038/sj.onc.1203277. [DOI] [PubMed] [Google Scholar]
- Christov K, Swanson SM, Guzman RC, Thordarson G, Jin E, Talamantes F, Nandi S. Carcinogenesis. 1993;14:2019–2025. doi: 10.1093/carcin/14.10.2019. [DOI] [PubMed] [Google Scholar]
- Christov KT, Guzman RC, Swanson SM, Thordarson G, Talamantes F, Nandi S. Carcinogenesis. 1996;17:1741–1746. doi: 10.1093/carcin/17.8.1741. [DOI] [PubMed] [Google Scholar]
- Clarke RB, Howell A, Potten CS, Anderson E. Cancer Res. 1997;57:4987–4991. [PubMed] [Google Scholar]
- Clevenger CV, Furth PA, Hankinson SE, Schuler LA. Endocr Rev. 2003;24:1–27. doi: 10.1210/er.2001-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke NE, Baxter JD. Nature. 1982;297:603–606. doi: 10.1038/297603a0. [DOI] [PubMed] [Google Scholar]
- DeOme KB, Faulkin LJ, Bern HA, Blair PB. Cancer Res. 1959;19:515–520. [PubMed] [Google Scholar]
- Edery M, Imagawa W, Larson L, Nandi S. Endocrinology. 1985;116:105–112. doi: 10.1210/endo-116-1-105. [DOI] [PubMed] [Google Scholar]
- Evan GI, Vousden KH. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- Goffin V, Touraine P, Pichard C, Bernichtein S, Kelly PA. Mol Cell Endocrinol. 1999;151:79–87. doi: 10.1016/s0303-7207(99)00023-4. [DOI] [PubMed] [Google Scholar]
- Gout PW, Beer CT, Noble RL. Cancer Res. 1980;40:2433–2436. [PubMed] [Google Scholar]
- Green JE, Shibata MA, Yoshidome K, Liu ML, Jorcyk C, Anver MR, Wigginton J, Wiltrout R, Shibata E, Kaczmarczyk S, Wang W, Liu ZY, Calvo A, Couldrey C. Oncogene. 2000;19:1020–1027. doi: 10.1038/sj.onc.1203280. [DOI] [PubMed] [Google Scholar]
- Grimm SL, Seagroves TN, Kabotyanski EB, Hovey RC, Vonderhaar BK, Lydon JP, Miyoshi K, Hennighausen L, Ormandy CJ, Lee AV, Stull MA, Wood TL, Rosen JM. Mol Endocrinol. 2002;16:2675–2691. doi: 10.1210/me.2002-0239. [DOI] [PubMed] [Google Scholar]
- Grippo PJ, Sandgren EP. Am J Pathol. 2000;157:805–813. doi: 10.1016/S0002-9440(10)64594-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankinson SE, Willett WC, Michaud S, Manson JE, Colditz GA, Longcope C, Rosner B, Speizer FE. J Natl Cancer Inst. 1999;91:629–634. doi: 10.1093/jnci/91.7.629. [DOI] [PubMed] [Google Scholar]
- Horseman ND, Zhao WZ, Montecino-Rodriguez E, Tanaka M, Nakashima K, Engle SJ, Smith F, Markoff E, Dorshkind K. EMBO J. 1997;16:6926–6935. doi: 10.1093/emboj/16.23.6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovey RC, Trott JF, Ginsburg E, Goldhar A, Sasaki MM, Fountain SJ, Sundararajan K, Vonderhaar BK. Dev Dyn. 2001;222:192–205. doi: 10.1002/dvdy.1179. [DOI] [PubMed] [Google Scholar]
- Hu Z-Z, Meng J, Dufau ML. J Biol Chem. 2001;276:41086–41094. doi: 10.1074/jbc.M102109200. [DOI] [PubMed] [Google Scholar]
- Humphreys RC, Hennighausen L. Cell Growth Differ. 1999;10:685–694. [PubMed] [Google Scholar]
