

Abstract
Brain atrophy, neurologic and psychiatric (NP) manifestations are common complications in the systemic autoimmune disease, lupus erythematosus (SLE). Here we show that the cerebrospinal fluid (CSF) from autoimmune MRL-lpr mice and a deceased NP-SLE patient reduce the viability of brain cells which proliferate in vitro. This detrimental effect was accompanied by periventricular neurodegeneration in the brains of autoimmune mice and profound in vivo neurotoxicity when their CSF was administered to the CNS of a rat. Multiple ionic responses with microfluorometry and protein peaks on electropherograms suggest more than one mechanism of cellular demise. Similar to the CSF from diseased MRL-lpr mice, the CSF from a deceased SLE patient with a history of psychosis, memory impairment, and seizures, reduced viability of the C17.2 neural stem cell line. Proposed mechanisms of cytotoxicity involve binding of intrathecally synthesized IgG autoantibodies to target(s) common to different mammalian species and neuronal populations. More importantly, these results indicate that the viability of proliferative neural cells can be compromised in systemic autoimmune disease. Antibody-mediated lesions of germinal layers may impair the regenerative capacity of the brain in NP-SLE and possibly, brain development and function in some forms of CNS disorders in which autoimmune phenomena have been documented.
Abbreviations: ACA, anti-cardiolipin antibodiesx; Abbreviations, anti-nuclear antibodies; CSF, cerebrospinal fluid; FJB, Fluoro Jade B stain; NP-SLE, neuropsychiatric systemic lupus erythematosus; PBS, phosphate buffered saline
1. Introduction
The overall prevalence of the autoimmune disease systemic lupus erythematosus (SLE) is estimated to range from 1:245 to 1:1000 individuals, and according to recent epidemiological studies, the number of affected people in North America alone may exceed 1 million (Lahita, 1995). Neurologic and psychiatric (NP) manifestations of unknown etiology are common in SLE and have been proposed to represent a more severe form of the disease, often denoting a graver prognosis (Bombardier et al., 1992; Rubin et al., 1985). Contemporary imaging techniques indicate that profound metabolic alterations and neuronal loss accompany neuropsychiatric lupus, or NP-SLE (Colamussi et al., 1995; Sibbitt et al., 1994). Periventricular lesions, cerebral atrophy, and ventricular enlargement of unknown etiology occur in up to 50% of patients (Baum et al., 1993; Bosma et al., 2000).
Similar to SLE in humans, an inbred strain of MRL/MpJ-Faslpr (MRL-lpr) mice develops an accelerated SLE-like condition (Theofilopoulos, 1992) accompanied by various neurological dysfunctions (Hess et al., 1993; Vogelweid et al., 1994), an anxious/depressive-like behavioral state (Sakic et al., 1994), ventricular enlargement (Denenberg et al., 1992), neuronal atrophy, and retarded brain growth (Sakic et al., 2000a; Sakic et al., 1998). Pathological abnormalities are also seen in the choroid plexus, a structure that synthesizes most of the cerebrospinal fluid (CSF) volume (Duprez et al., 2001; Schwartz and Roberts, 1983). Widespread damage of the blood–brain barrier in MRL-lpr mice is accompanied by lymphocyte and monocyte infiltration into the choroid plexus (Alexander et al., 1983; Farrell et al., 1997; Hess et al., 1993), in some cases as early as 8 weeks of age (Vogelweid et al., 1991). Areas around the third and lateral ventricles show enhanced neurodegeneration, as revealed by staining with Fluoro Jade B (Ballok et al., 2003), excessive DNA fragmentation (Sakic et al., 2000b) and expression of cell adhesion molecules (Zameer and Hoffman, 2003). More interestingly, the CSF from diseased MRL-lpr mice reduces the viability of pyramidal neurons in a primary neuron–astrocyte co-culture (Maric et al., 2001). Taken together, periventricular distribution of brain lesions led to the hypothesis that the viability of cells in the subependymal layer of the brain is compromised due to sustained interactions with soluble cytotoxic factors in the CSF. The pathogenic cascade may include drainage of circulating immune factors (e.g. activated cytotoxic lymphocytes and/or autoantibodies) into the CSF and their diffusion into neighboring interstitial tissue, with subsequent detrimental effects on cell function. Given that neural stem cells and progenitors in the periventricular zone constitute the largest pool of proliferative brain cells (Morshead and van der Kooy, 2001), one may expect profound consequences on brain development, repair capacity, and behavior if this cell population is affected by the autoimmune process, either during embryogenesis or during ontogeny.
As a first step in testing the above mechanism, we examine the viability and morphology of murine-derived, multipotent neural stem cells (C17.2) and neural stem cell/ progenitor preparations (neurospheres) after incubation with CSF from autoimmune animals. Given that the mammalian retina is a nucleus of the CNS and is composed of clearly defined neuronal layers, we also examined whether the CSF from MRL-lpr mice has neurotoxic effects in vivo by using the posterior rat eye chamber as a target tissue. This technically convenient approach also allowed us to examine whether CSF is indeed cytotoxic across species, as observed with the CSF from an NP-SLE patient administered to the mouse hippocampus (DeGiorgio et al., 2001), and whether previously reported in vitro cytotoxicity of CSF from MRL-lpr mice (Maric et al., 2001) can be observed in vivo. In addition, the death of one of our NP-SLE patients provided us with a unique opportunity to compare the cytotoxic effects of CSF obtained from animal and human forms of SLE using an in vitro system.
2. Materials and methods
2.1. Animals
MRL/MpJ-Faslpr (MRL-lpr) mice develop an accelerated form of an SLE-like disease, while age-matched congenic MRL/MpJ (MRL+/+) mice show only mild manifestations of systemic autoimmunity and inflammation (Theofilopoulos, 1992). The MRL-lpr males and congenic, age-matched MRL+/+ controls were obtained from The Jackson Laboratory (Bar Harbour, ME) and a local breeding colony. Healthy (non-autoimmune) CD1 males were purchased from Charles River Canada. Mice were housed in groups of four per cage and maintained under standard laboratory conditions. Sampling was performed on old (~4 months of age) and young animals (~5 weeks of age). The experimental protocols were approved by a local animal care committee and carried out in accordance with the rules and regulations of the Canadian Council of Animal Care.
2.2. Sample collection
Mice were anesthetized with an i.p. injection of Somnotol (65 mg/kg, 0.15–0.2 ml). The thoracic cavity was exposed to cut the inferior vena cava and quickly aspirate ~1 ml of blood with blood collecting tubes (Fisher Scientific, Nepean, ON). The animals were perfused intracardially with 40 ml of phosphate buffered saline (PBS) and processed for CSF collection, as described previously (Maric et al., 2001). Upon dissection of neck muscles a custom-made glass micropipette (Drummond Scientific, Broomall, PA; rinsed repeatedly with 70% EtOH) was inserted into the cisterna magna and 15–20 μl of aspirated CSF was transferred into autoclaved 50-μl plastic vials. They were centrifuged at 3000 r.p.m. over 3–5 s and examined under a brightly lit white background. The presence of red blood cell precipitate was used as a marker of contamination and a criterion for disposal (~15%–20% of the samples).
Several classes of leukocytes infiltrate the choroid plexus and meninges of MRL-lpr mice, including T- and B-cells (Zameer and Hoffman, 2004). Given that these cells are occasionally seen in the ventricle lumen (Sakic et al., 2000b), they were estimated in CSF samples from five MRL-lpr mice by immunostaining for T-cells (CD3-PE, Cat No. 550353 and CD8a-PE, Cat No. 553033, BD Biosciences Pharmingen, San Diego, CA) and B-cells (CD45R/ B220-FITC, Cat No. 553087), cytospinning (7000 r.p.m. for ~15 s), and counting on a hemocytometer. More B-cells than T-cells were seen, but they were rarely observed under regular microscopy (less than 10 in 20 μl CSF samples; the highest number observed in one sample was 5 cells/9 mm2).
Upon extraction, brains were extracted and immersed into 4% paraformaldehyde (PFA). Forty-eight hours later they were transferred into 30% sucrose in PBS. The weights of the fresh brain and spleen were taken on a digital scale (Mettler Toledo AB54-S, VWR Scientific of Canada).
2.3. Culturing of the C17.2 stem cells
The C17.2 stem cell line is originally derived from developing mouse cerebellum and immortalized with a recombinant retrovirus containing the v-myc oncogene. It is capable of differentiating in vivo into neurons and glial cells (Snyder et al., 1992). The C17.2 cells can be directed to differentiate into dopamine (Wagner et al., 1999), gamma aminobutyric acid and acetylcholine phenotypes (Doering and Snyder, 2000). In the present study, cells were grown on uncoated 10-cm tissue culture dishes (Corning, Corning, NY) in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS), 5% horse serum and 2 mM glutamine (GIBCO, Burlington, ON). Cells were passaged weekly (to a maximum of six passages), removed by trituration and their viability determined by Trypan blue exclusion. Approximately 2×104 cells were transferred to each well of Falcon 24-well plates and grown for 48 h on 18-mm glass cover slips (Bellco Glass, Vineland, NJ) coated with 0.1% poly-L-Ornithine (Sigma, St. Louis, MO) in DMEM. All tissue cultures were maintained in a standard, humidified (5% CO2) air incubator at 37 °C.
2.4. Neurosphere formation
To examine whether the CSF is toxic to primary stem and progenitor cells, ependymal/subependymal layers of the lateral ventricles from 4-month old MRL-lpr and CD1 mice were isolated by dissection under a microscope (Chiasson et al., 1999). The isolated tissue was manually dissociated by trituration with a Pasteur pipette and transferred into 1 ml of artificial cerebrospinal fluid supplemented with 0.13 mg kynurenic acid (Sigma, St. Louis, MO. 4-Hydroxyquinoline-2-carboxylic acid), 0.66 mg type 1-S hyaluronidase and 1.3 mg trypsin (Sigma) for 90 min in a shaking water bath set to 35 °C. The sample was triturated 30 times every 15 min. The single cell suspension was re-suspended in 1.66 μl of the trypsin inhibitor Ovomucoid (Sigma) and 4 ml of DMEM/F12 (GIBCO) in artificial cerebrospinal fluid, which consisted of 6.2% 2 M NaCl, 0.5% 1 M KCl, 0.32% 1 M MgCl2, 16.8% 155 mM NaHCO3, 1.0% 1 M glucose, 0.09% 108 mM CaCl2 and 75.1% dH2O. Cells were cultured and plated (5 cells/μl under clonal conditions) in uncoated 24, 48 or 96-well plates with serum-free media consisting of: 94.3 mL DMEM/F12 (GIBCO), 2 ml 30% glucose, 0.5 ml 1 M HEPES buffer, 0.1 mL progesterone, 1 ml putrescine (Sigma), 0.1 ml insulin– transferrin –sodium selenite (Roche, Laval, Quebec), 2 ml B27 Growth supplement (GIBCO), 20 μl epidermal growth factor, 20 μl basic fibroblast growth factor and 7.32 μl heparin (Sigma) (Weiss et al., 1996). The 24, 48 or 96-well plates contained a total of 500, 200, and 100 μl of suspension, respectively. The neurospheres were tested for the expression of nestin, an intermediate filament expressed in uncommitted neural stem/progenitor cells (Lendahl et al., 1990) with a mouse anti-rat nestin antibody (1:100, PharMingen, San Diego, CA). To assess the differentiation potential of the spheres, 10–15 spheres were transferred to individual wells of the Falcon 24-well plate described above. Two drops of media containing 10% FBS in DMEM was added to each well (Brannen and Sugaya, 2000), and cells were allowed to differentiate for three days before fixation in 4% PFA for 5 min. They were tested for immunoreactivity towards monoclonal antibodies specific for β-tubulin (1 : 100, Sigma), glial fibrillary acidic protein, GFAP (1 : 100, DAKO Diagnostics, Mississauga, ON), and anti-O4 (1 : 100, Chemicon, Temecula, CA) to mark neurons, astrocytes, and oligodendroglia, respectively. The total number of neurospheres was determined by inverted phase contrast microscopy at magnifications from ×340–×1300.
2.5. Cytotoxicity assay
The cytotoxicity assay was performed on the C17.2 cell culture and in several cases, on the neurospheres. In general, the volume of the culturing medium was reduced from 0.5 to 0.25 ml per well, and 10 μl of a sample (CSF or serum) was added within 2 min after collection. A concentration-dependent curve was obtained by diluting the CSF with the culturing medium (1:2, 1:10, 1:100, 1:500 and 1:1000). To examine whether infiltrated leukocytes are required for the cytotoxicity, twenty-five CSF samples were spun at 3000 r.p.m. for~10 s and supernatant was collected for the cytotoxicity assay with C17.2 cells. To examine whether fresh samples can be used exclusively, half the volume of four CSF samples were stored at 37 °C for 24 h and this was compared to the effects of freshly extracted CSF. An experimenter unaware of the sample origin assessed the cytotoxicity of animal and human samples by collecting the medium 24 h later and calculating a relative number of detached Trypan-stained cells. The number was expressed per 1 ml after manual counts on a hemocytometer (total number of cells on four fields×1.25×104/ 4). Using phase contrast microscopy, neurospheres were counted in the 24-well plate immediately before and 24 h after the addition of fresh 10 μl CSF sample. When bacterial contamination was observed in any of the wells (~15% cases), the sample was discarded.
