

Abstract
Complementary short-strand DNA homooligomers and methylthiourea-linked homonucleosides associate and form triplexes in solution. The melting temperatures, Tm, the association and dissociation kinetic and thermodynamic parameters, and activation energies were determined by UV thermal analysis for the triplexes of short-strand DNA homooligomers [d(pA)10–d(pA)23] and poly(dA) with the methylthiourea-linked nucleoside [5′-NH3+-d(Tmt)4-T-OH {DNmt5}]. Circular dichroism studies show evidence of triple-helical association dependent on the length of the target homooligomer. The melting and cooling curves exhibit hysteresis behavior in the temperature range of 10–95°C at 0.13 deg/min thermal rate. From these curves, the rate constants and the energies of activation for association (kon, Eon) and dissociation (koff, Eoff) were obtained. Tm decreases with the ionic strength and increases with increase in length of the monomers. The rate constants kon and koff at a given temperature (288 K–310 K) are dependent on the DNA strand length and also decrease and increase respectively with the ionic strength. The energies of activation for the association and dissociation processes are in the range of −18 to −38 kcal/mol and 3 to 18 kcal/mol, respectively. The equilibrium constant for the formation of the triplexes [5′-NH3+-d(Tmt)4-T-OH)2⋅d(pA)x, x = 10–23] is several orders of magnitude greater when compared with the triplexes of DNA. The number of base triplets in the nucleus of the DNmt2⋅DNA triple-helix (nucleation–zipping model) increases with decreased DNA oligomer length and with increased ionic strength. The values of ΔH° calculated from the activation parameters are between −30 and −50 kcal/(mol base) and the values of ΔG° are between −6 and −11 kcal/(mol base) for short-strand DNA.
Keywords: antisense, antigene, kinetics, triplex, cationic oligonucleotide
The negatively charged phosphodiester linkages of double- and triple-stranded DNA and RNA reside side by side, causing considerable charge–charge electrostatic repulsion. This is particularly so at the low ionic strength that is physiological. This feature, as well as the susceptibility of DNA and RNA to nuclease activity, limits the usefulness of RNA and DNA as antisense or antigene drugs. We have reported the replacement of the phosphate linkages in DNA and RNA by achiral guanido groups, which we identify as DNG and RNG (1–3) and more recently have designed the polycationic nucleotide linkage with methylisothiouronium salts, or methylated thioureas, abbreviated as DNmt (Scheme S1) (4, 5). In this study, we report the melting temperatures, Tm and circular dichroism curves, as well as the association and dissociation kinetic and thermodynamic parameters for the formation of triplexes of short-strand DNA homooligomers [d(pA)10, d(pA)15, d(pA)20, d(pA)23] with 5′-NH3+-d(Tmt)4-T-OH.
Scheme 1.

MATERIALS
The concentrations of nucleotide solutions were determined by using the extinction coefficients (per mol of nucleotide) calculated according to the nearest neighboring effects (6). For d(Tmt)5 we used ɛ268 = 8700 M−1 cm−1. All experiments were conducted in either (i) 0.015 M phosphate buffer, pH 7–7.5 or (ii) 0.008 M phosphate buffer at pH 6.85. The ionic strength, μ, was adjusted with KCl and is presented with the corresponding concentration of KCl. The concentration of nucleosides, expressed in M/base, was 4.0 × 10−5 M, and the ionic strength ranged from 0.03 to 0.12 M KCl. The nucleoside concentration referred to is the limiting component forming the triplex (e.g., a concentration of 4.0 × 10−5 M/base in the reaction of A + 2 T means [A] = 4.0 × 10−5 M/base and [T] = 8.0 × 10−5 M/base). All stock solutions were kept at 4°C between experiments.
CD, UV Spectroscopy, and Data Collection.
CD spectra were obtained on an OLIS (Jefferson, GA) RSM circular dichroism spectrophotometer. Scans were run from 320 nm to 190 nm. Measurements were recorded at every nanometer. Ten scans were recorded, averaged, and smoothed for each curve. Samples were held in a 1-cm path length cuvette at 25°C. UV spectra were recorded at λ = 260 nm on a Cary 1E UV/vis spectrophotometer equipped with temperature programming. Spectrophotometer stability and λ alignment were checked before initiation of each melting point experiment. For the Tm determinations, hypochromicity was used. Data were recorded every 1.0 degree. The samples were heated from 25°C to 95°C at 5 deg/min (Scheme S1), the annealing (95–10°C) and the melting (10–95°C) were conducted at 0.13 deg/min, and the samples were brought back to 25°C at a rate of 5 deg/min. The reaction solutions were equilibrated for 15 min at the highest and lowest temperatures.
Analysis of Kinetic Data for Triplex Formation.
The equation for triplex formation that was used (Eq. 1) describes the formation of the triplex (Tr) from the duplex (D) and the monomer (M), as individual strands. This equation could be interpreted as the reaction of double-strand D
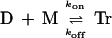 |
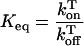 |
1 |
with single-strand M. However, the process of triplex formation may take place in different ways that we shall refer to as domino, normal, and slide (or dangling ends) (Scheme S2) (2).