- Huseby RA, Soares MJ, Talamantes F. Endocrinology. 1985;116:1440–1448. doi: 10.1210/endo-116-4-1440. [DOI] [PubMed] [Google Scholar]
- Imagawa W, Pedchenko VK, Helber J, Zhang HZ. J Steroid Biochem Mol Biol. 2002;80:213–230. doi: 10.1016/s0960-0760(01)00188-1. [DOI] [PubMed] [Google Scholar]
- Jamerson MH, Johnson MD, Dickson RB. Oncogene. 2000;19:1065–1071. doi: 10.1038/sj.onc.1203268. [DOI] [PubMed] [Google Scholar]
- Mahler JF, Stokes W, Mann PC, Takaoka M, Maronpot RR. Toxicol Pathol. 1996;24:710–716. doi: 10.1177/019262339602400606. [DOI] [PubMed] [Google Scholar]
- Medina D. Biochim Biophys Acta. 2002;1603:1. doi: 10.1016/s0304-419x(02)00053-7. [DOI] [PubMed] [Google Scholar]
- Mertani HC, Garcia-Caballero T, Lambert A, Gérard F, Palayer C, Boutin JM, Vonderhaar BK, Waters MJ, Lobie PE, Morel G. Int J Cancer. 1998;79:202–211. doi: 10.1002/(sici)1097-0215(19980417)79:2<202::aid-ijc17>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Muldoon TG. Endocrinology. 1987;121:141–149. doi: 10.1210/endo-121-1-141. [DOI] [PubMed] [Google Scholar]
- O’Neal KD, Yu-Lee L. J Biol Chem. 1994;269:26076–26082. [PubMed] [Google Scholar]
- Ormandy CJ, Camus A, Barra J, Damotte D, Lucas B, Buteau H, Edery M, Brousse N, Babinet C, Binart N, Kelly PA. Genes Dev. 1997a;11:167–178. doi: 10.1101/gad.11.2.167. [DOI] [PubMed] [Google Scholar]
- Ormandy CJ, Hall RE, Manning DL, Robertson JFR, Blamey RW, Kelly PA, Nicholson RI, Sutherland RL. J Clin Endocrinol Metab. 1997b;82:3692–3699. doi: 10.1210/jcem.82.11.4361. [DOI] [PubMed] [Google Scholar]
- Otten AD, Sanders MM, McKnight GS. Mol Endocrinol. 1988;2:143–147. doi: 10.1210/mend-2-2-143. [DOI] [PubMed] [Google Scholar]
- Qin W, Golovkina TV, Peng T, Nepomnaschy I, Buggiano V, Piazzon I, Ross SR. J Virol. 1999;73:368–376. doi: 10.1128/jvi.73.1.368-376.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds C, Montone KT, Powell CM, Tomaszewski JE, Clevenger CV. Endocrinology. 1997;138:5555–5560. doi: 10.1210/endo.138.12.5605. [DOI] [PubMed] [Google Scholar]
- Rose-Hellekant TA, Gilchrist K, Sandgren EP. Am J Pathol. 2002;161:1439–1447. doi: 10.1016/s0002-9440(10)64419-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosner A, Miyoshi K, Landesman-Bollag E, Xu X, Seldin DC, Moser AR, MacLeod CL, Shyamala G, Gillgrass AE, Cardiff RD. Am J Pathol. 2002;161:1087–1097. doi: 10.1016/S0002-9440(10)64269-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo J, Ao X, Grill C, Russo IH. Breast Cancer Res Treat. 1999;53:217–227. doi: 10.1023/a:1006186719322. [DOI] [PubMed] [Google Scholar]
- Schroeder MD, Rose-Hellekant T, Sandgren EPand Schuler LA. Mol Cell Endocrinol. 2001;175:173–183. doi: 10.1016/s0303-7207(01)00385-9. [DOI] [PubMed] [Google Scholar]