2.6. Neutralization of pro-inflammatory cytokines
Synthesis of the neuroactive cytokines interleukin-1 (IL-1), tumor necrosis factor-alpha (TNF-alpha), interferon gamma (IFN-gamma), and IL-6 are associated with advanced autoimmunity and CNS involvement in lupus patients (Tsai et al., 1994; Hirohata and Miyamoto, 1990) and MRL-lpr mice (Fitzpatrick et al., 1996; Tsai et al., 1995; Sakic et al., 1997; Tomita et al., 2001). To examine whether these cytokines contribute to toxicity, CSF samples from diseased MRL-lpr mice were pre-incubated for the purpose of neutralization with affinity purified rabbit anti-mouse polyclonal antibodies (pAb) to IL-1, TNF-alpha, and IL-6.
Half of each CSF sample was used to test for toxicity, while the second half was incubated for 1 h at 37 °C with 5 μl of reconstituted pAb (dose ~0.2 μg/ml) for the purpose of neutralization, as suggested by the manufacturer (Cedar Lane Laboratories Ltd., Hornby, ON). Following the pre-incubation period, the CSF was centrifuged briefly, aliquoted into a 24-well plate seeded with C17.2 cells and incubated for 24 h at 37 °C. This procedure was used to assess the effects of neutralization of each pAb alone, as well as the effect of a ‘‘cocktail’’ of pAbs on the survival of stem cells. Controls used were non-toxic CSF samples incubated with pAbs.
2.7. Autoantibody levels
To confirm serological differences in immune status, antinuclear (ANA) and anti-cardiolipin autoantibodies (ACA) were assessed in serum, as previously described (Sakic et al., 2000a; Hess et al., 1993). In addition, these markers were presently measured in 5 μl of CSF due to evidence that the blood–brain barrier is permeable to circulating immunoglobulins in diseased MRL-lpr mice (Vogelweid et al., 1991) and the possibility that ACA play an important role in the pathogenesis of NP-SLE (Hanly et al., 1999; Schwartz et al., 1998; Martinez-Cordero et al., 1997). Blood samples were left to coagulate in 1.5-ml plastic vials and centrifuged for 10 min at 3000 r.p.m. Serum was then separated from the clot, aliquoted, and stored at −20 °C until further analysis. Antinuclear antibody (ANA) concentration was measured using a sandwich Anti-Nuclear Antibody ELISA kit (Cat. #5200), according to the manufacturer’s instructions (Alpha Diagnostic International, San Antonio, TX) and the protocol previously described (Sakic et al., 2000a). In brief, serum samples were diluted 1:100 in diluent included in the kit, and applied to both experimental and control wells to assess the specificity of binding. The optical density of each well was determined using a microplate ELISA reader set to 450 nm. Similarly, a sandwich Anti-Cardiolipin Antibody ELISA kit (Cat. #5500) was used for semi-quantitative analysis of ACA levels according to the manufacturer’s instructions (Alpha Diagnostic International, San Antonio, TX).
2.8. Fluoro Jade B (FJB) staining
To examine whether CSF toxicity is associated with neuronal degeneration in situ (Schmued and Hopkins, 2000), brain sections were stained with the FJB dye (Histo-Chem Inc., Jefferson, AR), using the previously published protocol (Ballok et al., 2004a). Four brains from mice that largely differed in the extent of CSF toxicity were stored in 30% sucrose for at least 24 h before a frozen coronal section (cut at Bregma ~−1.0 mm) was processed (1 section/brain). Results were assessed with an epifluorescent microscope with blue (450–490 nm) excitation light (Diastar Fluoresence Microscope, Reichert Scientific, Buffalo, NY). The density of FJB-positive cells was quantified with NIH Image analysis software (Scion, Frederick, MD).
2.9. Intra-vitreous administration of CSF
The detailed protocol for intra-vitreous administration has been described elsewhere (van Adel et al., 2003). In brief, an experimenter blind to the origin of samples was given twenty vials (each containing 5 μl of CSF from either MRL-lpr or MRL+/+ mice) to inject into the left or right vitreous of ten adult Sprague–Dawley rats. The injection was made ~3 mm from the optic nerve exit to avoid the ciliary apparatus at the corneo–scleral junction (care was also taken not to touch the posterior capsule of the lens). The needle (WPI titanium) was held in place with a micromanipulator and the sclera was punctured with a 30-ga needle, which was further lowered and CSF was injected over 1 min. During the injection procedure, the cornea was protected with artificial tears (LacriLube) and the hole in the sclera was subsequently sealed with Vetbond. Five rats were euthanized either 2 or 4 days later and eyes were extracted for pathological analysis. Eyes were fixed by immersion in 4% phosphate buffered formaldehyde, and the cornea and lens removed. Gross changes in the eyes were documented by macroscopic photography and assessed by another experimenter blind to the origin of samples. Microscopic changes in the retinal layers were determined by staining the cellular layers with Yo-Pro 1. This is a DNA/Nuclear stain useful in labeling cellular layers, measuring the retinal thicknesses, and examining retinal flatmounts by confocal microscopy. Retinas were incubated in 250 μl of 10 μM Yo-Pro 1 (Molecular Probes, Eugene, OR) overnight and then washed 2 times in PBS for 20 min before whole mounting. The thickness of the retinal layers was determined from reconstructions in the orthogonal plane and measured using Zeiss LSM-510 software.
2.10. Microfluorometry
The calcium ions (Ca+ 2) have a principal role in cell death mechanisms (Demaurex and Distelhorst, 2003) including influx or release from internal stores (Orrenius et al., 2003). We aimed to assess this with Fura-2, but the cells from C17.2 culture loaded poorly with this indicator. Conversely, a pilot study with calcein AM (a useful indicator of cell viability and less specific for Ca+ 2) revealed good retention and fluorescence. This fluorochrome is retained better by viable cells, compared with other common indicators, and does not interfere with immunological processes (e.g. leukocyte chemotaxis or lymphocyte-target cell conjugation), and has minimal effects on cell function (De Clerck et al., 1994). Therefore, the calcein-release assay and fluorescence microfluorometry (Mwanjewe and Grover, 2004) were used to verify cytotoxicity and screen for qualitative changes in ionic metabolism. In brief, C17.2 cells were cultured in 35-mm petri dishes for 24 h, the center bottom having been removed and replaced with glass coverslips. They were then loaded for 15 min with a final concentration of 2 μM calcein AM (Molecular Probes, Eugene, OR), washed three times with physiological saline solution (PSS) and 1 ml of PSS was added. Fluorescence intensity in a field of 8–12 cells was measured by a Zeiss microscope (Axiovert S100) attached to an M-series dual-wavelength excitation system (Photon Technology International, Lawrenceville, NJ). The excitation wavelength used was 488 nm and the emitted fluorescent light was measured at 530 nm. To obtain basal levels, the fluorescence intensity was measured for 3 min prior to the addition of a 5 μl CSF sample. After the addition of CSF, fluorescence intensity was monitored for an additional 3 min. The signals were processed using ImageMaster software (Photon Technology International, Lawrenceville, NJ) and dynamic changes in fluorescence intensity within single cells were determined by defining regions of interest within a single cell, as per software instructions. Importance of the NR2 system was presently tested by blocking the NMDA receptor with MK801 (100 μM, Sigma, Oakville, ON). Ryanodine (100 μM, Sigma, Oakville, ON) and the IP3 receptor blocker 2-Aminoethoxydiphenylborate, APB (100 μM, Calbiochem, San Diego, CA) were used in an attempt to block intracellular Ca+ 2 release (Pal et al., 2001; Bouron et al., 2004; Fernandez et al., 2003).
2.11. Activity of ionic channels
Autoantibodies to ion channels have been proposed to play a role in the pathology of several neurological and psychiatric disorders (Vincent et al., 1999, 2003). Using the patch–clamp technique, we examined whether the CSF from MRL-lpr mice were capable of altering ion channel function in C17.2 cells over a period of 5 min. Dishes with C17.2 cells were transferred to a recording chamber (200 μl) mounted on the stage of an Axiovert S100 microscope (Zeiss, Toronto, ON). Whole-cell patch–clamp recordings were made using patch electrodes of resistance 4–7 MΩ. Patch electrodes were fabricated from 1.5/0.75 mm o.d./i.d. borosilicate glass (World Precision Instruments, Sarasota, FL) using a P-97 Brown–Flaming horizontal electrode puller (Sutter Instrument Company, Novato, CA). To record K+ currents, electrodes were filled with a solution containing 5 mM NaCl, 135 mM KCl, 10 mM HEPES, 11 mM EGTA, 1 mM CaCl2, and 2 mM Na2ATP (pH 7.2 with KOH). Cells were perfused with a solution containing 135 mM NaCl, 5 mM KCl, 1.2 mM MgCl2, 5 mM HEPES, 2.5 mM CaCl2, and 10 mM D-glucose (pH 7.4 with NaOH). To record Ca2+ currents, cells were perfused with a solution composed of 95 mM NaCl, 5 mM CsCl, 0.6 mM MgC12, 5 or 10 mM BaCl2, 5 mM HEPES, 10 mM D-glucose, 20 mM TEA-Cl (21–24 °C, pH adjusted to 7.4 with NaOH) and patch electrodes were filled with a solution containing 120 mM CsCl, 20 mM TEA-Cl, 2 mM MgC12, 10 mM EGTA, 10 mM HEPES, 2 mM ATP (pH adjusted to 7.2 with CsOH). Experiments were performed at 21–24 °C. Cells were voltage clamped at −60 mV (−80 mV for Ca2+ currents), and currents evoked by step depolarizations to the required test potential for 100 ms at a frequency of 0.1 Hz. Traces were filtered at 5 kHz, digitized at 10 kHz and stored on computer for later analysis. Capacitative transients were minimized by analogue means. Analyses and voltage protocols were performed using an EPC 9 amplifier (HEKA, Mahone Bay, NS) with an integrated AD/DA converter, ITC-16 interface, and Pulse software (HEKA). Steady-state outward and inward currents were measured as the average current between 90 and 99 ms of the voltage step. Data were analyzed using Pulsefit software (HEKA). Six cells were exposed to 10 μl of individual CSF samples for the recording of K+ currents, in a volume of 1 ml of extracellular perfusate for a period of 3 min. Recordings were made prior to and following the addition of CSF and the subsequent current amplitudes were compared. In these studies, the experimenter was blinded to the source of the CSF.
2.12. Imaged capillary isoelectric focusing (cIEF)
To assess the pI values of proteins abundant in murine and human CSF, we used the one-step cIEF Convergent Bioscience (Toronto, ON) iCE280 instrument equipped with fused-silica focusing capillary (Janini et al., 2002). The capillary was 5 cm long (ID =100 μm, OD=200 μm) and the inner surface was coated with a fluorocarbon J and W Scientific FC coating. The capillary in a cartridge was separated from the catholyte (100 mM NaOH) and anolyte (80 mM H3PO4) reservoirs by two pieces of hollow fiber membrane. A 280 nm light beam from a deuterium lamp was focused on the capillary by a bundle of optical fibers and a set of lenses. After the introduction of the sample (diluted 5 times in a running buffer) and carrier ampholyte mixture into the column through an HPLC sample introduction valve with a 2.5 μL sample loop, a 3 kV dc voltage (0.6 kV/cm) was applied through electrodes immersed in the electrolyte reservoirs. The focusing (~6 min) was monitored by taking the whole capillary absorption image by a charge-coupled device camera every 30 s. All reagents used were of analytical grade. Methyl-cellulose (Sigma, St. Lois, 0.35%, 1500 cP for 2% aqueous solution at 20 °C), and Pharmalyte carrier ampholytes (8%, pH 3–10) were purchased from Sigma (St. Louis, MO). The pI markers (~5 μg/ml, pI =5.3 and pI =8.6) were from BioRad (Hercules, CA). The UV absorption image and marker peaks were aligned with EZChrom software (EZChrom Chromatography Data System 6.7, Scientific Software Inc., San Ramon, CA). Murine samples (pooled from 10 animals/ group) were run in triplicate and human samples (from the deceased NP-SLE patient and from a deceased drug user) were run in quadruplicate.