Scheme 2.

The expression of kinetic equations for the reactants and products (Eq. 2) can be accomplished by expressing the concentrations of strands in molar per base (M/base). The theory behind the “on” and “off” rates of the dissociation/association of the triplexes and the derivation of kinetic equations has been described in detail (2, 7). In Eq. 1, a triplex is formed from and dissociates to a duplex and a third monomer strand with rate constants kon and koff. The associated rate for this reaction is given in Eq. 2.
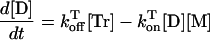 |
2 |
Letting Dtot = [D] + [Tr] and Mtot = [M] + [Tr], where the subscript “tot” stands for the total concentration and superscript T stands for temperature, the monitored absorbance is a weighted combination of the absorbances of the trimer, dimer, and monomer (Eq. 3), where α = [Tr]/Dtot.
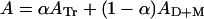 |
3 |
The expressions for kon and koff were solved as previously reported (2, 7). The rate constants kon and koff are functions of temperature and, therefore, can be expressed as Arrhenius equations (2, 7):
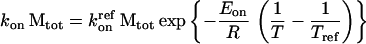 |
4a |
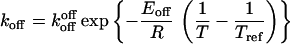 |
4b |
where R = 1.98 cal/mol⋅K, and Tref is the reference temperature at which the rate constant kref applies. In a plot of ln(kon/off) vs. 1/T − 1/Tref, the thermodynamic parameters Eon/offR are obtained as the slope and the kinetic konMtot, koff parameter as the intercept. The limitations of this approach have been reported previously (2, 7–9).
Data Analysis.
The raw data from the melting point determinations was subjected to Gaussian smoothing by using matlab 4. Calculations of thermodynamic parameters were performed by using the resulting database.
RESULTS
Melting Curves and Their Hysteresis Behavior.
Melting studies of triplexes formed from short-strand DNA homooligomers and 5′-NH3+-d(Tmt)4-T-OH were carried out by using UV spectroscopy at 260 nm. The ratio between 5′-NH3+-d(Tmt)4-T-OH and d(pA)x was 2:1. We have found that all melting curves exhibit hysteresis (Fig. 1). The divergence of the heating and cooling curves are functions of the rates of heating and cooling.
Figure 1.

Hysteresis curve for the triplex denaturation and renaturation of d(pA)20 with 5′-NH3+-d(Tmt)4-T-OH (Tmt⋅Ap⋅Tmt) at 4.0 × 10−5 M/base of single-strand DNA respectively at μ = 0.03 and 0.13 deg/min heating (lower curve)/cooling (upper curve) rate.
Stoichiometry of the Binding.
Job plots were constructed from absorbances of solutions containing 5′-NH3+-d(Tmt)4-T-OH and the DNA homooligomers d(pA)10–23 and poly(dA) to assess whether the hystereses generally seen in the heating and cooling curves (Fig. 2) are associated with triplex denaturation. As experienced with our previously reported studies of DNmt binding to poly(dA) and poly(rA) (4), the Job plots clearly establish a minimum at ca. 67% 5′-NH3+-d(Tmt)4-T-OH, which corresponds to the formation of a 2:1 (5′-NH3+-d(Tmt)4-T-OH)2⋅(DNA) complex. The same results were obtained at three wavelengths (202, 260, and 284 nm) (4).
Figure 2.

Job plots of d(pA)20 with 5′-NH3+-d(Tmt)4-T-OH in a concentration of 4.0 × 10−5 M/base at 260 nm (●) and 284 nm (■).
Circular Dichroism Spectroscopy of Complementary DNA Oligomers with DNmt.
The CD spectrum of an oligomer solution can give valuable information about the conformation of the oligomers as single strands or in association with other DNA oligomers. The CD spectrum of the complex formed between pentameric thymidyl DNA oligomer 5′-NH3+-d(Tmt)4-T-OH and poly(dA) is shown in Fig. 3. When DNmt is associated with complementary DNA, the CD spectrum of the complex does not match the spectrum calculated from the weighted sums of the CD spectra for the constituent oligomers. Fig. 4 shows the difference CD spectrum of the triple helical complex of DNmt with d(pA)x subtracted from the spectrum obtained for d(pA)x alone.
Figure 3.

CD spectra of poly(dA) compared with DNmt2⋅poly(dA) triplex. Solution conditions: 1.0 × 10−5 M in oligomer, 0.03 M KCl, 10 mM phosphate buffer, pH 6.8 at 25°C.
Figure 4.

Difference CD spectra of poly(dA), d(pA)30 and d(pA)25 binding to DNmt. The ratio of d(pA)x to DNmt was 1:2. Spectrum of poly(dA) refers to the difference spectra [poly(dA) − DNmt2⋅poly(dA) triplex]. Solution conditions: 1.0 × 10−5 M in oligomer, 0.03 M KCl, 10 mM phosphate buffer, pH 6.8 at 25°C.
