

Abstract
Many important enzyme activities are regulated by Ca2+-dependent interactions with calmodulin (CaM). Some of the most important targets for CaM action are in the nucleus, and Ca2+-dependent CaM translocation into this organelle has been reported. Hormone-evoked cytosolic Ca2+ signals occur physiologically as oscillations, but, so far, oscillations in CaM concentration have not been described. We loaded fluorescent-labeled CaM into pancreatic acinar cells and monitored the fluorescence in various regions by confocal microscopy. Sustained high concentrations of the hormone cholecystokinin or the neurotransmitter acetylcholine evoked a transient movement of cytosolic CaM from the basal nonnuclear area into the secretory granule region and, thereafter, a more substantial and prolonged translocation of CaM into the nucleoplasm. About 50% of the CaM that bound Ca2+ translocated. At a lower hormone concentration, evoking Ca2+ oscillations, regular spikes of increased CaM concentration were seen in the secretory granule region with mirror image spikes of decreased CaM concentration in the basal nonnuclear region. The nucleus was able to integrate the Ca2+ spike-evoked pulses of CaM translocation into a sustained elevation of the nucleoplasmic concentration of this protein.
Calmodulin (CaM) is a ubiquitous and abundant Ca2+-binding protein that is involved in the control of many cellular processes, and many important enzymes are regulated directly by Ca2+-dependent interactions with CaM (1–5). Ca2+ binding to CaM results in a conformational change, exposing hydrophobic sites that can interact with binding partners (6–8). There has been particular interest in Ca2+-dependent regulation of gene expression via CaM kinase-triggered phosphorylation of the transcription factor cyclic AMP response element-binding protein (CREB) (9–15). CaM may act both as a mobile Ca2+ buffer and Ca2+ sensor. Translocation of CaM determines availability of the protein for binding to targets at specific cellular locations, and translocation of CaM into the nucleus can occur under different conditions (16–18). Recently, Ca2+-mediated movement of CaM into the nucleus has been demonstrated in several systems (19, 20).
The pancreatic acinar cell is a particularly interesting system in which to investigate Ca2+-induced CaM translocation. This cell is structurally polarized with an apical part dominated by secretory (zymogen) granules (SG) and a basal part containing the nucleus surrounded by endoplasmic reticulum (ER). Secretion (exocytosis) occurs exclusively across the apical membrane (21). Agonist-evoked cytosolic Ca2+ signals are initiated in the apical pole, and, depending on stimulus strength and duration, they can be oscillating (low-agonist concentration) or sustained (high-agonist concentration) and can be concentrated in the apical pole (local signals) or spread throughout the cell including the nucleus (global signals) (22). The Ca2+ signals primarily are due to release from the ER. The ER functions as a unit specialized for Ca2+ uptake via Ca2+ ATPase pumps in the basal part and release via inositol 1,4,5-trisphosphate or cyclic ADP ribose receptors in the fine terminals invading the granule-rich apical pole. The ER acts as a tunnel, absorbing Ca2+ entering the basal pole through store-operated Ca2+ channels and releasing Ca2+ in the apical pole from where the signal, depending on agonist concentration and duration of stimulation, can or cannot spread to the rest of the cell (22–25).
Isolated pancreatic acinar cells remain polarized (24). We have taken advantage of this by monitoring the movement of fluorescent-labeled CaM loaded into acinar cells via a patch clamp pipette (whole-cell configuration). Stimulation with the neurotransmitter acetylcholine (ACh), the circulating hormone cholecystokinin (CCK), or a Ca2+ ionophore (ionomycin) evoked translocation of CaM into the apical pole and the nucleus. Translocation into the apical granule-rich area occurred relatively quickly, but as soon as the cytosolic Ca2+ concentration ([Ca2+]i) declined, CaM left the apical pole to move back into the basal pole. Translocation into the nucleus, on the other hand, usually occurred after a delay of more than 30 s, but CaM that had entered the nucleoplasm was able to leave the nucleus only very slowly. The result of these different characteristics of CaM movement is that short-lasting stimuli favor CaM translocation to the apical pole, whereas prolonged stimulation allows substantial CaM accumulation in the nucleus. At a CCK concentration evoking regular [Ca2+]i oscillations, there were regular CaM oscillations in the apical pole with mirror-like oscillations in the basal nonnuclear region, whereas the nucleus was able to integrate the pulsatile CaM movements into the nucleoplasm into a sustained rise in its CaM concentration. Our results indicate that only oscillating Ca2+ signals can evoke a sustained rise in the nuclear CaM concentration.