2.13. CSF fractionation
To examine the possibility that IgG autoantibodies mediate the cytotoxic effect (DeGiorgio et al., 2001), a simple fractionation protocol was used. Protein A Sepharose CL-4B (Pharmacia Biotech, Cat.# 17-0780-01) beads were swelled in PBS overnight at 4 °C. They were then washed 3 times with 100 volumes/wash of PBS, which was subsequently decanted. One hundred microliters of Protein A Sepharose beads were added two times to 200 μl of CSF pooled from lupusprone or control mice. After mixing for 2 h at RT, the mixture was centrifuged for 30 s at 14,000 r.p.m. The beads from two depletions were merged and washed 10 times with 5 volumes of PBS/wash at RT. The beads were incubated twice with 200 μl of 0.1 M glycin pH 2.5 for 2 min at RT. Two eluted solutions were combined and immediately neutralized to pH 6.5 by adding 1 M NaOH. IgG-rich (eluted from Protein A Sepharose beads) and IgG-depleted CSF fractions were subjected to the cytotoxicity assay (described above) and electrophoresis (described below).
2.14. Two-dimensional gel electrophoresis
CSF samples were initially centrifuged at 2000 g for 10 min to remove cell and/or cellular debris. Subsequently, they were mixed with 600 μl of ice-cold acetone and centrifuged at 10,000 g for 10 min. The resulting pellet was dissolved in 10 μl of buffer A [10% SDS (w/v), 2.3 DTT (w/ v)] and the sample heated at 95 °C for 10 min. The sample was then diluted in 60 μl of solubilization buffer (7 M urea, 2 M thiourea, 0.8% pharmalyte, pH 3–10, 2% CHAPS and 1% DTT). IEF was conducted using 7 cm immobilized pH gradient (IPG) strips with a non-linear pH gradient of 3–10. Solubilized samples were combined with rehydration buffer [7 M urea, 2M thiourea either 2% CHAPS, 15 mM DTT, 0.2% (w/v) Pharmalyte 3–10, and trace amounts of Bromophenol Blue] to a final volume of 125 μl. The resulting sample preparations were loaded into the IPG strip holder and subsequent rehydration of the IPG strips proceeded for 8 h at 20 °C followed by 1 h at 30 V. The protocol for IEF was as follows: 150 V for 1 h min, 300 V for 1 h, 1000 V for 1 h, 2500 V for 1 h, from 2500 V to 8000 V for 1 h, 8000 V for 3 h for a total 34,000 V h. Following IEF, strips were incubated for 15 min at room temperature with continuous shaking in equilibration buffer [6M urea, 30% (w/v) glycerol and 2% (w/v) SDS in 0.05 M Tris HCl pH 8.8 and a trace of bromophenol].
IPG mini-strips, molecular weight markers, glycerol, Pharmalytes 3–10, as well as isoelectric focusing unit (IPGphor), and SDS-PAGE apparata (mini-gel casting unit, cassettes, and electrophoresis chamber) were purchased from Amersham Pharmacia (Piscataway, NJ). Acrylamide solutions, 1.5 M Tris (pH 8.8), SDS, Urea, DTT, CHAPS, and Sypro Ruby fluorescent stain were from Genomics Solutions (Chelmsford, MA). All other chemicals were of analytical grade.
2.15. Human tissues
A 58-year old female SLE patient with a 15-year history of NP-SLE characterized by memory impairment, psychosis, and complex seizures presented in status epilepticus (managed with phenytoin and diazepam) one day before her death. She remained comatose despite aggressive immuno-suppression, including high dose methylprednisolone, plasmapheresis, and cyclophosphamide; terminally, she developed disseminated intravascular coagulation. Brain tissue was extracted and CSF collected within 4 h of death. Another CSF sample was obtained from a 27-year old woman with American College of Rheumatology-confirmed SLE (malar rash, photosensitivity, and nephritis), but without any history of prior CNS involvement. Three days prior to CSF sampling, however, she complained of headache, neck pain and neck swelling, accompanied by disorientation and confusion. On admission, she was found to have a blood pressure of 240/160. A brain CT revealed two hypodense lesions, consistent with hypertensive encephalopathy or vasculitis. Lumbar puncture revealed elevated CSF protein. Control CSF samples were also obtained from a 33-year old male suffering from post-traumatic chronic headaches and an 87-year old woman with suspected meningitis. Given that novel, potentially neurodestructive proteins can be detected in the CSF 6 h after brain death (Lescuyer et al., 2004), we addressed the possibility that the CSF from our NP-SLE patient became neurotoxic within 3–4 h post mortem. For this purpose we included the CSF sample from a 27-year old drug addict who died from a drug overdose and was subjected for autopsy at a time comparable to the NP-SLE patient. In all instances patients or their family members signed consent forms for protocols approved by a local Human Ethics Committee.
The brain of the NP-SLE patient was examined macroscopically after fixation in 10% buffered formalin. Coronal, serial sections were paraffin wax embedded and stained with luxol fast blue/haematoxylin and eosin. Other special stains used were Beilshowsky silver and congo red. Using standard immunohistochemical methods, sections were stained for tau protein, alpha synuclein, beta-amyloid, ubiquitin, leucocyte common antigen (LCA), T-cell markers (CD3, CD4, CD8), B-cell marker (CD20), and macrophage marker (CD68). These sections were examined by light microscopy.
2.16. Statistical analysis
Data was analyzed by an ANOVA with group as the between-group factors and Bonferroni’s test in the post hoc analysis. Significance level was set at p <0.05 and all computations were performed using the SPSS 11.0 statistical package. Non-linear correlations were graphed and computed with the Fig P 2.7 (Biosoft, Cambridge, UK) statistical page as described below. Graphs show means SEM, with *p ≤0.05; **p ~0.01; and ***p ~0.001 between the groups. Sample size of each group is shown as n.
3. Results
3.1. CSF samples from autoimmune mice and NP-SLE patient are toxic to proliferating brain cells
After a 48-h pre-incubation period, the culture obtained from the C17.2 cell line was a mixture of spherical cells and cells with neuronal-like processes (Fig. 1A). The first cohort (n =6 samples/strain) of CSF samples from aged MRL-lpr mice, MRL+/+ and CD1 males was screened for cytotox-icity by gross observation under the microscope. The addition of CSF samples from autoimmune MRL-lpr mice generally induced detachment of the cells from the culture surface, a change in cell morphology (Fig. 1B), and significant lysis ~24-h later (Fig. 1C). The second cohort of MRL-lpr mice was used to quantify the effect. The toxicity was present in 27 out of 31 CSF samples (~87%), which induced ~70% cell loss. Conversely, CSF samples from asymptomatic (4-week old) MRL-lpr mice, and CSF and serum from non-autoimmune CD1 mice did not significantly affect cell survival in the colony (Fig. 2A), suggesting that toxicity is associated with the progress of autoimmunity. Interestingly, serum from diseased MRL-lpr mice did not appear to be cytotoxic, suggesting that CSF toxicity is due to intrathecally activated or synthesized factors. When allowed to grow, spherical accumulations of self-dividing cells (neurospheres) were able to differentiate into several populations of brain cells, including neurons, astrocytes, and oligodendrocytes (Fig. 3). Similar to C17.2 cells, neurospheres obtained from MRL-lpr and CD1 brains were in most cases obliterated only by the CSF from diseased MRL-lpr mice (Fig. 2B). Similar toxic effects on the C17.2 colony (as evidenced by increased number of Trypan-positive cells) were seen with CSF from the deceased NP-SLE patient (Table 1). The dose–response relationship was estimated as a regression function (first order monoexponential decay with residual, df =2). The obtained curves indicated that CSF samples from autoimmune mice (n =3) and CSF from the NP-SLE patient (run in triplicate) were profoundly toxic up to dilutions of 1:10 and 1:100, respectively (Fig. 2C). No attenuation in toxicity was seen with supernatants obtained by spinning (Fig. 2D), suggesting that infiltrated cytotoxic leukocytes do not directly compromise the viability of cells obtained from the C17.2 culture.
Fig. 1.
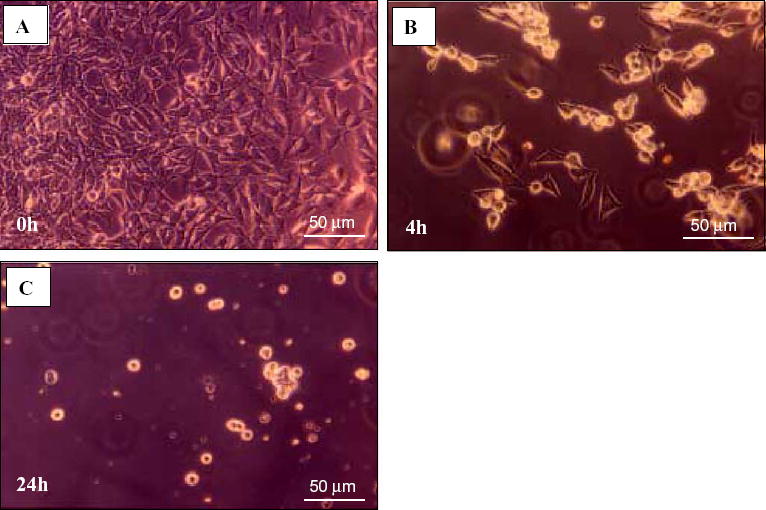
Representative photos of the C17.2 neural stem cell line during incubation with CSF from a diseased MRL-lpr mouse. A) The cell layer was confluent at 0 h, i.e. before CSF was added. B) Adhered cells lost their processes and became rounded within 4 h, detached from surface, and C) most of them degenerated within 24 h.
Fig. 2.
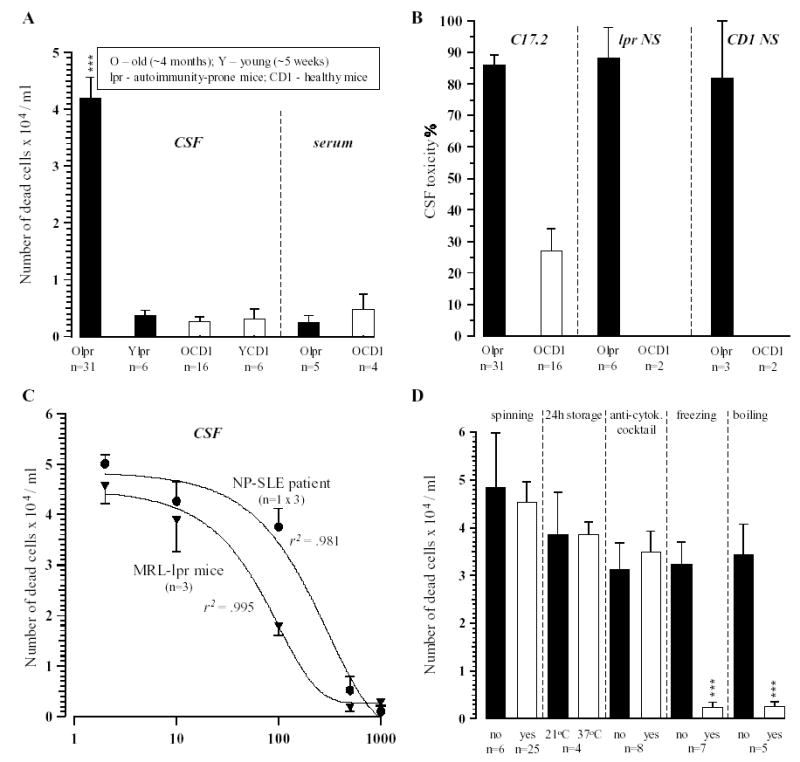
Cytotoxicity assays with proliferating brain cells in culture. A) Comparison of CSF and serum samples on the viability of C17.2 cells revealed the significant toxic effect of the CSF, but not serum from old MRL-lpr mice. B) The comparison of effects on primary stem cells (grown as neurospheres, NS) and the C17.2 stem cell line revealed that the CSF from old, diseased MRL-lpr mice induced similar extensive degeneration of the neurospheres generated from either MRL-lpr or CD1 mice. C) The dose–response curve revealed significant toxic capacity of CSF from MRL-lpr mice in 1:10 dilution and in NP-SLE patient in up to a 1:100 dilution. D) The use of supernatant, overnight storage of samples at RT, and pre-incubation with monoclonal antibodies to pro-inflammatory cytokines did not significantly reduce the toxic activity of CSF samples from diseased MRL-lpr mice. However, when the other half of the same sample was exposed to extreme temperatures the effect was abolished, suggesting that conformational state of the cytotoxic factors is important in accounting for their biological activity. (Note: ‘‘no’’—half of the sample was not exposed; ‘‘yes’’—other half of the same sample was exposed to a given condition).
Fig. 3.