The Effect of Ionic Strength on Tm.
At 4.0 × 10−5 M concentration in base A and a 2:1 ratio between the T bases and A bases (in 10 mM phosphate buffer pH 7.0), we saw only one Tm transition (at 260 nm) after annealing 5′-NH3+-d(Tmt)4-T-OH with d(pA)10, d(pA)15, d(pA)20, and d(pA)23 (Fig. 1). We assigned this transition to the triplex denaturation. In contrast, interaction of 5′-NH3+-d(Tmt)4-T-OH with poly(dA) exhibits the two transitions corresponding to triplex and duplex melting, as described previously (4). With increase in ionic strength (μ), the value of Tm decreases (Table 2). Above μ = 0.12, the melting curves become shallow (i.e., as ΔA260 becomes smaller), and the Tm determination is less accurate.
Table 2.
Activation parameters for the triplexes of d(pA)x with DNmt
| μ | d(pA)x, x = | Eon, kcal/mol | Eoff, kcal/mol | ΔH, kcal/mol*† | Tm, °C‡ |
|---|---|---|---|---|---|
| 0.03 | 10 | −26.87 | 10.37 | −37.24 | 36.03 |
| 0.03 | 15 | −24.37 | 10.85 | −35.22 | 52.74 |
| 0.03 | 20 | −18.92 | 18.73 | −37.65 | 64.22 |
| 0.03 | 23 | −21.7 | 16.65 | −38.35 | 60.31 |
| 0.08 | 10 | −37.73 | 3.129 | −40.859 | 34.7 |
| 0.08 | 15 | −38.01 | 11.52 | −49.53 | 45.16 |
| 0.08 | 20 | −33.67 | 17.087 | −50.757 | 49.6 |
| 0.08 | 23 | −31.34 | 11.68 | −43.02 | 49.7 |
Margin of errors: kon⋅Mtot = ±0.25 × 10−4 s−1; koff ±0.25 × 10−6 s−1; Eon and Eoff = ±0.5 kcal/mol.
From Eon − Eoff5;
±0.2 kcal/mol;
±1°C.
Kinetics of the Association and Dissociation.
An expression that provides the temperature dependence of the rate constants for association (kon) and dissociation (koff) is provided in Eq. 4. As the rate of the heating and cooling increases, the rate of equilibration of the species lags such that the hysteresis becomes more marked. The four ramps depicted in Fig. 5 show the variation of the absorbance (A260) vs. temperature. At equilibrium, both heating and cooling curves coincide, satisfying the mathematical condition dα/dt = 0 (α = fraction of duplex engaged in the triplex). Eq. 4 provides konref and koffref as well as the activation Eon and Eoff parameters at a given reference temperature. Examination of plots of ln(kon) and ln(koff) vs. 1/T show that in each case there is a linear and a scattered region. The scattered region is associated with the initial and final portions of the triplex melting curve where there is little change in the absorbance, i.e., dα/dt = 0. For our studies, plots of ln(k) vs. 1/T were found to be linear between the temperature range of 14°C–40°C. An exception was d(pA)10, which showed linearity before 35°C because of the lower Tm compared with longer DNA oligomers. From the intercepts of plots of ln(kon) and ln(koff) vs. 1/T − 1/Tref, we obtain konrefMtot and koff, respectively. Eon/R and Eoff/R are determined from the respective slopes from Fig. 6 (Table 1). Numerical values calculated from Fig. 6 by using Eqs. 4a and 4b are:
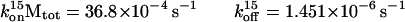 |
 |
 |
Inspection of Table 1 shows that the values of kon15°CMtot, for short-strand DNA oligonucleotides, vary between 7.52 × 10−4 and 49.04 × 10−4 s−1 and those of koff15°C vary between 1.45 × 10−6 and 20.01 × 10−6 s−1. The energies of activation Eon15°C for short-strand oligonucleotides were between −18 and −38 kcal/mol and Eoff15°C between 3.1 and 18.7 kcal/mol. Table 1 shows the values of koff15°C, koff15°C, and Keq at different ionic strengths and temperatures. Reference temperatures (15°C, 25°C, and 37°C) were chosen to make relevant comparisons with other published works (15°C, 25°C) (2, 3, 7, 10) and their biological relevance (37°C).
Figure 5.

Effect of the heating/cooling rate on the extent of association/dissociation of the triplex of d(pA)20 with 5′-NH3+-d(Tmt)4-T-OH at 4.0 × 10−5 M/base monomer concentration. 1, fast heating (5 deg/min); 2, slow annealing (0.13 deg/min); 3, slow melting (0.13 deg/min); 4, fast cooling (5 deg/min).
Figure 6.

Dependence of ln(konMT) and ln(koff) on 1/T for the denaturation/renaturation of triplex of (5′-NH3+-d(Tmt)4-T-OH)2⋅d(pA)20 at 4.0 × 10−5 M/base and μ = 0.03 and dT/dt = 0.13 deg/min.
Table 1.