MATERIALS AND METHODS
Cell Isolation and Solutions.
Isolated pancreatic acinar cells and small clusters of pancreatic acinar cells (two or three cells) were obtained from isolated mouse pancreata by collagenase treatment as described previously (22, 26). The extracellular solution used in our experiments contained 140 mM NaCl/4.7 mM KCl/1.13 mM MgCl2/1 mM CaCl2/10 mM glucose/10 mM Hepes, pH 7.2. Secretagogues (ACh or CCK) or ionomycin was added to this solution to achieve the concentrations stated for particular series of experiments. The “intracellular” patch pipette solution contained 140 mM KCl/1.13 mM MgCl2/10 mM glucose/0.1 mM EGTA/1 mM ATP/10 mM Hepes, pH 7.2. All experiments were performed at room temperature. 2-Chloro-(ɛ-amino-Lys75)-(6-(4-N,N-diethylamino-phenyl)-1,3,5-triazin-4-yl)-CaM (TA-CaM) and 5-(4,6-dichlorotriazinyl)-amino-fluorescein-CaM (DTAF-CaM) were produced by K.T. (27, 28).
Cell Loading with Fluorescent Calmodulin.
TA-CaM or DTAF-CaM was loaded into the cells by using a patch clamp pipette. TA-CaM and DTAF-CaM were dissolved in “intracellular” solution to achieve concentrations of 100 μM and 50 or 100 μM, respectively. DTAF-CaM or TA-CaM was loaded into the tip of a glass pipette, which then was backfilled with the normal “intracellular” solution. The pipette was positioned on the basal part of the cell, a gigaseal was obtained, and the membrane under the pipette was destroyed by suction to achieve the patch clamp whole-cell recording configuration. The pipette was left in place for 3–5 min to allow diffusion of the chosen substance into the cytosol. To avoid continuous flux of labeled CaM into the cell (which could mask the redistribution process), the pipette was withdrawn before the start of confocal measurements in each experiment. The concentration of DTAF-CaM inside the cell was estimated by comparing the intensity of fluorescence recorded from the cells with the intensity of fluorescence recorded from small droplets of “intracellular” solution with a calibrated concentration of DTAF-CaM (50 μM). The estimated intracellular concentrations were in the range of 20–30 μM, which is of the same order of magnitude as the estimated normal CaM concentration in rat pancreas (29). Correction for bleaching was done by using parts of traces obtained before agonist application. The specificity of CaM translocation was tested in experiments with fluorescein-labeled dextrans (Mr 10,000). Cells were loaded with fluorescent dextrans via a patch pipette and stimulated with 1 nM CCK. In eight experiments of this type we found no indications of translocation.
Confocal Microscopy and Video Imaging.
Fluorescent images were obtained by using a laser-scanning confocal microscope, ODYSSEY (Noran Instruments, Middleton, WI), with objective ×40, n.a. 1.3 oil (Nikon). For DTAF-CaM, the excitation and emission wavelengths were 488 nm and above 515 nm, respectively, whereas for TA-CaM they were 364 nm and above 400 nm, respectively. A linear color scale was used to color code the intensity of fluorescence of different cellular compartments.
[Ca2+]i measurements were made in separate experiments by using video imaging (QuantiCell; Applied Imaging, Sunderland, U.K.) and Fura-2 as a calcium indicator, as described previously (24).