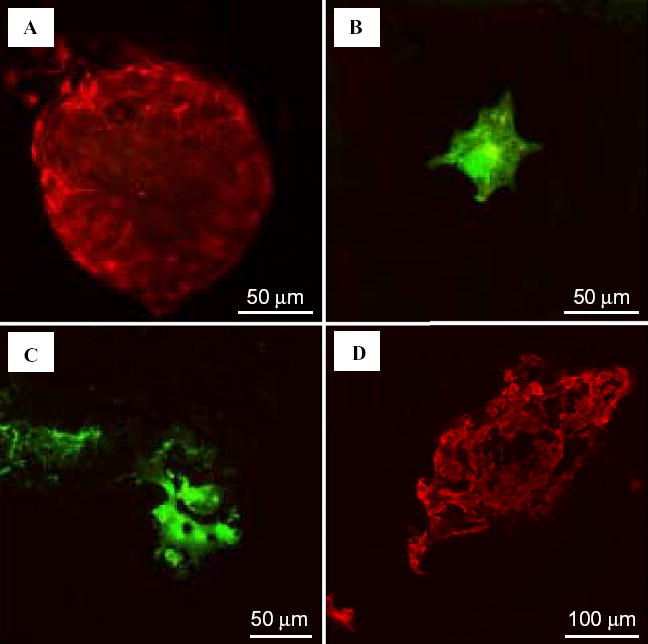
Generation of neuronal and glial progeny from neurospheres isolated from a MRL-lpr mouse. A) A neurosphere stained with anti-nestin. B) An astrocyte stained with anti-GFAP. C) Oligodendrocytes stained with the O4 antibody. D) Neurons stained with anti-β-tubulin.
Table 1.
The relative number of dead/dying C17.2 cellsa which detached from plate surface after 24-h incubation with 10 μl of CSF from live (L) and deceased (D) patients
| Headache L | Meningitis L | SLE L | Drug overdose D | NP-SLE D |
|---|---|---|---|---|
| 3125 | 3125 | 12500 | 6250 | 43750 |
Assessed by Trypan blue exclusion after ~2×104 cells/0.5 ml were grown for 48 h on 18-mm glass cover slips coated with 0.1% poly-L-Ornithine in DMEM.
3.2. Circulating immune factors do not account for cytotoxic effects of CSF
No attenuation in cytotoxicity was observed after pre-incubation with pAbs to individual pro-inflammatory cytokines (data not shown), when the mixture (cocktail) of pAbs was used, or when samples were stored in the incubator for 24 h (Fig. 2D), suggesting that cytokines are not direct mediators of toxicity. A complete prevention of toxicity was obtained when CSF samples were exposed to −20 °C (overnight) and +100 °C (over 10 min). Since many proteins lose their biological activity if exposed to temperature extremes, this implicated that autoantibodies (rather than reactive oxygen species or cytokines) underlie the biological effects of CSF from autoimmune mice. Although high levels of ANA and ACA were detected in the serum of MRL-lpr mice, ANA were not present in their CSF and ACA levels were not significantly different from CSF ACA levels in age-matched MRL+/+ mice (Fig. 4). This suggested that these two major classes of autoantibodies do not account for the cytotoxic properties of CSF from autoimmune MRL-lpr mice.
Fig. 4.
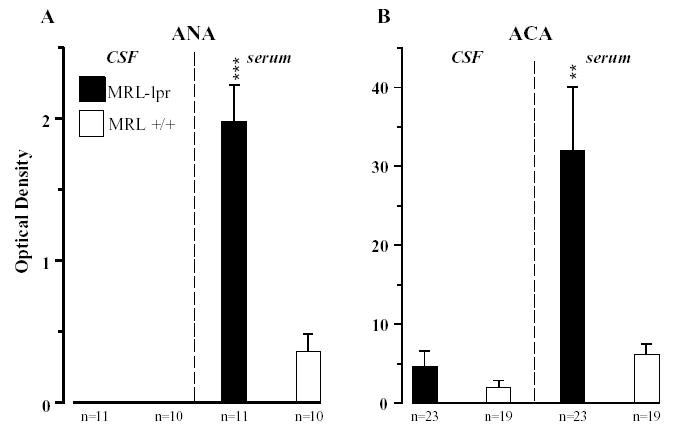
Serological markers of autoimmunity in age-matched, 4-month old MRL mice. A) Although abundant in serum, ANA could not be detected in the CSF from MRL-lpr mice. B) Low levels of ACA were present in CSF samples from both MRL substrains. Similar to ANA, high levels of ACA in the serum of MRL-lpr mice confirmed severe lupus-like disease in this substrain.
3.3. The relationship between CSF cytotoxicity and neurodegeneration
The severity of autoimmune symptoms in the MRL-lpr group was confirmed by enlarged spleens and a reduced brain weight (Fig. 5A). The number of FJB+ cells in brain sections from four MRL-lpr mice correlated exponentially with CSF cytotoxicity, suggesting that toxic CSF induces neurodegeneration in the periventricular areas (Fig. 5B).
Fig. 5.
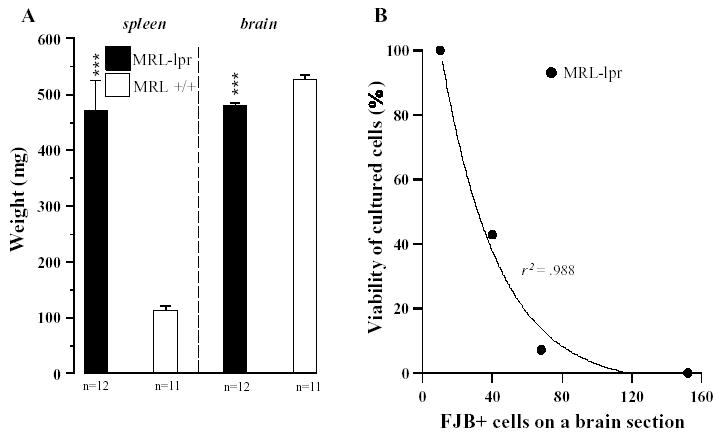
Organ involvement in 4-month old MRL mice. A) As expected, the systemic disease in the MRL-lpr strain was also accompanied by splenomegaly (t21 =6.223, p <.001) and lower brain mass (t21 =5.275, p <.001). B) Significant exponential correlation between the CSF toxicity and FJB staining in the periventricular region of four MRL-lpr mice suggested that profound CSF toxicity is associated with severe degeneration of neurons in the vicinity of lateral ventricles.
Upon injection to the rat retinal eye chamber of CSF from MRL-lpr mice, inflammatory damage was marked within 2 days and profound within 4 days post-injection. No toxic effects were seen on the opposite eye injected with CSF from control MRL+/+ mice. Eyes extracted 4 days later could be distinguished by gross observation from eyes injected with the control CSF. In comparison to the preserved retinal morphology after injection of CSF from MRL+/+ mice (Fig. 6A), the cellular layers in eyes extracted 2 days later were severely disrupted in retinas injected with CSF from MRL-lpr mice, showing decreased thickness in nuclear and synaptic layers (Fig. 6B). However, the above observations could not provide us with an answer on whether the non-specific cellular death was caused by CSF directly (causing necrosis), or indirectly, by recruiting inflammatory cells, such as cytotoxic T-lymphocytes, macrophages, or NK-cells.
Fig. 6.
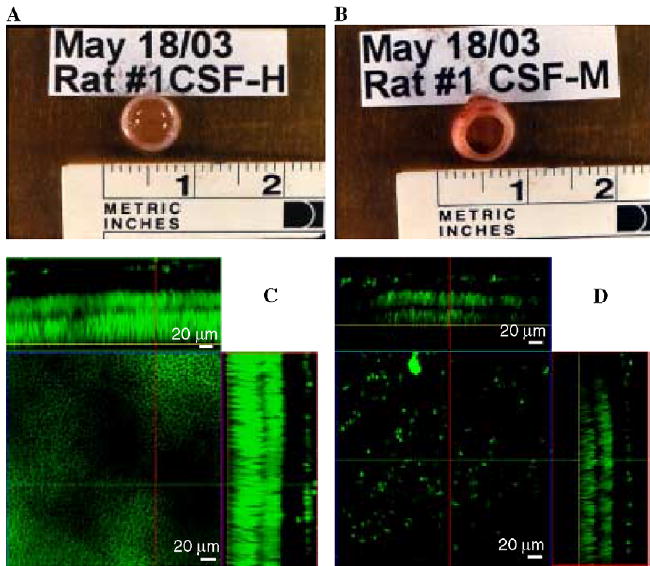
Eyes extracted from a rat four days after a coded intravitreal injection (one eye each) of 5 μl of CSF collected from control MRL+/+ (A) or diseased MRL-lpr mouse (B). Necrotic changes were detected in 7 out of 10 rats after administration of CSF from diseased MRL-lpr mice. Images of retinas exposed to CSF from MRL+/+ (C) and MRL-lpr mouse (D), viewed under confocal microscopy. Necrotic changes were detected in 7 out of 10 rats after administration of CSF from diseased MRL-lpr mice. The panel above the main square represents the ‘‘X’’ plane of the optically reconstructed section of the retina, whereas the panel on the right represents the ‘‘Y’’ plane of the section. The center squares display transverse planes within the distal photoreceptor nuclear layer (yellow line in the optical sections) of the two treatment groups. The retina treated with CSF from a MRL-lpr mouse showed severe thinning of the overall retinal thickness, as well as that of the individual layers.
3.4. Molecular mechanisms of cytotoxicity
On loading the cells with calcein and challenging them with the CSF from MRL-lpr mice or the NP-SLE patient, the fluorescence of calcein intensified. Such an effect was not seen with control samples (Fig. 7A,B). This increase however, was not uniform; in most cases it was gradual, in same cases it showed an oscillatory pattern with the tendency for an increase (data not shown), while in two samples it was immediate, resulting in overt cell death within 4–5 min. The latter observation was rather dramatic, as evidenced by spikes in fluorescence and rapid disintegration of 17 out of 18 cells in the field (Fig. 7C). To test if the increased fluorescence was due to an external Ca+ 2 influx, cells were washed with PBS containing 500 μM EDTA, which chelated the external Ca+ 2 before treatment with CSF (Fig. 7D). The above effect was still observed, suggesting an involvement of calcium from internal stores. In a recent study, neurotoxic antibodies in the CSF from a SLE patient were proposed to bind to and activate NR2 receptors (DeGiorgio et al., 2001). In our study, neither MK801 (NR2 receptor blocker) nor APB and ryanodine were able to block cell death 24 h later. However, the mixture of the two latter drugs significantly abrogated the increase in fluorescence within 3 min of CSF administration (Fig. 7D), suggesting that functional IP3 and ryanodine channels are required (at least in the initial phase) for the release of Ca+ 2 from internal stores.
Fig. 7.
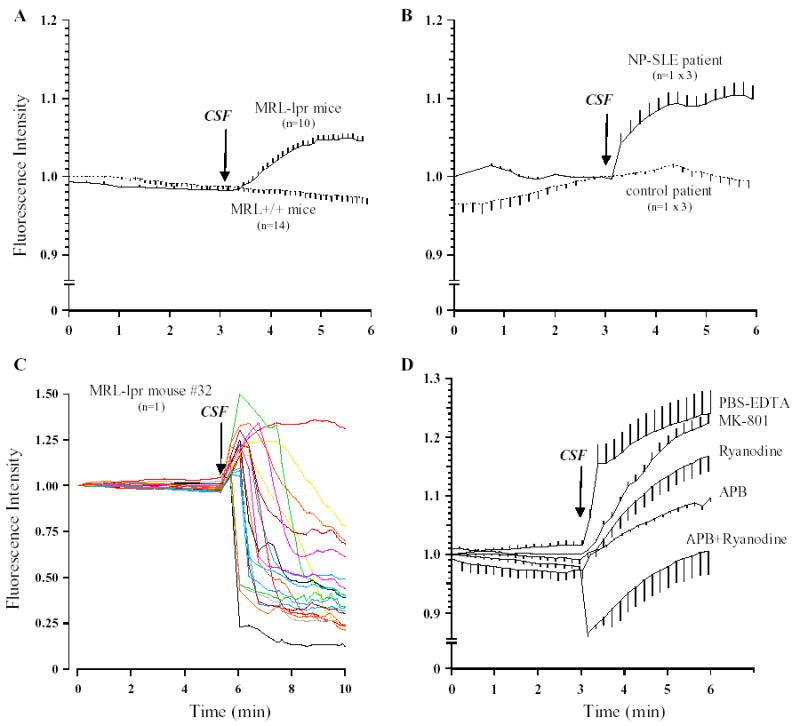
Microfluorometry. C17.2 cells loaded with calcein were monitored for the first 3 min to determine basal fluorescence. CSF was added at the times indicated by the arrow. Background fluorescence determined from an area with no cells was subtracted and this value at time 0 min was taken as 100%. The fluorescence values at all the other times were determined relative to the ‘‘0 time’’ intensity. Typically, values of fluorescence intensity shown are for mean± SEM from 8–10 cells per field. A) In comparison to CSF samples from control MRL+/+ mice, the addition of 5 μl CSF from MRL-lpr mice led to a gradual increase in fluorescence intensity over 3 min. B) Similar effects were observed in three repetitions with CSF from the NP-SLE patient. C) Individual cell fluorescence recordings from one of the highly toxic CSF samples obtained from an autoimmune mouse. D) The lack of permanent blockade of cytotoxicity induced by murine CSF samples. C17.2 cells washed of external Ca+ 2 (using PBS-EDTA), or pre-treated with NR2, IP3 receptor and ryanodine receptor channels antagonists were still sensitive to detrimental effects of CSF from a diseased MRL-lpr mouse. However, a combination of IP2/ryanodine receptor blockade with APB and ryanodine (100 μM) abrogated the increase of fluorescence activity within 3 min.