Kinetic parameters for the triplex melting of d(pA)x with DNmt
| T, °C | μ | d(pA)x, x = | kon⋅MT⋅104 | koff·106 | Keq⋅10−5 |
|---|---|---|---|---|---|
| 15 | 0.03 | 10 | 20.23 | 11.88 | 42.572 |
| 0.03 | 15 | 38.21 | 9.54 | 100.13 | |
| 0.03 | 20 | 36.68 | 1.451 | 631.98 | |
| 0.03 | 23 | 49.04 | 2.176 | 563.42 | |
| 0.08 | 10 | 7.52 | 20.01 | 9.3953 | |
| 0.08 | 15 | 32.14 | 12.2 | 65.861 | |
| 0.08 | 20 | 36.23 | 4.382 | 206.7 | |
| 0.08 | 23 | 14.56 | 11.93 | 30.511 | |
| 25 | 0.03 | 10 | 2.73 | 19.95 | 3.4211 |
| 0.03 | 15 | 9.108 | 18.06 | 12.608 | |
| 0.03 | 20 | 11.95 | 4.36 | 68.521 | |
| 0.03 | 23 | 13.6 | 5.79 | 58.722 | |
| 0.08 | 10 | 1.7 | 27.7 | 1.5343 | |
| 0.08 | 15 | 3.39 | 24.04 | 3.5254 | |
| 0.08 | 20 | 5 | 11.975 | 10.438 | |
| 0.08 | 23 | 5.79 | 23.72 | 6.1024 | |
| 37 | 0.03 | 15 | 1.838 | 36.84 | 1.2473 |
| 0.03 | 20 | 3.45 | 14.94 | 5.7731 | |
| 0.03 | 23 | 3.246 | 17.284 | 4.6951 | |
| 0.08 | 15 | 0.27 | 51.44 | 0.13122 | |
| 0.08 | 20 | 0.547 | 36.832 | 0.37128 | |
| 0.08 | 23 | 0.28 | 51.1 | 0.13699 |
Influence of the DNA Length on kon, koff, and Keq.
The rate constant for the association (kon) of 5′-NH3+-d(Tmt)4-T-OH to d(pA)x increases with increase in chain length (x = 10–20), while the rate constant for the dissociation (koff) decreases (Table 1 and Fig. 7). On going from d(pA)20 to d(pA)23, there is a slight drop in the value of kon (except at μ = 0.03, 15°C), whereas koff increases slightly. This is best explained by looking at the Keq values. At every ionic strength and temperature, Keq increases from d(pA)10 to d(pA)20 and decreases slightly from d(pA)20 to d(pA)23 (Table 1, Fig. 7).
Figure 7.

Dependence of kon15 and koff15 with the DNA length for the dissociation of the triplex (5′-NH3+-d(Tmt)4-T-OH)2⋅d(pA)x at 4.0 × 10−5 M/base monomer concentration, μ = 0.03 and dT/dt = 0.13 deg/min.
Dependence of Kinetic and Activation Parameters on Ionic Strength.
By increasing the ionic strength, kon increases while koff decreases (Table 1). For d(pA)10 at 4.0 ×10−5 M/base concentration using 15°C as Tref, ΔkonMtot = 13 × 10−4 s−1 and Δkoff = 8.2 × 10−6 s−1. For d(pA)10, with the ionic strength decrease (0.08 to 0.03), Eon decreases from −37 to −26 kcal/mol, whereas Eoff increases from 3.12 to 10.37 kcal/mol (Table 2).
Dependence of Activation Parameters on DNA Length.
At μ = 0.03, an increase in DNA length from d(pA)10 to d(pA)20 while maintaining the 2:1 ratio between 5′-NH3+-d(Tmt)4-T-OH and DNA results in an increase in Eoff from 10.37 to 18.73 kcal/mol, whereas Eon decreases from −26 kcal/mol to −18 kcal/mol or becomes less negative at μ = 0.03 (Table 2). Eoff values decrease (similar to increase in koff) on going from d(pA)20 to d(pA)23.
DISCUSSION
Our previous work on DNmt binding (4, 5) to polynucleotides has shown that (i) thymidyl DNmt (5′-NH3+-d(Tmt)4-T-OH) has much stronger affinity for DNA and RNA, because of electrostatic attractions, than DNA for RNA or vice versa; (ii) thymidyl DNmt is specific for its complementary tracts of adenine bases and does not interact with guanylic, cytidylic, or uridylic tracts; (iii) the thermal stability of DNmt⋅RNA and DNmt⋅DNA structures is attenuated by increasing salt concentrations; (iv) DNmt forms triple helical structures from 15–60°C that are very stable under physiological ionic strength conditions. The effect of ionic strength on melting is more pronounced with 5′-NH3+-d(Tmt)4-T-OH interacting with d(pA)x and has an opposite effect as compared with DNA complexes with DNA or RNA. This is because of electrostatic interactions being attenuated by increasing salt concentrations.