Calculation of Ca2+-Calmodulin Binding.
The proportion of CaM that binds Ca2+ as a result of agonist application was estimated by using the following formula:
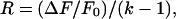 |
where R is the ratio of the concentration of TA-CaM that binds Ca2+ as a result of stimulation and the total concentration of TA-CaM in the cell. ΔF represents the change in intensity of the whole-cell fluorescence as a result of agonist application, and Fo denotes the initial intensity of fluorescence. k represents the ratio of the maximal intensity of fluorescence of TA-CaM (Ca2+ bound form) to the minimal intensity of fluorescence (Ca2+ free-form). For our experiments the value of k was 2.3. The spectra of TA-CaM (both Ca2+ free and Ca2+ bound) are quite broad, but there is a slight shift of the maximal excitation wavelength for TA-CaM upon Ca2+ binding (27). The effect of this spectral shift on the amplitude of the signal is, however, quite small at an excitation wavelength of 364 nm. We estimate that at 364 nm the spectral shift could account for a 4.7% change in the amplitude of the fluorescence intensity. As a result, the proportion of Ta-CaM that binds Ca2+ could be overestimated by approximately 1%.
Cellular Localization of the Nucleus.
At the end of the experiments, nuclear staining with Hoechst 33342 was used to verify the position of nuclei. The fluorescence of Hoechst 33342 was excited by using the laser line at 364 nm; emission was recorded at wavelengths above 400 nm. Nuclear staining was performed by short (2- to 3-min) incubation of cells with Hoechst 33342. The Hoechst 33342 concentration in the extracellular solution was 50 μM. Hoechst 33342 and ionomycin were purchased from Molecular Probes, collagenase was purchased from Worthington, and other reagents were obtained from Sigma.
Detection of Endogenous Calmodulin by Immunocytochemistry.
Cells in suspension were aliquoted onto poly-l-lysine-coated glass coverslips in a multiwell plate and were immersed in the standard extracellular solution without or with 1 nM CCK for 150 s. These solutions then were replaced with cold (4°C) fixative [4% paraformaldehyde (Sigma) dissolved in 0.1 M PBS (0.1 M phosphate buffer containing 0.9% NaCl [wt/vol], pH 7.4) for 5 min]. After 2 × 10-min washes in PBS, cells were permeabilized with 0.1% Triton X-100 (octyl phenoxy polyethoxyethanol dissolved in PBS; Sigma) for 5 min. After washing in PBS, coverslips then were pretreated with PBS containing 2% BSA and 10% normal goat serum to prevent nonspecific binding of antibodies. Mouse monoclonal anticalmodulin (clone 6D4; Sigma), used alone or in combination with a second monoclonal anticalmodulin (TCS Biologicals, Buckingham, U.K.), was diluted with PBS containing 2% BSA and 0.25% sodium azide (final dilution, 1:40). Stimulated and unstimulated cells were incubated in the antibody solution overnight at 4°C; control cells were incubated in diluting medium only. Finally, all cells were incubated in a solution containing a secondary antibody, Cy3-conjugated donkey anti-mouse IgG (1:50 dilution, Jackson ImmunoResearch), for 1 hr at room temperature and then rinsed in distilled water before being placed on slides in a droplet of Vectashield mounting medium (Vector Laboratories). Cells were examined and photographed on a fluorescent microscope (Leitz Dialux 20, Orthomat camera; Leitz), by using Kodak Ektachrome 35-mm color film. Positives were scanned into Adobe photoshop to obtain measurements of relative fluorescence intensity in the nuclear area compared with that in the whole cell. Only cells with sharp boundaries and a well defined nucleus in the transmitted light image were considered.
RESULTS
Calmodulin Translocation Evoked by Cholecystokinin.