Despite the above observations, the calcein-releasing assay presently described does not conclusively show involvement of Ca+ 2 because calcein has a similar affinity for Mg2+ (Chiu and Haynes, 1977). A well-characterized ratiometric dye should be used in future studies to confirm Ca+ 2 involvement and quantify intracellular concentrations during CSF-induced cytotoxicity.
3.5. Lack of effects of CSF on membrane ionic currents in C17.2 stem cells
Using recording solutions to isolate currents through K+ channels, which regulate cell membrane potential and thus Ca2+ entry through voltage-gated channels, step depolarizations evoked sustained outward currents in C17.2 cells. On the addition of 10 μl of either normal CSF (n =3) or CSF isolated from MRL-lpr mice (n = 3) to the recording chamber, the amplitude of these outward currents was not significantly altered (data not shown). When using solutions to isolate voltage-gated Ca2+ currents themselves, step depolarization evoked no discernible inward currents, even when using a high concentration of Ba2+ ions (10 mM) as charge carriers in the extracellular solution (data not shown). Similar to undifferentiated neural progenitor cells isolated from the rat hippocampus (Hogg et al., 2004), the present data demonstrates the lack of voltage-dependent Ca2+ channels in C17.2 cells, thus making Ca2+ entry an unlikely pathway to account for elevated [Ca2+]i in response to toxic CSF. It should be noted that in most cases, cell death was apparent 3–4 min after application of the toxic CSF. Taken together, these data suggest that cytotoxic effects of CSF from autoimmune mice do not involve alterations in voltage-gated ion channel activity on the cell surface, findings consistent with those of store-mediated elevation of [Ca2+]i described above.
3.6. Intrathecally synthesized immunoglobulins as terminal cytotoxic factors
Capillary isoelectric focusing (cIEF) of pooled CSF samples revealed unique peak signals in the MRL-lpr group (vs. the sample obtained from MRL+/+ mice) and in the CSF from the NP-SLE patient, in comparison to CSF from a deceased control (Fig. 8A). The protein peaks pI ~6.9 (in the MRL-lpr sample) and pI ~5.85 (in the NP-SLE patient) may appear somewhat lower than expected, but were still within the range common for IgGs. The lower values can be accounted for by a nonlinear relationship (steep on the basic side and shallow on the acidic side) between the pH gradient and detection time for pI markers during cIEF separation (Shimura et al., 2000). Most of the high peaks were in the acidic region (including native albumin with pI of ~4), although likely reflecting artifacts induced by salt content. Although the present samples were not fresh or desalted, and a relatively broad range interval (pH 3–10) was used for the screening purposes, intraday peak reproducibility (expressed as the relative standard deviation, RSD) was within the range of the iCE280 instrument (Janini et al., 2002). For the MRL-lpr sample, the RSD was 2.41%, the MRL+/+ sample 1.58%, NP-SLE patient 1.39% and for the deceased drug user it was 2.57%.
Fig. 8.
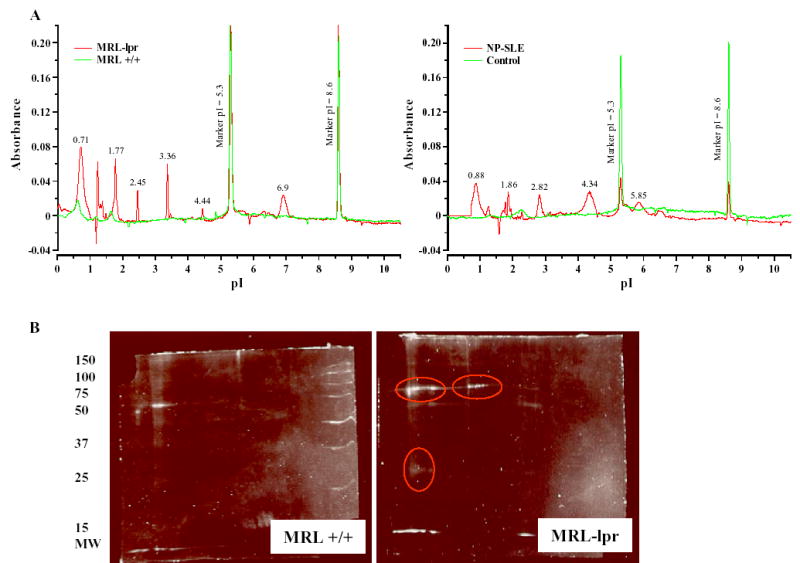
A) Typical electropherograms of major protein fractions obtained by loading iCE280 instrument with CSF samples stored at RT. The data from animals (left) were obtained by pooling individual CSFs from 10 mice/group. The CSF samples from a deceased NP-SLE patient and deceased drug user (right) were run 3–4 weeks upon death. The peaks in the acidic region suggest a general increase in protein content in CSF from autoimmune subjects, including IgG peaks (~6.9 and ~5.85) between the pI markers and the peak typical for native albumin (~4). B) 2-D protein profiles from affinity purified IgG fractions (separated by SDS-PAGE mini-gel electrophoresis) of pooled CSF from MRL+/+ controls and MRL-lpr mice. The gels were stained with SyroRuby fluorescent stain with image capture obtained through the ImageMaster 2-D software package. Protein spots of interest in the lupus-prone MRL-lpr group are circled in red.
Fractionation of pooled CSF samples from MRL-lpr mice and CSF from the NP-SLE patient revealed that IgG-rich eluates were as toxic as the CSF itself (data not shown). However, they differed in that the IgG-depleted fraction (‘‘run-through’’) from MRL-lpr mice was inactive, while the same fraction from human CSF showed cytotoxic capacity which was weaker then the one observed with the IgG-rich fraction (data not shown). This discrepancy can be a result of higher concentrations of toxic IgG in human than in murine samples (as suggested by the superimposed dose–response curve, Fig. 2C), no binding affinity of Protein A for human IgG3 isotype (as specified by manufacturer, Cedarlane Laboratories Ltd, ON, Canada), or presence of immunoglobulin classes other than IgG. 2-Dimensional gel electrophoresis (2DE) was used to further separate proteins based on their size and charge. The comparison of the 2DE-protein profiles revealed distinct differences between IgG-rich CSF fractions from MRL-lpr and MRL+/+ mice (Fig. 8B). In the MRL-lpr sample, several protein spots with the molecular size of IgG subunits (heavy and light chain) were present, including ~55 kDa spot and a faint spot at ~25 kDa. These spots were comparable to the size and charge of protein spots obtained when purified IgG was loaded on the gel (data not shown). The identity of these spots in individual CSF samples has been confirmed with MS/ MALDI-TOF methodology in our recent study (Sidor et al., 2005).
3.7. Brain pathology in the NP-SLE patient
A more detailed pathological analysis of the brain from the NP-SLE patient has been reported elsewhere (Ballok et al., 2004b). In brief, routine neuropathological analysis revealed dilatation of the third and lateral ventricles and thinning of surrounding cortex. Scattered inflammatory cells were seen within the choroid plexus. Consistent with the evidence on neuronal and astrocytic damage in NP-SLE (Trysberg et al., 2003), areas around the lateral and third ventricles showed loss of neurons, gliosis, and focal axonal injury.
4. Discussion
The present study reveals that a large proportion of CSF samples from aged, autoimmune MRL-lpr mice and a CSF sample from a deceased NP-SLE patient are toxic to cultured proliferative brain cells. Consistent with a potent toxic activity at high dilutions, small volumes of CSF from MRL-lpr mice induced profound, non-selective demise of rat retinal neurons in vivo. This suggests that self-renewing cell populations are not exclusive, but merely one of the targets of the autoimmune attack. Extreme temperatures blocked the cytotoxic effect, pointing to the possibility that a specific conformational state of toxic proteins (e.g., cryoglobulins) accounts for biological properties of the CSF from MRL-lpr mice (Fulpius et al., 1993) and the lupus patient (Winfield, 1983; Manger et al., 2002; Garcia-Carrasco et al., 2001). The notion that soluble proteins (and not immunocompetent cells) account for the cytotoxicity was corroborated by the observation that CSF supernatant produced effects comparable to non-centrifuged CSF samples. An IgG-rich fraction accounted for the cytotoxicity in our animal model and partially in the CSF sample from one of two SLE patients. Previous evidence on toxicity of human CSF to the mouse brain (DeGiorgio et al., 2001), and our observations that murine CSF affects rat brain tissue and that human CSF reduces the viability of a murine cell line, imply that molecular mechanisms are not dependent on major histocompatibility antigens.
The present study did not exclude the possibility that fungal and/or bacterial CNS infections, which occur in NP-SLE (Hung et al., 2005), contribute to overall CSF toxicity. Although the media pH remained unchanged in the present study (suggested by constancy of culturing media color), live bacteria were commonly seen in C17.2 cell cultures 24 h after CSF addition. However, this mechanism is not consistent with cases in which addition of CSF led to a rapid release of calcein and cell death, or when presence of bacteria in culturing medium was not accompanied by cell death. Along the same line, the discrepancy between biological activities of IgG-rich and IgG-depleted fractions of a pooled CSF sample is difficult to explain by bacterial/ fungal factors. As previously shown with a primary neuronal/astrocyte co-culture (Maric et al., 2001), serum from diseased MRL-lpr was not cytotoxic in the present study. This implies that a class of intrathecally synthesized autoantibodies accounts better for the CNS effect than circulating autoantibodies (at least from ANA and ACA classes). Consistent with this notion is the evidence that ACA levels in the CSF did not differ between the MRL substrains, but largely differed in the serum. Similarly, although abundant in the sera from aged MRL-lpr mice, ANA could not be detected in CSF samples by a standard ELISA kit. Nevertheless, the possibility that the concentration of cytotoxic antibodies in the serum was very low (and thus biologically ineffective) cannot be denied. As shown by patch–clamp technique, voltage-gated ion channel activity on the cell surface does not seem to account for cytotoxic effects of CSF from autoimmune mice. Conversely, the gradual increase in calcein fluorescence suggests release of intracellular ions (e.g. Ca+ 2) after cells from C17.2 culture were exposed to CSF from autoimmune mice. Moreover, a rapid increase and then decrease in calcein fluorescence may indicate massive, unregulated cellular damage (e.g., direct disruption of lipid membrane integrity) followed by elevation of intracellular Ca+ 2. The cells from C17.2 culture loaded poorly with Fura-2. The use of other neural stem cell lines, more specific indicators (e.g. Fura-2, Indo-1 and Fluo-3), and specific receptor blockers (e.g. thapsigargin) would definitely help in future assessments of intracellular Ca+ 2 involvement. Despite the lack of a clear understanding as to a common molecular mechanism, multiple pI peaks and protein spots on 2D-gels may suggest a rather complex involvement of various autoantibodies and antigen targets.
The obtained results are in agreement with the evidence that neural stem cells, although relatively resistant to metabolic insults (Morshead and van der Kooy, 1992; Morshead et al., 1994) are sensitive to the inflammatory process (Monje et al., 2003), which is an integral aspect of systemic autoimmune disease. Autoantibodies in the CSF and immunocompetent cells in the choroid plexus of mice (Sakic et al., 2000b; Vogelweid et al., 1991) and the NP-SLE patient are further evidence that the blood–brain barrier in SLE becomes permeable to circulating factors (Abbott et al., 2003). Previous clinical studies had engendered the hypothesis that various brain-reactive antibodies (Denburg et al., 1993), cytokines, and toxic metabolites (Svenungsson et al., 2001; Trysberg et al., 2000) play principal roles in induction of SLE-induced brain damage in general, and gray matter in particular (Steens et al., 2004). With respect to the origin of autoantibodies, one of the important issues is whether they passively diffuse through the damaged blood–brain barrier or are synthesized intrathecally (i.e., within the brain). Although the former possibility is viable when serum antibodies (originally directed towards peripheral antigens) are considered, our studies and clinical evidence (Lai and Lan, 2000) suggest that neuroactive antibodies are synthesized intrathecally. Similar to the present lack of discriminative value in the MRL model, in some clinical studies ACA could not be detected in the CSF of patients with NP-SLE, even when found in their sera (Jedryka-Goral et al., 2000). With respect to pro-inflammatory cytokines measured in the present study, we infer that they do not play a key role in cytotoxicity towards proliferating cells. Clinically, the diagnostic value of serum and CSF cytokines is limited in NP-SLE, probably due to the heterogeneity of disease pathogenesis (Jonsen et al., 2003). However, we cannot exclude the possibility that cytokines play a facilitatory role in extravasation of circulating lymphocytes (Munoz-Fernandez and Fresno, 1998; Seabrook and Hay, 2001), thus indirectly contributing to the neuropathogenic process.