The melting studies of complexes formed from short-strand DNA homooligomers and 5′-NH3+-d(Tmt)4-T-OH showed that all melting curves exhibit hysteresis (Fig. 1) at the rate of heating–cooling used (0.13 deg/min). This points to the formation of triplexes between 5′-NH3+-d(Tmt)4-T-OH and d(pA)x. Previous melting studies with poly(dA) and 5′-NH3+-d(Tmt)4-T-OH established the existence of two transitions corresponding to melting of triplex and duplex (4). The triplex formation between 5′-NH3+-d(Tmt)4-T-OH and d(pA)10–23 under our experimental conditions leads to only one transition between 5°C and 95°C, similar to results obtained with DNG melting curves with DNA homooligomers (2). Further evidence for triplex formation comes from the use of the continuous variation method. The minimum observed in Fig. 2 occurs at ca. 67% 5′-NH3+-d(Tmt)4-T-OH, demonstrating the existence of the (5′-NH3+-d(Tmt)4-T-OH)2⋅(DNA) complex. To further study the nature of (5′-NH3+-d(Tmt)4-T-OH)2⋅(DNA) triplex, we used circular dichroism (Figs. 3 and 4). Fig. 3 shows the binding of 5′-NH3+-d(Tmt)4-T-OH to poly(dA) in a 2:1 fashion at a concentration of 4.0 × 10−5 M/base poly(dA). As expected, the molar ellipticity of poly(dA) is substantially changed on formation of the triplex. Fig. 4 shows the difference spectra of 5′-NH3+-d(Tmt)4-T-OH binding to poly(dA), d(pA)30, and d(pA)25. Clear differences can be seen indicating that different degrees of structural changes have taken place in the three oligomers because of their association. For the complementary poly(dA), the difference spectra show the largest changes of all the samples. As the length of the oligomer drops, difference spectra drop in amplitude, indicating that the degree of association is weakening. For the complementary oligomer d(pA)10 (data not shown), the difference spectra are very flat, indicating little association that reorganizes the bases of the nucleosides.
At 15°C, the second order rate constant, kon, for the triplex formation with short-strand (10–23) oligonucleotides is 17–122 M−1 s−1 (the molarity is expressed in per base) and the first-order rate constant koff is 0.14–2.54 × 10−5 s−1 in the range of μ = 0.03–0.08. These kon values for DNmt5, like DNG5 (kon = 30–200 M−1 s−1, koff = 0.10–3.19 × 10−5 s−1) are, on average, one order of magnitude larger than those found for a DNA triplex having the length of 22 bp at [NaCl] = 0.02–0.30 M. On the other hand, the koff values are of the same order of magnitude as seen with a DNA triplex having the length of 22 bp (7). This comparison is qualitative only because the ionic strength acts in the opposite direction for DNmt relative to DNA, and the composition of the 22-mer DNA oligonucleotide differs from the short-strand DNA oligonucleotides in this study.
As in the case of DNA duplexes and triplexes, the association of the strands decreases as the oligomer strand becomes shorter. The second order rate constant for reaction 5′-NH3+-d(Tmt)4-T-OH with d(pA)x, kon, increases with the increased length of the DNA strand, while the first order rate constant, koff, decreases (Fig. 7 and Table 1). This is not so for the transition from d(pA)20 to d(pA)23. At every ionic strength and temperature, Keq increases from d(pA)10 to d(pA)20 and decreases slightly from d(pA)20 to d(pA)23. This is best explained by the domino effect of DNmt5 binding to d(pA)23 (Scheme S2). The kinetic equations get very complicated if the presence of unoccupied sites is considered [e.g., d(pA)23 + 5 × DNmt5, Scheme S2]. Even in the case of d(pA)10, 15, 20, where we expect to have fully occupied (−) sites by two molecules of 5′-NH3+-d(Tmt)4-T-OH, there can be slides of the strands (Scheme S2) (2). Species other than full associations are neglected (all-or-none model) in the kinetic and thermodynamic studies. Whereas studies of DNG⋅DNA triplexes showed an increase in kon values with an increase in DNA length (5 through 7) (2), increase in the length of triplex from DNmt2⋅d(pA)20 to DNmt2⋅d(pA)23 shows a decrease in kon and increase in koff values (Table 1), illustrating, a little more selectivity in the binding of DNmt to its target. In contrast to DNG⋅DNA triplexes, DNmt complexes with oligomers d(pA)5–8 did not show appreciable hypochromicity on binding. This shows that DNmt binds less strongly than DNG, as previously reported.
As expected, by decreasing the ionic strength kon increases, while koff decreases (Table 1). We find that the sensitivity of kon and koff to μ is similar to that observed for DNG⋅DNA triplexes (2). On the other hand, koff values for DNA⋅DNA triplexes are largely independent of ionic strength (7).