Acutely isolated pancreatic acinar cells retain their polarity, and in nonconfocal transmitted light images the secretory granule region can be seen (Fig. 1) (24, 30). The position of the nucleus can be verified by using the membrane-permeable nuclear stain Hoechst 33342 (30). Fig. 1 shows the effect of a supramaximal CCK concentration on the movement of fluorescent DTAF-CaM, which does not change its fluorescence when Ca2+ binds (31), introduced into one of the cells of a doublet via a patch pipette (patch clamp whole-cell configuration). DTAF-CaM fluorescence was monitored in the basal nonnuclear region (Fig. 1, green trace), in the nucleus (Fig. 1, blue trace), and in the apical SG region (Fig. 1, red trace). CCK evoked a steep rise in the fluorescence from the SG region followed by a more substantial but slower increase in the nucleus (n = 14). The DTAF-CaM concentration in the basal nonnuclear region decreased. In all experiments, the total averaged cellular fluorescence (when corrected for bleaching) remained unchanged, indicating that the local changes are caused by redistribution. The CCK-induced increase of nuclear fluorescence was 11.0 ± 1.5% (SE; n = 14), and the increase in the SG region was 8.4 ± 1.1% (n = 14). The CCK-evoked decrease of fluorescence in the basal nonnuclear region was 12.3 ± 1.9% (n = 14). CCK evokes a movement of CaM from the cytosol in the basal nonnuclear region to mainly the nucleus but, also and more transiently, to the cytosol in the SG region. The peak of the fluorescence intensity in the SG region occurred after 44 ± 12 s (n = 14), whereas the nuclear maximum was reached 168 ± 24 s (n = 14) after start of CCK stimulation. In 9 of 14 cells stimulated with CCK, the nuclear response was delayed as compared with the change in the SG region. Before the stimulation and at the end of the recovery process, the fluorescence of DTAF-CaM in the nucleus usually was somewhat higher (20–40%) than in other parts of the cell. This has been observed previously for the distribution of other fluorescent probes (30) and probably results from the absence of organelles in the nucleoplasm. In contrast, the basal nonnuclear region is densely packed with ER (which effectively decreases the “free cytosol” in this region).
Figure 1.

CaM undergoes intracellular redistribution after application of CCK. The presence of CCK in the extracellular solution is indicated by the bar. (A) Relative changes in fluorescence in several regions of interest. The CCK-evoked rapid transient increase in the fluorescence signal from the SG region (red trace) is followed by a delayed, but stronger, signal from the nucleus (blue trace). These effects are accompanied by a corresponding decrease in the fluorescence intensity from the basal nonnuclear area (green trace). The traces were corrected for bleaching. Arrowheads at the top of the graph show the time points at which the five confocal images (b– f) in B were taken. (B, a) Nonconfocal-transmitted light picture of the pancreatic acinar doublet selected. DTAF-CaM was loaded into the cell on the left via a patch clamp pipette. The boxes over the nucleus (blue), SG region (red), and basal nonnuclear area (green) show where the fluorescence intensity changes were measured. (Bar = 10 μm.) (B, b– f) Color-coded images of fluorescence intensity (linear scale). (b) Before application of CCK. Note that the injected cell is stained brightly with DTAF-CaM, whereas the neighbor contains no fluorescent dye. DTAF-CaM is present throughout the cytosol, but is more concentrated in the area corresponding to the nucleus. (c– f) Series of images revealing the intracellular redistribution of DTAF-CaM evoked by CCK. (c) Before the peak response to CCK, DTAF-CaM appears to be accumulating initially in the SG region (increased intensity of yellow and red in this area as compared with b). (d) At the peak of the response the nuclear area is bright red, indicating a substantial accumulation of DTAF-CaM in this region. Note that the nonnuclear basal region is blue rather than green, indicating a low CaM concentration. (e) The fluorescence signal from the SG region is now reduced significantly (compared with d). (f) The distribution of fluorescence is similar to what was observed before stimulation (b). The overall intensity is somewhat lower than in b because of bleaching.