- Schroeder MD, Symowicz J, Schuler LA. Mol Endocrinol. 2002;16:45–57. doi: 10.1210/mend.16.1.0762. [DOI] [PubMed] [Google Scholar]
- Schwertfeger KL, Richert MM, Anderson SM. Mol Endocrinol. 2001;15:867–881. doi: 10.1210/mend.15.6.0663. [DOI] [PubMed] [Google Scholar]
- Shafie S, Brooks SC. Cancer Res. 1977;37:792–799. [PubMed] [Google Scholar]
- Shiu RPC, Elsholtz HP, Tanaka T, Friesen HG, Gout PW, Beer CT, Noble RL. Endocrinology. 1983;113:159–165. doi: 10.1210/endo-113-1-159. [DOI] [PubMed] [Google Scholar]
- Shoker BS, Jarvis C, Clarke RB, Anderson E, Hewlett J, Davies MP, Sibson DR, Sloane JP. Am J Pathol. 1999;155:1811–1815. doi: 10.1016/S0002-9440(10)65498-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyamala G, Chou YC, Louie SG, Guzman RC, Smith GH, Nandi S. J Steroid Biochem Mol Biol. 2002;80:137–148. doi: 10.1016/s0960-0760(01)00182-0. [DOI] [PubMed] [Google Scholar]
- Silberstein GB. Breast Cancer Res. 2001;3:218–223. doi: 10.1186/bcr299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoesz SPand Gould MN. Oncogene. 1995;11:2233–2241. [PubMed] [Google Scholar]
- Touraine P, Martini JF, Zafrani B, Durand JC, Labaille F, Malet C, Nicolas A, Trivin C, Postel-Vinay MC, Kuttenn F, Kelly PA. J Clin Endocrinol Metab. 1998;83:667–674. doi: 10.1210/jcem.83.2.4564. [DOI] [PubMed] [Google Scholar]
- Tseng YH, Kessler MA, Schuler LA. Mol Cell Endocrinol. 1997;128:117–127. doi: 10.1016/s0303-7207(97)04028-8. [DOI] [PubMed] [Google Scholar]
- Vomachka AJ, Pratt SL, Lockefeer JA, Horseman ND. Oncogene. 2000;19:1077–1084. doi: 10.1038/sj.onc.1203348. [DOI] [PubMed] [Google Scholar]
- Vonderhaar BK. In: Endocrine Oncology. Ethier SP, editor. Humana Press; Totowa, NJ: 2000. pp. 101–120. [Google Scholar]
- Welsch CW, Nagasawa H. Cancer Res. 1977;37:951–963. [PubMed] [Google Scholar]
- Wennbo H, Gebre-Medhin M, Gritli-Linde A, Ohlsson C, Isaksson OGPand Törnell J. J Clin Invest. 1997;100:2744–2751. doi: 10.1172/JCI119820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennbo H, Törnell J. Oncogene. 2000;19:1072–1076. doi: 10.1038/sj.onc.1203349. [DOI] [PubMed] [Google Scholar]
- Wiseman BS, Werb Z. Science. 2002;296:1046–1049. doi: 10.1126/science.1067431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarus S, Hadsell D, Rosen JM. Genet Eng. 1996;18:57–81. doi: 10.1007/978-1-4899-1766-9_5. [DOI] [PubMed] [Google Scholar]
- Yoshidome K, Shibata MA, Couldrey C, Korach KS, Green JE. Cancer Res. 2000;60:6901–6910. [PubMed] [Google Scholar]
- Yu-Lee L. Mol Cell Endocrinol. 1990;68:21–28. doi: 10.1016/0303-7207(90)90165-5. [DOI] [PubMed] [Google Scholar]
- Zeps N, Bentel JM, Papadimitriou JM, D’Antuono MF, Dawkins HJ. Differentiation. 1998;62:221–226. doi: 10.1046/j.1432-0436.1998.6250221.x. [DOI] [PubMed] [Google Scholar]