The same trend has been found for the energies of activation, Eon and Eoff. For d(pA)10, with decrease in ionic strength, Eon decreases from −37.73 to −26.87 kcal/mol, whereas Eoff increases from 3.12 to 10.37 kcal/mol (Table 2, Fig. 8). A more negative Eon at high ionic strength and a less positive value of Eoff favors a better association at low μ values. The negative values for Eon (activation energy for kon) are obtained since the rate of triple helical formation (kon) decreases with temperature leading to a positive slope (−Eon/R) of ln(kon) vs. 1/T (Fig. 6). This is similar to the negative activation energies obtained for association of double and triple helical DNA complexes (2, 7). However, an elementary kinetic step cannot have an activation energy less than zero. Therefore, kon (and koff) must represent composites of rate constants for individual steps. As proposed for DNA⋅DNA complexes, the negative activation energies rule out the formation of the first base pair as rate limiting (7, 11). The development of the nucleation-zipping model, as applied previously to triple-helical DNAs, can be used to explain this large negative value of Eon (7, 11). The helix formation begins with two or three bases pairing and unpairing in rapid but unfavorable equilibrium. On formation of the critical intermediate, a helix nucleus is formed, which zips up to form the fully bonded helix more rapidly than it dissociates to single strands. The equilibrium constant K = kon/koff = βsn, where β is the equilibrium constant for nucleation of the triplex (formation of the first base triplet). The chain growth parameter s = kf/kb, where kf and kb are the first order rate constants for the formation and breakage of the base triplet at the end of a triplex segment, and n is the number of base triplets being formed. If ν is the number of base triplets in the nucleus, which is in rapid equilibrium with the separated duplex + third strand, the activation energy Eon equals the sum of one activation energy, Ekf, and ν + 1 reaction enthalpies for base triplet reactions, ΔHβ + νΔHs:
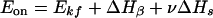 |
5 |
The first term is small and positive but the enthalpies are negative, such that Eon becomes negative with its magnitude increasing with ν, and Eoff (Eoff = Ekf − (n − ν) ΔHs) is largely positive. As a first approximation (Ekf + ΔHβ ≈ 0), if the temperature dependence of kinetic parameters rests on the growing chain factor s, we obtain Eq. 6 (from ΔH° = Eon − Eoff).
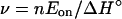 |
6 |
By using Table 2 and n = 5, the following values of ν are obtained:
Figure 8.

Dependence of Eon and Eoff on chain length for denaturation/renaturation of the triplex formed from d(pA)x (4.0 × 10−5 M/base) with 5′-NH3+-d(Tmt)4-T-OH at dT/dt = 0.13 deg/min.
| d(pA)x | d(pA)10 | d(pA)15 | d(pA)20 | d(pA)23 |
| ν (μ = 0.03) | 3.6 | 3.4 | 2.5 | 2.8 |
| ν (μ = 0.08) | 4.6 | 3.8 | 3.3 | 3.6 |
ν must be an integer value, so it increases from 2 to 4 bases [almost to 5 in d(pA)10]. In other words, more than 50% of the DNmt5 strand is required for nucleation with these homooligomers, and yet we are able to form stable triplexes that exceed the association rates of DNA triplexes of 22 bp (ν = 2–4) and longer. The drop in ν from d(pA)20 to d(pA)23 is anticipated because of the incomplete association of DNmt in the domino fashion, as previously explained. The lower value of ν at lower ionic strength again reinforces the role of ionic attractions of oppositely charged backbones in the formation and stability of the helices. We notice from Table 2 that with an increase in ionic strength, Eon values become more negative [for d(pA)20, Eon goes from −18.9 kcal/mol (μ = 0.03) to −33.6 kcal/mol (μ = 0.08)]. This is in stark contrast to previous studies with DNG⋅DNA triplexes, where Eon was found to increase with decreased ionic strength (2). Whereas ΔHs/base pair (Eq. 5) would tend to decrease with an increase in ionic strength for DNG as well as DNmt, the value of ν increases with increased ionic strength. It is the balance between ΔHs and ν that determines the overall value of Eon and further the value of ΔH°. In the case of DNmt2⋅DNA triplexes, the increase in ν compensates for the decrease in ΔHs. In other words, the number of bases required for nucleation in DNmt2⋅DNA triplexes increases with increase in μ and decreases with increase in the size of d(pA)x. On the other hand, for DNG5 binding to d(pA)10, the value of ν (2.8, calculated from ref. 2) remains virtually unchanged in going from an ionic strength of 0.03 to 0.22. The decrease of ν with length of d(pA)x studied also explains why the Eon values become less negative as we go from d(pA)10 to d(pA)20, since the extent of ΔHs/base pair would be expected to remain constant with the change in length of d(pA)x. The difference in DNG and DNmt binding to DNA can be ascribed to the relative independence of ν to μ and d(pA)x length in case of DNG⋅DNA triplexes.
The thermodynamic parameters can be extracted from the kinetic data or experimentally determined by calorimetric methods (2, 8, 9). In most cases, these data obtained by different methods are in good agreement. Eq. 7 shows the relationship between the thermodynamic parameters ΔH° and ΔS° and the equilibrium constant Keq at a given temperature.