We also tried to resolve the relatively slow nuclear translocation of CaM by using immunostaining. In all cells stained with antibodies against CaM, the protein was present throughout the cytosol and was particularly concentrated in the apical region. In the majority of unstimulated cells the degree of CaM labeling of the nuclear region was close to that of the neighboring basal regions of the cytosol (Fig. 2A). The brightness of the nuclear region divided by the averaged brightness of the whole cell was 0.98 ± 0.03 (n = 52). Stimulation with CCK (1 nM) resulted in a considerable increase in nuclear CaM staining (Fig. 2B). In this situation the ratio of nuclear to whole-cell brightness was 1.9 ± 0.2 (n = 27). Control cells that were incubated with a secondary antibody, but not anti-CaM antibodies, showed only a vague hint of uniform background staining because of the Cy3 marker (see Materials and Methods). These results confirm that CCK can evoke a marked translocation of CaM from the cytosol to the nucleus.
Figure 2.

Immunocytochemical staining of endogenous calmodulin in isolated pancreatic acinar cell. (Bar = 20 μm.) (A) Two unstimulated cells stained for calmodulin by using antibody clone 6D4 from Sigma, followed by a secondary antibody labeled with a Cy3 fluorescent marker. In both cells, calmodulin labeling can be seen throughout the cytosol and SG region. (B) An example of two cells that have been stimulated with 1 nM CCK for 150 s and then incubated in anticalmodulin antibody; the nuclei in both cells are labeled strongly for calmodulin.
Calmodulin Translocation Evoked by Acetylcholine.
Stimulation with ACh (1 μM) resulted in the same pattern of DTAF-CaM redistribution (Fig. 3) (n = 7) as evoked by CCK. The rise and fall of the CaM concentration in the SG region were considerably faster than in the nucleus (Fig. 3). On the time scale relevant to our experiments (Fig. 3), [Ca2+]i appears to rise simultaneously in all three regions of interest, although it is well documented that the response starts in the SG region and then spreads within 0.5–1 s toward the base (32, 33). The [Ca2+]i rise peaked in all three regions within a few seconds, but the CaM concentration rise in the SG region peaked 33 ± 5 s (n = 7) after the start of the [Ca2+]i response. The nuclear CaM rise began only after a delay of 37 ± 7 s (n = 7) and continued to rise while the CaM concentration was declining in the SG region and remained at a reduced level in the basal nonnuclear area. The peak was reached 165 ± 18 s (n = 7) after the Ca2+ signal initiation, and then the CaM concentration in the nucleus declined slowly while the CaM concentration returned to the resting level in the basal nonnuclear area. The [Ca2+]i response to sustained supramaximal ACh stimulation is composed of a major initial transient followed by a smaller, sustained elevation (Fig. 3). The fall in the CaM concentration in the SG region after the peak level followed a time course very similar to the declining phase of the initial [Ca2+]i transient, whereas the nuclear CaM concentration declined much more slowly.
Figure 3.

A rapid, global [Ca2+]i rise triggers CaM redistribution. The ACh stimulation periods are indicated by the bars. (A) Typical [Ca2+]i signal in response to 1 μM ACh (measured by using Fura-2) in three regions of interest: nucleus (blue trace), SG region (red), and basal nonnuclear area (green). (B) ACh-evoked changes of DTAF-CaM fluorescence in the nucleus (blue), SG region (red), and basal nonnuclear area (green). A and B were recorded in separate experiments.
Because the agonist-evoked rise in CaM concentration initially is much steeper in the SG region than in the nucleus, shorter pulses of stimulation should favor CaM translocation into the SG region. Therefore, we tested the effect of short pulses of ACh (1 μM) stimulation (Fig. 4). There was a brief, but clearly resolved transient rise in the CaM concentration in the SG region (n = 11). In 5 of these 11 experiments a small nuclear rise (always smaller than the SG response) was seen.
Figure 4.