The present study further supports the hypothesis that brain morphology and function are compromised by the local inflammatory/autoimmune process. If antibodies reactive to neurons and proliferating cells are indeed specific to brain tissue, the source of their production (at least in MRL-lpr mice) are likely the activated B-cells in the choroid plexus and ventricular lumen (Farrell et al., 1997; Sakic et al., 2000b; Zameer and Hoffman, 2004). Circulating leukocytes may cross the blood–brain barrier which is damaged by systemic inflammation (Vogelweid et al., 1991) and become activated upon encountering brain-specific antigens around the ventricles. This interaction may lead to intrathecal production of antibodies to neurons and stem cells in the subependymal layer, and subsequently, a reduction in their viability. It is important to emphasize that this type of autoimmunity-induced brain damage may represent an additional insult after a major imbalance in the immuno-neuroendocrine network at the onset of systemic auto-immune disease. Namely, accumulating evidence suggests that glucocorticoids are critical for regulation of the immune system and maintenance of homeostasis (McEwen et al., 1997). In autoimmune animals, the neuroendocrine network is significantly affected with the onset of autoimmune disease (Wick et al., 1993). The MRL-lpr strain develops an SLE-like disease at a young age and profound changes in responsiveness of the hypothalamus–pituitary–adrenal axis occur concurrently with the first autoimmune symptoms. Sustained increases in baseline corticosterone levels (Lechner et al., 1996) and its imbalanced circadian rhythm (Lechner et al., 2000) could represent one of the very first metabolic insults to the brains of autoimmune animals. This possibility is based on the evidence that sustained exposure to glucocorticoids renders pyramidal neurons vulnerable to other metabolic insults (Sapolsky, 2000; Armanini et al., 1990). Along the same line, we could not exclude the possibility that prolonged immunosuppressive therapy contributed to brain pathology and ventricular enlargement in the SLE patient who died from CNS complications. With respect to cyclophosphamide, the dose given to the NP-SLE patient (1–2 mg/kg/day i.v.) was relatively small in comparison to the dose (100 mg/kg i.p./week) we used in our previous studies with lupus-prone mice. The evidence that chronic treatment with cyclophosphamide abolished CSF toxicity in MRL-lpr mice (Ballok et al., 2004a) and is effective in the treatment of NP manifestations (Trysberg et al., 2003; Ramos et al., 1996; West, 1996) suggest that cyclophosphamide and its active metabolites do not have a detrimental effect on the viability of immature neuronal cells. However, despite the results obtained with the IgG-rich fraction of CSF from the NP-SLE patient, one may propose that sustained steroid treatment contributed to the overall brain atrophy.
An important step towards the identification of functionally important autoantibodies in NP-SLE was the recent identification of anti-DNA antibodies which cross-react with central NMDA receptors (DeGiorgio et al., 2001). These antibodies also produced neuronal apoptosis when injected into the mouse hippocampus, and were proposed to have psychotomimetic properties in patients with NP-SLE. Antibodies to other enzymatic and receptor systems (such as ion channels) have been consistently reported in neuro-immunological diseases of the peripheral nervous system (Skeie et al., 2003; Loirand et al., 1992). The modulatory effect of ryanodine/IP3 blockers in the present study raises the possibility that IgGs to the Ca+ 2 regulatory system, as reported in myasthenia gravis (Mygland et al., 1993; Skeie et al., 2003), can be synthesized centrally and interfere with the proliferative capacity of brain cell progenitors. In addition to NP-SLE, activated B-cells and anti-neuronal antibodies were reported in CSF from patients with multiple sclerosis (DeVries, 2004; Corcione et al., 2004; Silber et al., 2002). Although highly speculative at this point, one may wonder whether antibody-mediated depletion of proliferative brain cells contributes to impaired capacity of oligodendrocytes to regenerate and repair damaged axons.
With respect to neurodevelopmental aspects of SLE, it is known that IgGs from mothers with SLE can affect the fetus by crossing the blood–placental barrier and even have toxic effects on cultured rat embryos (Nadler et al., 1995). Although neonatal lupus is often a self-limiting condition, brain imaging analyses of infants reveal subclinical CNS disease, including vasculopathy (Cabanas et al., 1996), attenuations in white matter, and mild ventricular enlargement (Prendiville et al., 2003). The supportive evidence for autoimmunity-induced brain damage during embryogenesis and behavioral dysfunction in adulthood has been obtained by transfer of fertilized ova from healthy animals to the uterus of autoimmune females (Denenberg et al., 1991). If brain-reactive antibodies can indeed affect brain morphology and function, it is tempting to hypothesize that proliferating brain cells are a target during embryogenesis or early post-natal life (i.e. until maternal antibodies are cleared from the infant’s circulation). This possibility further leads to a notion that some highly toxic autoantibodies (as proposed in a few CSF samples from MRL-lpr mice) may have long-lasting consequences on behavior. The increased prevalence of autism spectrum disorders is a novel medical conundrum which involves autoreactivity to brain antigens (Todd et al., 1988; Singh et al., 1998, 2002; Silva et al., 2004) and other autoimmune phenomena (Singh and Rivas, 2004; Sweeten et al., 2003; Croonenberghs et al., 2002). Together with immunological changes in schizophrenia spectrum disorders (Jones and Cannon, 1998) and epilepsies (Rogers et al., 1996), the above correlations beg for a deeper understanding of the relationship between autoimmunity and the developing brain.
In the present study we made preliminary efforts to identify and characterize the IgG species in pooled CSF samples and, as shown in clinical studies (Hiraoka et al., 2002), the cIEF may become a useful tool in screening for unique CSF proteins in subjects with behavioral manifestations. However, to achieve more definitive data our future studies will focus on proteomic analysis of individual CSF samples, biochemical characterization of cytotoxic antibodies and their targets, as well as electrophysiological measurements of Ca+ 2-dependent intracellular mechanisms. Future experimental studies should involve other cell lines (including human) and assess neural stem cell proliferation rates in vivo along the progress of lupus-like disease. Along the same line, clinical studies would benefit from comparing CSF biological activity between larger cohorts of NP-SLE and SLE patients. However, our short-term goal is to replicate behavioral deficits in healthy mice by administering CSF IgG fractions from diseased MRL-lpr animals. Aberrant behavior and/or neuromorphological changes would be instrumental in confirming that autoantibodies to antigens on immature and mature neural cells are a key factor in the etiology of autoimmunity-associated CNS dysfunction.
Acknowledgments
We thank Drs. Ian Q. Whishaw, Ashok Grover, Margaret Fahnestock, Monalisa Sur, and Jack Rosenfeld for helpful feedbacks in designing the experiments and data interpretation. Our deepest thanks go to Brian van Adel for technical assistance, and Dr. Jiaqi Wu and Mr. Arthur H. Watson from Convergent Bioscience (Toronto, Ontario, Canada), who analyzed the CSF samples using their iCE280 system. Finally, we are thankful to the Cicci family and the Lupus Society of Hamilton for supporting research and allowing the human studies to take place. This work was supported by funds to B. Sakic from the Canadian Institutes of Health Research (MOP 38065) and National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH (1R21 AR49163-01). B. Sakic is a recipient of the FSORC career development award.
References
- Abbott NJ, Mendonca LL, Dolman DE. The blood–brain barrier in systemic lupus erythematosus. Lupus. 2003;12:908–915. doi: 10.1191/0961203303lu501oa. [DOI] [PubMed] [Google Scholar]
- Alexander EL, Murphy ED, Roths JB, Alexander GE. Congenic autoimmune murine models of central nervous system disease in connective tissue disorders. Ann Neurol. 1983;14:242–248. doi: 10.1002/ana.410140211. [DOI] [PubMed] [Google Scholar]
- Armanini MP, Hutchins C, Stein BA, Sapolsky RM. Glucocorticoid endangerment of hippocampal neurons is NMDA-receptor dependent. Brain Res. 1990;532:7–12. doi: 10.1016/0006-8993(90)91734-x. [DOI] [PubMed] [Google Scholar]
- Ballok DA, Millward JM, Sakic B. Neurodegeneration in autoimmune MRL-lpr mice as revealed by Fluoro Jade B staining. Brain Res. 2003;964:200–210. doi: 10.1016/s0006-8993(02)03980-x. [DOI] [PubMed] [Google Scholar]
- Ballok DA, Earls AM, Krasnik C, Hoffman SA, Sakic B. Autoimmune-induced damage of the midbrain dopaminergic system in lupus-prone mice. J Neuroimmunol. 2004a;152:83–97. doi: 10.1016/j.jneuroim.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Ballok DA, Woulfe J, Sur M, Cyr M, Sakic B. Hippocampal damage in mouse and human forms of systemic autoimmune disease. Hippocampus. 2004b;14:649–661. doi: 10.1002/hipo.10205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum KA, Hopf U, Nehrig C, Stover M, Schorner W. Systemic lupus erythematosus: neuropsychiatric signs and symptoms related to cerebral MRI findings. Clin Neurol Neurosurg. 1993;95:29–34. doi: 10.1016/0303-8467(93)90088-x. [DOI] [PubMed] [Google Scholar]
- Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the SLEDAI. A disease activity index for lupus patients The Committee on Prognosis Studies in SLE. Arthritis Rheum. 1992;35:630–640. doi: 10.1002/art.1780350606. [DOI] [PubMed] [Google Scholar]
- Bosma GP, Rood MJ, Zwinderman AH, Huizinga TW, van Buchem MA. Evidence of central nervous system damage in patients with neuropsychiatric systemic lupus erythematosus, demonstrated by magnetization transfer imaging. Arthritis Rheum. 2000;43:48–54. doi: 10.1002/1529-0131(200001)43:1<48::AID-ANR7>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Bouron A, Mbebi C, Loeffler JP, De WM. The beta-amyloid precursor protein controls a store-operated Ca2+ entry in cortical neurons. Eur J Neurosci. 2004;20:2071–2078. doi: 10.1111/j.1460-9568.2004.03680.x. [DOI] [PubMed] [Google Scholar]
- Brannen CL, Sugaya K. In vitro differentiation of multipotent human neural progenitors in serum-free medium. Neuroreport. 2000;11:1123–1128. doi: 10.1097/00001756-200004070-00042. [DOI] [PubMed] [Google Scholar]
- Cabanas F, Pellicer A, Valverde E, Morales C, Quero J. Central nervous system vasculopathy in neonatal lupus erythematosus. Pediatr Neurol. 1996;15:124–126. doi: 10.1016/0887-8994(96)00159-2. [DOI] [PubMed] [Google Scholar]
- Chiasson BJ, Tropepe V, Morshead CM, van der Kooy D. Adult mammalian forebrain ependymal and subependymal cells demonstrate proliferative potential, but only subependymal cells have neural stem cell characteristics. J Neurosci. 1999;19:4462–4471. doi: 10.1523/JNEUROSCI.19-11-04462.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu VC, Haynes DH. High and low affinity Ca2+ binding to the sarcoplasmic reticulum: use of a high-affinity fluorescent calcium indicator. Biophys J. 1977;18:3–22. doi: 10.1016/S0006-3495(77)85592-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colamussi P, Giganti M, Cittanti C, Dovigo L, Trotta F, Tola MR, Tamarozzi R, Lucignani G, Piffanelli A. Brain single-photon emission tomography with Tc-99m-HMPAO in neuropsychiatric systemic lupus erythematosus: relations with EEG and MRI findings and clinical manifestations. Eur J Nucl Med. 1995;22:17–24. doi: 10.1007/BF00997243. [DOI] [PubMed] [Google Scholar]