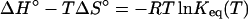 |
7 |
At the melting temperature, Tm, Keq = 2/Mtot and at 288 K, Keq is given by konref/koffref. From Eq. 7 one can calculate the standard molar entropies for the triplex formation and the free energies of formation, ΔG° (Table 2, Fig. 8).
The standard molar enthalpies have larger negative values at higher ionic strength than at low ionic strength in accord with the observation that at lower μ values the value of ν of 5′-NH3+-d(Tmt)4-T-OH binding with d(pA)x is lower. The standard molar enthalpies ΔH°(288) = Eon − Eoff are between −35 and − 50 kcal/(mol base) for short strands (10–23 base pairs, Table 2) as compared with −120 kcal/mol for a 22 base pair DNA triplex (7), and −40 to −60 kcal/(mol base) for DNG5⋅DNA triplexes (2). The thermodynamic parameters for the RNA/DNA hybrid duplexes have been studied and pentameric hybrid duplexes with different mixed base sequences have been shown to have ΔH° of ca. −45 kcal/(mol strand) (12, 13). These large and negative values of ΔH°(288) indicate a better stabilization of the (5′-NH3+-d(Tmt)4-T-OH)2⋅(DNA) complex as compared with the DNA triplexes with at least one order of magnitude on the enthalpy scale even with the entropy compensation. With an increase in temperature, the entropic contribution, ΔS°, of formation of the triplexes gets more negative as the triplex begins to dissociate, leading to an increase in entropy (Table 3). Triplex formation of 5′-NH3+-d(Tmt)4-T-OH with d(pA)x is associated with values of ΔG°(288) ranging between −8 and −11 kcal/mol with large negative values at low ionic strengths (Table 3). The free energies of triplex formation have also been calculated at 25°C and 37°C and show a gradual decrease as the temperature is raised (Table 3, Fig. 9). (For 5′-NH3+-d(Tmt)4-T-OH2⋅d(pA)20 triplex, ΔG° decreases from −10.2 kcal/mol (288 K) to −8.9 kcal/mol (298 K) and further to −7.5 kcal/mol (310 K). This denotes the decreasing stabilization of triplex with increase in temperature, which is expected.
Table 3.
Thermodynamic parameters for the triplexes of d(pA)x with DNmt
| T, °C | μ | d(pA)x, x = | ΔS°,§ cal/mol⋅K | ΔG°,¶ kcal/mol |
|---|---|---|---|---|
| 15 | 0.03 | 10 | −99.015 | −8.7087 |
| 0.03 | 15 | −90.312 | −9.1967 | |
| 0.03 | 20 | −95.097 | −10.248 | |
| 0.03 | 23 | −97.753 | −10.182 | |
| 0.08 | 10 | −114.57 | −7.8467 | |
| 0.08 | 15 | −140.8 | −8.9577 | |
| 0.08 | 15 | −140.8 | −8.9577 | |
| 0.08 | 20 | −142.8 | −9.6102 | |
| 0.08 | 23 | −119.73 | −8.5187 | |
| 25 | 0.03 | 10 | −104.01 | −7.2703 |
| 0.03 | 15 | −94.414 | −8.0145 | |
| 0.03 | 20 | −99.496 | −8.9803 | |
| 0.03 | 23 | −102.23 | −8.8922 | |
| 0.08 | 10 | −118.15 | −6.8128 | |
| 0.08 | 15 | −146.6 | −7.2874 | |
| 0.08 | 20 | −148.71 | −7.9067 | |
| 0.08 | 23 | −122.92 | −7.6005 | |
| 37 | 0.03 | 10 | — | — |
| 0.03 | 15 | −98.995 | −6.6946 | |
| 0.03 | 20 | −104.39 | −7.5688 | |
| 0.03 | 23 | −107.23 | −7.4509 | |
| 0.08 | 10 | — | — | |
| 0.08 | 15 | −153.12 | −5.4099 | |
| 0.08 | 20 | −155.31 | −6.0033 | |
| 0.08 | 23 | −130.44 | −5.4344 |
±1 cal/mol⋅K;
±0.2 kcal/mol.
Figure 9.

Dependence of ΔG° on temperature for the triplex (5′-NH3+-d(Tmt)4-T-OH)2⋅d(pA)20 at μ = 0.03 (⧫), μ = 0.08 (●) and triplex (5′-NH3+-d(Tmt)4-T-OH)2⋅d(pA)15 at μ = 0.03 (▴), μ = 0.08 (■).
CONCLUSIONS
Replacement of the phosphodiester linkages of DNA with methylthiourea linkages provides a polycation deoxyribonucleic methylthiourea (DNmt). The polycation 5′-NH3+-d(Tmt)4-T-OH binds to homooligomers d(pA)x with high affinity and with base pair specificity to provide triple-stranded helices. The electrostatic attraction between polycation 5′-NH3+-d(Tmt)4-T-OH and polyanion d(pA)x stabilizes the triple helical hybrid structures. At a concentration of 4.0 × 10−5 M/(base A) and at a stoichiometry of 2:1 T/A, one observes triplexes with d(pA)15 and d(pA)20 possessing Tm values of 52.74°C and 64.22°C, respectively.