Short applications of ACh (indicated by bars) trigger transient rises in CaM concentration in the SG region. In A the corresponding fall in the fluorescence from the basal nonnuclear region is barely resolved, whereas in B there is a clear decrease in the basal nonnuclear CaM concentration during the rise in the SG region. Relative changes of DTAF-CaM fluorescence in the nucleus (blue), SG region (red), and basal nonnuclear area (green) are shown.
Calmodulin Translocation Evoked by Calcium Ionophore.
To bypass plasma membrane receptor activation, we also investigated the effect of the Ca2+ ionophore, ionomycin. At the submaximal concentration (2 μM) used in this study, ionomycin evoked a transient cytosolic Ca2+ signal (n = 4) similar to the ACh or CCK response. The CaM redistribution response to ionomycin was very similar to that described for CCK and ACh (Fig. 5). Initially, ionomycin evoked a rise in the CaM concentration in the SG region followed by a much larger and more prolonged rise in the nucleus (n = 4). The CaM concentration in the basal nonnuclear region declined and returned slowly to the prestimulation level during the phase when the CaM concentration decreased in the nucleus. The response in the SG region reached its peak in 22 ± 8 s (n = 4). As with CCK and ACh stimulation, the nuclear rise was much slower and attained its maximum only after 260 ± 45 s (n = 4). In two of the four experiments with ionomycin, there was a clear delay (13 and 38 s, respectively) in the beginning of the nuclear fluorescence rise as compared with the rise in the SG region.
Figure 5.

The calcium ionophore ionomycin (applied during period indicated by the bar) induces redistribution of CaM. Relative changes of DTAF-CaM fluorescence in nucleus (blue), SG region (red), and basal nonnuclear region (green) are shown.
Oscillations of Intracellular Calmodulin Concentrations.
Using a CCK concentration, which evokes repetitive [Ca2+]i spikes, it was possible to resolve oscillations of CaM concentration with different patterns in the nucleus, the SG region, and the basal nonnuclear area. At 50 pM, CCK evokes a simple pattern of repetitive global [Ca2+]i spikes rising from the basal level, with each transient lasting about 20–50 s (34). Typically, the first spike is two to four times longer than each of the following spikes (34). Fig. 6 shows the effect of 50 pM CCK on [Ca2+]i and on the CaM concentrations in different regions of the cell. The [Ca2+]i and CaM concentration changes were monitored in separate cells. The repetitive [Ca2+]i and CaM spikes in the SG region (Fig. 6) are similar in shape. In contrast, there was a sustained elevation of the nuclear CaM concentration with superimposed oscillations, which were delayed and incomplete compared with those in the SG region. This pattern was clearly different from the pattern of Ca2+ oscillations in the nucleus (Fig. 6). The CaM concentration in the basal nonnuclear region also oscillated, and the pattern that eventually established itself was a mirror image of the oscillations in the SG region (Fig. 6) (n = 7). In the basal part of the cell, spikes of increasing Ca2+ concentration induced spikes of decreasing CaM concentration. The total cellular fluorescence from DTAF-CaM remained unchanged when corrected for bleaching in all the experiments, indicating that the local oscillations are due to redistribution of a constant amount of the protein.
Figure 6.

CCK (50 pM) (periods of stimulation indicated by bars) induces uniform [Ca2+]i oscillations but different types of oscillations of CaM concentration in different regions of pancreatic acinar cells. The point of agonist addition also is shown by the positions of short, vertical bars on the x axes. The [Ca2+]i and CaM concentrations were monitored in separate cells. (A) Calcium oscillations in three regions of interest: nucleus (blue), secretory region (red), and basal nonnuclear region (green). (B) Oscillations of calmodulin concentration (oscillations of intensity of DTAF-CaM fluorescence) in similarly selected regions of interest: nucleus (blue), SG region (red), and basal nonnuclear region (green).
Monitoring the Calcium–CaM Reaction.