- Corcione A, Casazza S, Ferretti E, Giunti D, Zappia E, Pistorio A, Gambini C, Mancardi GL, Uccelli A, Pistoia V. Recapitulation of B cell differentiation in the central nervous system of patients with multiple sclerosis. Proc Natl Acad Sci U S A. 2004;101:11064–11069. doi: 10.1073/pnas.0402455101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croonenberghs J, Bosmans E, Deboutte D, Kenis G, Maes M. Activation of the inflammatory response system in autism. Neuro-psychobiology. 2002;45:1–6. doi: 10.1159/000048665. [DOI] [PubMed] [Google Scholar]
- De Clerck LS, Bridts CH, Mertens AM, Moens MM, Stevens WJ. Use of fluorescent dyes in the determination of adherence of human leucocytes to endothelial cells and the effect of fluorochromes on cellular function. J Immunol Methods. 1994;172:115–124. doi: 10.1016/0022-1759(94)90384-0. [DOI] [PubMed] [Google Scholar]
- DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med. 2001;7:1189–1193. doi: 10.1038/nm1101-1189. [DOI] [PubMed] [Google Scholar]
- Demaurex N, Distelhorst C. Cell biology. Apoptosis—the calcium connection. Science. 2003;300:65–67. doi: 10.1126/science.1083628. [DOI] [PubMed] [Google Scholar]
- Denburg SD, Denburg JA, Carbotte RM, Fisk JD, Hanly JG. Cognitive deficits in systemic lupus erythematosus. Rheum Dis Clin North Am. 1993;19:815–831. [PubMed] [Google Scholar]
- Denenberg VH, Mobraaten LE, Sherman GF, Morrison L, Schrott LM, Waters NS, Rosen GD, Behan PO, Galaburda AM. Effects of the autoimmune uterine/maternal environment upon cortical ectopias, behavior and autoimmunity. Brain Res. 1991;563:114–122. doi: 10.1016/0006-8993(91)91522-3. [DOI] [PubMed] [Google Scholar]
- Denenberg VH, Sherman GF, Rosen GD, Morrison L, Behan PO, Galaburda AM. A behavior profile of the MRL/Mp lpr/lpr mouse and its association with hydrocephalus. Brain Behav Immun. 1992;6:40–49. doi: 10.1016/0889-1591(92)90058-v. [DOI] [PubMed] [Google Scholar]
- DeVries GH. Cryptic axonal antigens and axonal loss in multiple sclerosis. Neurochem Res. 2004;29:1999–2006. doi: 10.1007/s11064-004-6873-1. [DOI] [PubMed] [Google Scholar]
- Doering LC, Snyder EY. Cholinergic expression by a neural stem cell line grafted to the adult medial septum/diagonal band complex. J Neurosci Res. 2000;61:597–604. doi: 10.1002/1097-4547(20000915)61:6<597::AID-JNR3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Duprez T, Nzeusseu A, Peeters A, Houssiau FA. Selective involvement of the choroid plexus on cerebral magnetic resonance images: a new radiological sign in patients with systemic lupus eryxthematosus with neurological symptoms. J Rheumatol. 2001;28:387–391. [PubMed] [Google Scholar]
- Farrell M, Sakic B, Szechtman H, Denburg JA. Effect of cyclophosphamide on leucocytic infiltration in the brain of MRL/lpr mice. Lupus. 1997;6:268–274. doi: 10.1177/096120339700600310. [DOI] [PubMed] [Google Scholar]
- Fernandez SF, Huang MH, Davidson BA, Knight PR, III, Izzo JL., Jr Modulation of angiotensin II responses in sympathetic neurons by cytosolic calcium. Hypertension. 2003;41:56–63. doi: 10.1161/01.hyp.0000047513.75459.7e. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick JM, Koh JS, Hartwell D, Beller DI, Levine JS. Dysregulated cytokine expression in vivo in prediseased and diseased autoimmune-prone MRL mice. Autoimmunity. 1996;23:217–229. doi: 10.3109/08916939608995345. [DOI] [PubMed] [Google Scholar]
- Fulpius T, Spertini F, Reininger L, Izui S. Immunoglobulin heavy chain constant region determines the pathogenicity and the antigen-binding activity of rheumatoid factor. Proc Natl Acad Sci U S A. 1993;90:2345–2349. doi: 10.1073/pnas.90.6.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Carrasco M, Ramos-Casals M, Cervera R, Trejo O, Yague J, Siso A, Jimenez S, de la RG, Font J, Ingelmo M. Cryoglobulinemia in systemic lupus erythematosus: prevalence and clinical characteristics in a series of 122 patients. Semin Arthritis Rheum. 2001;30:366–373. doi: 10.1053/sarh.2001.20265. [DOI] [PubMed] [Google Scholar]
- Hanly JG, Hong C, Smith S, Fisk JD. A prospective analysis of cognitive function and anticardiolipin antibodies in systemic lupus erythematosus. Arthritis Rheum. 1999;42:728–734. doi: 10.1002/1529-0131(199904)42:4<728::AID-ANR16>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Hess DC, Taormina M, Thompson J, Sethi KD, Diamond B, Rao R, Feldman DS. Cognitive and neurologic deficits in the MRL/lpr mouse: a clinicopathologic study. J Rheumatol. 1993;20:610–617. [PubMed] [Google Scholar]
- Hiraoka A, Tominaga I, Hori K. One-step capillary isoelectric focusing of the proteins in cerebrospinal fluid and serum of patients with neurological disorders. J Chromatogr. 2002;A961:147–153. doi: 10.1016/s0021-9673(02)00174-7. [DOI] [PubMed] [Google Scholar]
- Hirohata S, Miyamoto T. Elevated levels of interleukin-6 in cerebrospinal fluid from patients with systemic lupus erythematosus and central nervous system involvement. Arthritis Rheum. 1990;33:644–649. doi: 10.1002/art.1780330506. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Chipperfield H, Whyte KA, Stafford MR, Hansen MA, Cool SM, Nurcombe V, Adams DJ. Functional maturation of isolated neural progenitor cells from the adult rat hippocampus. Eur J Neurosci. 2004;19:2410–2420. doi: 10.1111/j.0953-816X.2004.03346.x. [DOI] [PubMed] [Google Scholar]
- Hung JJ, Ou LS, Lee WI, Huang JL. Central nervous system infections in patients with systemic lupus erythematosus. J Rheumatol. 2005;32:40–43. [PubMed] [Google Scholar]
- Janini G, Saptharishi N, Waselus M, Soman G. Element of a validation method for MU-B3 monoclonal antibody using an imaging capillary isoelectric focusing system. Electrophoresis. 2002;23:1605–1611. doi: 10.1002/1522-2683(200206)23:11<1605::AID-ELPS1605>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Jedryka-Goral A, Zabek J, Wojciechowski B, Zaborski J, Chwalinska-Sadowska H, Czlonkowska A. Evaluation of cerebrospinal fluid for the presence of anticardiolipin antibodies (aCL) in NP-SLE patients. Clin Rheumatol. 2000;19:306–310. doi: 10.1007/s100670070051. [DOI] [PubMed] [Google Scholar]
- Jones P, Cannon M. The new epidemiology of schizophrenia. Psychiatr Clin North Am. 1998;21:1–25. doi: 10.1016/s0193-953x(05)70358-0. [DOI] [PubMed] [Google Scholar]
- Jonsen A, Bengtsson AA, Nived O, Ryberg B, Truedsson L, Ronnblom L, Alm GV, Sturfelt G. The heterogeneity of neuropsychiatric systemic lupus erythematosus is reflected in lack of association with cerebrospinal fluid cytokine profiles. Lupus. 2003;12:846–850. doi: 10.1191/0961203303lu472sr. [DOI] [PubMed] [Google Scholar]
- Lahita RG. Special report: adjusted lupus prevalence. Results of a marketing study by the Lupus Foundation of America. Lupus. 1995;4:450–453. doi: 10.1177/096120339500400605. [DOI] [PubMed] [Google Scholar]
- Lai NS, Lan JL. Evaluation of cerebrospinal anticardiolipin antibodies in lupus patients with neuropsychiatric manifestations. Lupus. 2000;9:353–357. doi: 10.1191/096120300678828415. [DOI] [PubMed] [Google Scholar]
- Lechner O, Hu Y, Jafarian-Tehrani M, Dietrich H, Schwarz S, Herold M, Haour F, Wick G. Disturbed immunoendocrine communication via the hypothalamo–pituitary–adrenal axis in murine lupus. Brain Behav Immun. 1996;10:337–350. doi: 10.1006/brbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Lechner O, Dietrich H, Oliveira dos SA, Wiegers GJ, Schwarz S, Harbutz M, Herold M, Wick G. Altered circadian rhythms of the stress hormone and melatonin response in lupus-prone MRL/MP-fas(Ipr) mice. J Autoimmun. 2000;14:325–333. doi: 10.1006/jaut.2000.0375. [DOI] [PubMed] [Google Scholar]
- Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- Lescuyer P, Allard L, Zimmermann-Ivol CG, Burgess JA, Hughes-Frutiger S, Burkhard PR, Sanchez JC, Hochstrasser DF. Identification of post-mortem cerebrospinal fluid proteins as potential biomarkers of ischemia and neurodegeneration. Proteomics. 2004;4:2234–2241. doi: 10.1002/pmic.200300822. [DOI] [PubMed] [Google Scholar]
- Loirand G, Faiderbe S, Baron A, Geffard M, Mironneau J. Autoanti-phosphatidylinositide antibodies specifically inhibit noradrenaline effects on Ca2+ and Cl-channels in rat portal vein myocytes. J Biol Chem. 1992;267:4312–4316. [PubMed] [Google Scholar]
- Manger K, Manger B, Repp R, Geisselbrecht M, Geiger A, Pfahlberg A, Harrer T, Kalden JR. Definition of risk factors for death, end stage renal disease, and thromboembolic events in a monocentric cohort of 338 patients with systemic lupus erythematosus. Ann Rheum Dis. 2002;61:1065–1070. doi: 10.1136/ard.61.12.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maric D, Millward JM, Ballok DA, Szechtman H, Barker JL, Denburg JA, Sakic B. Neurotoxic properties of cerebrospinal fluid from behaviorally impaired autoimmune mice. Brain Res. 2001;920:183–193. doi: 10.1016/s0006-8993(01)03060-8. [DOI] [PubMed] [Google Scholar]
- Martinez-Cordero E, Rivera Garcia BE, Aguilar Leon DE. Anticardiolipin antibodies in serum and cerebrospinal fluid from patients with systemic lupus erythematosus. J Investig Allergol Clin Immunol. 1997;7:596–601. [PubMed] [Google Scholar]
- McEwen BS, Biron CA, Brunson KW, Bulloch K, Chambers WH, Dhabhar FS, Goldfarb RH, Kitson RP, Miller AH, Spencer RL, Weiss JM. The role of adrenocorticoids as modulators of immune function in health and disease: neural, endocrine and immune interactions. Brain Res Rev. 1997;23:79–133. doi: 10.1016/s0165-0173(96)00012-4. [DOI] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- Morshead CM, van der Kooy D. Postmitotic death is the fate of constitutively proliferating cells in the subependymal layer of the adult mouse brain. J Neurosci. 1992;12:249–256. doi: 10.1523/JNEUROSCI.12-01-00249.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morshead CM, van der Kooy D. A new ‘spin’on neural stem cells? Curr Opin Neurobiol. 2001;11:59–65. doi: 10.1016/s0959-4388(00)00174-4. [DOI] [PubMed] [Google Scholar]
- Morshead CM, Reynolds BA, Craig CG, McBurney MW, Staines WA, Morassutti D, Weiss S, van der Kooy D. Neural stem cells in the adult mammalian forebrain: a relatively quiescent subpopulation of subependymal cells. Neuron. 1994;13:1071–1082. doi: 10.1016/0896-6273(94)90046-9. [DOI] [PubMed] [Google Scholar]
- Munoz-Fernandez MA, Fresno M. The role of tumour necrosis factor, interleukin 6, interferon-gamma and inducible nitric oxide synthase in the development and pathology of the nervous system. Prog Neurobiol. 1998;56:307–340. doi: 10.1016/s0301-0082(98)00045-8. [DOI] [PubMed] [Google Scholar]
- Mwanjewe J, Grover AK. Role of transient receptor potential canonical 6 (TRPC6) in non-transferrin-bound iron uptake in neuronal phenotype PC12 cells. Biochem J. 2004;378:975–982. doi: 10.1042/BJ20031187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mygland A, Tysnes OB, Aarli JA, Matre R, Gilhus NE. IgG subclass distribution of ryanodine receptor autoantibodies in patients with myasthenia gravis and thymoma. J Autoimmun. 1993;6:507–515. doi: 10.1006/jaut.1993.1042. [DOI] [PubMed] [Google Scholar]