The triplex forms and dissociates to a duplex and a third monomer strand with rate constants kon and koff. At 15°C and in the range of ionic strengths of 0.03–0.12, the second order rate constant (kon) for the triplex formation with short-strand oligonucleotides [d(pA)x, where x = 10, 15, 20, and 23] is 17–22 M−1 s−1 (the molarity is expressed in per bases) and the first order rate constant (koff) is 0.14–2 × 10−5 s−1. The value of kon for reaction of 5′-NH3+-d(Tmt)4-T-OH with d(pA)x increases with increase in length of DNA strand while the rate constant koff decreases. Similar trends have been found for the activation parameters, Eon and Eoff. For d(pA)10–23, there is a decrease in Eon from −38.0 to −18.9 kcal/mol with decrease in ionic strength, whereas Eoff increases from 3.1 to 18.7 kcal/mol. A less negative Eon at low ionic strength and a more positive value of Eoff favors association, since ν decreases with increase in μ. The difference in DNG and DNmt binding to DNA is ascribed to the dependence of ν (number of base triplets in the nucleus of the forming helix) to μ (ionic strength) and length of oligomer d(pA)x in case of DNmt⋅DNA triplexes. The standard molar enthalpies, ΔH° (288) = Eon − Eoff are between −30 and −50 kcal/(mol base) and the standard free energies ΔG° (288) are between −8 and −11 kcal/(mol base) for short strands (10–23 base pairs). In comparison, the standard free energies ΔG° (288) reported for DNG binding to short-strand DNA (5–15 base pairs) is between −9 and −13 kcal/mol. The entropies ΔS° and free energies ΔG° of triplex formation show a gradual decrease as the temperature is raised. The ΔH° (288) and ΔG° (288) values indicate a better stabilization of the [5′-NH3+-d(Tmt)4-T-OH]2⋅d(pA)x complex as compared with triplexes of DNA, but less than that of DNG triplexes to DNA. A better understanding of physical parameters responsible for the equilibration of these positively charged triple helices (DNmt2⋅DNA and DNG2⋅DNA) is thus obtained. DNmt, a positively charged analogue of DNG, is unique in its strong binding strength (kon, koff comparable to DNG2⋅DNA triple helix) yet resembling DNA2⋅DNA triple helices in their nucleation behavior. Further studies of DNmt2⋅DNA triple helices involving complementary mismatch sequences coupled with synthetic efforts to design longer DNmt sequences should help in the development of a novel class of putative antisense/antigene agents.
Acknowledgments
We express our gratitude to Helgi Adalsteinsson (this lab) and Dr. Andrei Blasko (Roche BioScience) for their help and suggestions in improving the manuscript. This study was supported by the National Institutes of Health (Grant BK09171).
References
- 1.Dempcy R O, Browne K, Bruice T C. J Am Chem Soc. 1995;117:6140–6141. [Google Scholar]
- 2.Blasko A, Dempcy R O, Minyat E E, Bruice T C. J Am Chem Soc. 1996;118:7892–7899. [Google Scholar]
- 3.Blasko A, Dempcy R O, Minyat E E, Bruice T C. Biochemistry. 1997;36:7821–7831. doi: 10.1021/bi970064v. [DOI] [PubMed] [Google Scholar]
- 4.Arya D P, Bruice T C. J Am Chem Soc. 1998;120:12419–12427. [Google Scholar]
- 5.Arya D P, Bruice T C. J Am Chem Soc. 1998;120:6619–6620. [Google Scholar]
- 6.Gray D M, Hung S H, Johnson K H. Methods in Enzymology. Vol. 246. New York: Academic; 1995. pp. 19–48. [DOI] [PubMed] [Google Scholar]
- 7.Rougee M, Faucon B, Mergny J L, Barcelo F, Giovannageli C, Garestier T, Helene C. Biochemistry. 1992;31:9269–9278. doi: 10.1021/bi00153a021. [DOI] [PubMed] [Google Scholar]
- 8.Breslauer K. In: Methods for Obtaining Thermodynamic Data on Oligonucleotide Transitions. Hinz H-J, editor. Berlin: Springer; 1986. pp. 403–427. [Google Scholar]
- 9.Marky L A, Breslauer K J. Bioploymers. 1987;26:1607–1621. doi: 10.1002/bip.360260912. [DOI] [PubMed] [Google Scholar]
- 10.Maher L J, I, Dervan P B, Wold B J. Biochemistry. 1990;29:8820–8826. doi: 10.1021/bi00489a045. [DOI] [PubMed] [Google Scholar]
- 11.Cantor C R, Schimmel P R. Biophysical Chemistry. San Francisco: Freeman; 1980. [Google Scholar]
- 12.Sugimoto, N., Shintani, Y. & Sasaki, M. (1991) Chem. Lett. 11287–1290.
- 13.Sugimoto N, Shintani Y, Tanaka A, Sasaki M. Bull Chem Soc Jpn. 1992;65:535–539. [Google Scholar]