- Nadler DM, Klein NW, Aramli LA, Chambers BJ, Mayes M, Wener MH. The direct embryotoxicity of immunoglobulin G fractions from patients with systemic lupus erythematosus. Am J Reprod Immunol. 1995;34:349–355. doi: 10.1111/j.1600-0897.1995.tb00963.x. [DOI] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Pal S, Sun D, Limbrick D, Rafiq A, DeLorenzo RJ. Epileptogenesis induces long-term alterations in intracellular calcium release and sequestration mechanisms in the hippocampal neuronal culture model of epilepsy. Cell Calcium. 2001;30:285–296. doi: 10.1054/ceca.2001.0236. [DOI] [PubMed] [Google Scholar]
- Prendiville JS, Cabral DA, Poskitt KJ, Au S, Sargent MA. Central nervous system involvement in neonatal lupus erythematosus. Pediatr Dermatol. 2003;20:60–67. doi: 10.1046/j.1525-1470.2003.03014.x. [DOI] [PubMed] [Google Scholar]
- Ramos PC, Mendez MJ, Ames PRJ, Khamashta MA, Hughes GRV. Pulse cyclophosphamide in the treatment of neuro-psychiatric systemic lupus erythematosus. Clin Exp Rheumatol. 1996;14:295–299. [PubMed] [Google Scholar]
- Rogers SW, Twyman RE, Gahring LC. The role of auto-immunity to glutamate receptors in neurological disease. Mol Med Today. 1996;2:76–81. doi: 10.1016/1357-4310(96)88742-9. [DOI] [PubMed] [Google Scholar]
- Rubin LA, Urowitz MB, Gladman DD. Mortality in systemic lupus erythematosus: the bimodal pattern revisited. Q J Med. 1985;55:87–98. [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Talangbayan H, Denburg SD, Carbotte RM, Denburg JA. Disturbed emotionality in autoimmune MRL-lpr mice. Physiol Behav. 1994;56:609–617. doi: 10.1016/0031-9384(94)90309-3. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Braciak TA, Richards CD, Gauldie J, Denburg JA. Reduced preference for sucrose in autoimmune mice: a possible role of interleukin-6. Brain Res Bull. 1997;44:155–165. doi: 10.1016/s0361-9230(97)00107-x. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Denburg JA, Gorny G, Kolb B, Whishaw IQ. Progressive atrophy of pyramidal neuron dendrites in autoimmune MRL-lpr mice. J Neuroimmunol. 1998;87:162–170. doi: 10.1016/s0165-5728(98)00085-x. [DOI] [PubMed] [Google Scholar]
- Sakic B, Kolb B, Whishaw IQ, Gorny G, Szechtman H, Denburg JA. Immunosuppression prevents neuronal atrophy in lupus-prone mice: evidence for brain damage induced by autoimmune disease? J Neuroimmunol. 2000a;111:93–101. doi: 10.1016/s0165-5728(00)00364-7. [DOI] [PubMed] [Google Scholar]
- Sakic B, Maric I, Koeberle PD, Millward JM, Szechtman H, Maric D, Denburg JA. Increased TUNEL-staining in brains of autoimmune Fas-deficient mice. J Neuroimmunol. 2000b;104:147–154. doi: 10.1016/s0165-5728(99)00277-5. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM. Glucocorticoids and hippocampal atrophy in neuro-psychiatric disorders. Arch Gen Psychiatry. 2000;57:925–935. doi: 10.1001/archpsyc.57.10.925. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Schwartz MM, Roberts JL. Membranous and vascular choroid-opathy: two patterns of immune deposits in systemic lupus erythematosus. Clin Immunol Immunopathol. 1983;29:369–380. doi: 10.1016/0090-1229(83)90040-5. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Rochas M, Weller B, Sheinkman A, Tal I, Golan D, Toubi N, Eldar I, Sharf B, Attias D. High association of anticardiolipin antibodies with psychosis. J Clin Psychiatry. 1998;59:20–23. doi: 10.4088/jcp.v59n0105. [DOI] [PubMed] [Google Scholar]
- Seabrook TJ, Hay JB. Intracerebroventricular infusions of TNF-alpha preferentially recruit blood lymphocytes and induce a perivascular leukocyte infiltrate. J Neuroimmunol. 2001;113:81–88. doi: 10.1016/s0165-5728(00)00429-x. [DOI] [PubMed] [Google Scholar]
- Shimura K, Wang Z, Matsumoto H, Kasai K. Synthetic oligopeptides as isoelectric point markers for capillary isoelectric focusing with ultraviolet absorption detection. Electrophoresis. 2000;21:603–610. doi: 10.1002/(SICI)1522-2683(20000201)21:3<603::AID-ELPS603>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Sibbitt WL, Haseler LJ, Griffey RH, Hart BL, Sibbitt RR, Matwiyoff NA. Analysis of cerebral structural changes in systemic lupus erythematosus by proton MR spectroscopy. AJNR Am J Neuroradiol. 1994;15:923–928. [PMC free article] [PubMed] [Google Scholar]
- Sidor M, Sakic B, Malinowski P, Ballok DA, Oleschuk CJ, Macri J. Elevated immunoglobulin levels in the cerebrospinal fluid from lupus-prone mice. J Neuroimmunol. 2005;165:104–113. doi: 10.1016/j.jneuroim.2005.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silber E, Semra YK, Gregson NA, Sharief MK. Patients with progressive multiple sclerosis have elevated antibodies to neurofilament subunit. Neurology. 2002;58:1372–1381. doi: 10.1212/wnl.58.9.1372. [DOI] [PubMed] [Google Scholar]
- Silva SC, Correia C, Fesel C, Barreto M, Coutinho AM, Marques C, Miguel TS, Ataide A, Bento C, Borges L, Oliveira G, Vicente AM. Autoantibody repertoires to brain tissue in autism nuclear families. J Neuroimmunol. 2004;152:176–182. doi: 10.1016/j.jneuroim.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Singh VK, Rivas WH. Prevalence of serum antibodies to caudate nucleus in autistic children. Neurosci Lett. 2004;355:53–56. doi: 10.1016/j.neulet.2003.10.026. [DOI] [PubMed] [Google Scholar]
- Singh VK, Lin SX, Yang VC. Serological association of measles virus and human herpesvirus-6 with brain autoantibodies in autism. Clin Immunol Immunopathol. 1998;89:105–108. doi: 10.1006/clin.1998.4588. [DOI] [PubMed] [Google Scholar]
- Singh VK, Lin SX, Newell E, Nelson C. Abnormal measles–mumps–rubella antibodies and CNS autoimmunity in children with autism. J Biomed Sci. 2002;9:359–364. doi: 10.1007/BF02256592. [DOI] [PubMed] [Google Scholar]
- Skeie GO, Mygland A, Treves S, Gilhus NE, Aarli JA, Zorzato F. Ryanodine receptor antibodies in myasthenia gravis: epitope mapping and effect on calcium release in vitro. Muscle Nerve. 2003;27:81–89. doi: 10.1002/mus.10294. [DOI] [PubMed] [Google Scholar]
- Snyder EY, Deitcher DL, Walsh C, Arnold-Aldea S, Hartwieg EA, Cepko CL. Multipotent neural cell lines can engraft and participate in development of mouse cerebellum. Cell. 1992;68:33–51. doi: 10.1016/0092-8674(92)90204-p. [DOI] [PubMed] [Google Scholar]
- Steens SC, dmiraal-Behloul F, Bosma GP, Steup-Beekman GM, Olofsen H, Le CS, Huizinga TW, van Buchem MA. Selective gray matter damage in neuropsychiatric lupus. Arthritis Rheum. 2004;50:2877–2881. doi: 10.1002/art.20654. [DOI] [PubMed] [Google Scholar]
- Svenungsson E, Andersson M, Brundin L, van Vollenhoven R, Khademi M, Tarkowski A, Greitz D, Dahlstrom M, Lundberg I, Klareskog L, Olsson T. Increased levels of proinflammatory cytokines and nitric oxide metabolites in neuropsychiatric lupus erythematosus. Ann Rheum Dis. 2001;60:372–379. doi: 10.1136/ard.60.4.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeten TL, Bowyer SL, Posey DJ, Halberstadt GM, McDougle CJ. Increased prevalence of familial autoimmunity in probands with pervasive developmental disorders. Pediatrics. 2003;112:e420-. doi: 10.1542/peds.112.5.e420. [DOI] [PubMed] [Google Scholar]
- Theofilopoulos AN. Murine models of lupus. In: Lahita RG, editor. Systemic Lupus Erythematosus. Churchill Livingstone; New York: 1992. pp. 121–194. [Google Scholar]
- Todd RD, Hickok JM, Anderson GM, Cohen DJ. Antibrain antibodies in infantile autism. Biol Psychiatry. 1988;23:644–647. doi: 10.1016/0006-3223(88)90012-1. [DOI] [PubMed] [Google Scholar]
- Tomita M, Holman BJ, Santoro TJ. Aberrant cytokine gene expression in the hippocampus in murine systemic lupus erythematosus. Neurosci Lett. 2001;302:129–132. doi: 10.1016/s0304-3940(01)01679-2. [DOI] [PubMed] [Google Scholar]
- Trysberg E, Carlsten H, Tarkowski A. Intrathecal cytokines in systemic lupus erythematosus with central nervous system involvement. Lupus. 2000;9:498–503. doi: 10.1177/096120330000900704. [DOI] [PubMed] [Google Scholar]
- Trysberg E, Nylen K, Rosengren LE, Tarkowski A. Neuronal and astrocytic damage in systemic lupus erythematosus patients with central nervous system involvement. Arthritis Rheum. 2003;48:2881–2887. doi: 10.1002/art.11279. [DOI] [PubMed] [Google Scholar]
- Tsai CY, Wu TH, Tsai ST, Chen KH, Thajeb P, Lin WM, Yu HS, Yu CL. Cerebrospinal fluid interleukin-6, prostaglandin E2 and autoantibodies in patients with neuropsychiatric systemic lupus erythematosus and central nervous system infections. Scand J Rheumatol. 1994;23:57–63. doi: 10.3109/03009749409103028. [DOI] [PubMed] [Google Scholar]
- Tsai CY, Wu TH, Huang SF, Sun KH, Hsieh SC, Han SH, Yu HS, Yu CL. Abnormal splenic and thymic IL-4 and TNF-alpha expression in MRL-lpr/lpr mice. Scand J Immunol. 1995;41:157–163. doi: 10.1111/j.1365-3083.1995.tb03548.x. [DOI] [PubMed] [Google Scholar]
- van Adel BA, Kostic C, Deglon N, Ball AK, Arsenijevic Y. Delivery of ciliary neurotrophic factor via lentiviral-mediated transfer protects axotomized retinal ganglion cells for an extended period of time. Hum Gene Ther. 2003;14:103–115. doi: 10.1089/104303403321070801. [DOI] [PubMed] [Google Scholar]
- Vincent A, Lily O, Palace J. Pathogenic autoantibodies to neuronal proteins in neurological disorders. J Neuroimmunol. 1999;100:169–180. doi: 10.1016/s0165-5728(99)00210-6. [DOI] [PubMed] [Google Scholar]
- Vincent A, Dalton P, Clover L, Palace J, Lang B. Antibodies to neuronal targets in neurological and psychiatric diseases. Ann N Y Acad Sci. 2003;992:48–55. doi: 10.1111/j.1749-6632.2003.tb03137.x. [DOI] [PubMed] [Google Scholar]
- Vogelweid CM, Johnson GC, Besch-Williford CL, Basler J, Walker SE. Inflammatory central nervous system disease in lupus-prone MRL/lpr mice: comparative histologic and immunohistochemical findings. J Neuroimmunol. 1991;35:89–99. doi: 10.1016/0165-5728(91)90164-3. [DOI] [PubMed] [Google Scholar]
- Vogelweid CM, Wright DC, Johnson JC, Hewett JE, Walker SE. Evaluation of memory, learning ability, and clinical neurologic function in pathogen-free mice with systemic lupus erythematosus. Arthritis Rheum. 1994;37:889–897. doi: 10.1002/art.1780370617. [DOI] [PubMed] [Google Scholar]
- Wagner J, Akerud P, Castro DS, Holm PC, Canals JM, Snyder EY, Perlmann T, Arenas E. Induction of a midbrain dopaminergic phenotype in Nurr1-overexpressing neural stem cells by type 1 astrocytes. Nat Biotechnol. 1999;17:653–659. doi: 10.1038/10862. [DOI] [PubMed] [Google Scholar]
- Weiss S, Dunne C, Hewson J, Wohl C, Wheatley M, Peterson AC, Reynolds BA. Multipotent CNS stem cells are present in the adult mammalian spinal cord and ventricular neuroaxis. J Neurosci. 1996;16:7599–7609. doi: 10.1523/JNEUROSCI.16-23-07599.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West SG. Lupus and the central nervous system. Curr Opin Rheumatol. 1996;8:408–414. doi: 10.1097/00002281-199609000-00004. [DOI] [PubMed] [Google Scholar]
- Wick G, Hu Y, Schwarz S, Kroemer G. Immunoendocrine communication via the hypothalamo–pituitary–adrenal axis in auto-immune diseases. Endocr Rev. 1993;14:539–563. doi: 10.1210/edrv-14-5-539. [DOI] [PubMed] [Google Scholar]
- Winfield JB. Cryoglobulinemia. Hum Pathol. 1983;14:350–354. doi: 10.1016/s0046-8177(83)80121-x. [DOI] [PubMed] [Google Scholar]
- Zameer A, Hoffman SA. Increased ICAM-1 and VCAM-1 expression in the brains of autoimmune mice. J Neuroimmunol. 2003;142:67–74. doi: 10.1016/s0165-5728(03)00262-5. [DOI] [PubMed] [Google Scholar]
- Zameer A, Hoffman SA. B and T cells in the brains of autoimmune mice. J Neuroimmunol. 2004;146:133–139. doi: 10.1016/j.jneuroim.2003.10.052. [DOI] [PubMed] [Google Scholar]