To observe the Ca2+-CaM reaction, we employed another form of fluorescently labeled CaM, TA-CaM, a compound that increases its fluorescence upon Ca2+ binding (27, 28). Using supramaximal agonist stimulation [1 μM ACh (n = 6) and 1 nM CCK (n = 11)], the fluorescence of TA-CaM increased. We estimated the proportion of CaM that binds Ca2+ as a result of agonist stimulation by comparing the TA-CaM fluorescence response to the agonist with the change of TA-CaM fluorescence induced by saturating the probe with Ca2+ (see Materials and Methods). We estimate that 24 ± 3% (n = 11) of TA-CaM binds Ca2+ as a result of supramaximal agonist stimulation. If one corrects for the slight spectral shift (see Materials and Methods), this value decreases to 23%. By comparing this result with the results of the DTAF-CaM experiments, we estimate that approximately 50% of the CaM that binds Ca2+ translocates. About 0.01 fmol of CaM per cell, corresponding to a total cellular concentration of 2–3 μM, can be redistributed inside a cell as a result of a maximal agonist-evoked Ca2+ signal. We also tried to estimate the extent of CaM translocation after hormonal stimulation by using antibodies against CaM (Fig. 2). The antibody approach indicates a relatively more substantial redistribution, but in the experiments with fluorescent CaM, we inevitably are increasing the total CaM concentration in the cytosol, and the translocation mechanism may not be capable of moving all the additional CaM.
DISCUSSION
Fig. 7 summarizes our main findings. A global [Ca2+]i rise induces movement of CaM from the nonnuclear basal area into the SG region and more slowly into the nucleus (nucleoplasm). Because of the slow exit of CaM from the nucleoplasm, the nucleus can integrate Ca2+ spike-driven pulses of CaM entry, allowing a steady, incremental build-up of its CaM concentration. In contrast, the exit of CaM from the SG region is relatively fast and the CaM concentration in this part of the cytosol therefore will oscillate in synchrony with the global Ca2+ spikes. The duration of the individual pulses of [Ca2+]i elevation and their frequency will determine the relative magnitudes of CaM movements into the SG granule region and the nucleus. Short pulses favor CaM movements into the SG zone, whereas longer pulses enhance nuclear CaM accumulation.
Figure 7.

Different types of Ca2+ and CaM oscillations induced by CCK in various regions of a pancreatic acinar cell; schematic diagram indicating the hormone-evoked movements of CaM induced by global [Ca2+]i spikes. Application of CCK evokes initially a very long [Ca2+]i transient and, thereafter, repetitive, regular global Ca2+ spikes (34). The first movement of CaM (1) is into the secretory granule region. CaM leaves this region as soon as [Ca2+]i falls (2). There is a delayed but substantial movement of CaM into the nucleus (3). The exit of CaM from the nucleus (4) is very slow, allowing integration of the pulses of CaM influx to give a sustained rise in the nuclear CaM concentration.
The relatively slow rise of the nuclear CaM concentration evoked by agonists and Ca2+ ionophore (Figs. 1–5) probably reflects transfer through the nuclear pore complexes (NPC). Supramaximal agonist stimulation depletes the ER-nuclear envelope store of Ca2+ to about the same extent as total inhibition of the ER Ca2+ ATPase by thapsigargin (25, 35). Inositol 1,4,5-trisphosphate and cyclic ADP ribose release Ca2+ from the nuclear envelope store (36, 37). Such depletion of internal stores inhibits transport of many macromolecules through the NPCs and changes the conformation of these pores (38). Our results indicate that Ca2+ depletion of the ER-nuclear envelope store does not inhibit CaM transfer through the NPC. There are at least two types of macromolecule movements through the NPC: the well characterized, GTP-dependent mode and a GTP-independent, but Ca2+-CaM-dependent, transport. The GTP-dependent transport is inhibited by Ca2+ store depletion, whereas the Ca2+-CaM-dependent transport is activated under such conditions (39). CaM itself may be imported into the nucleoplasm by this Ca2+-CaM-dependent pathway.
A Ca2+ rather than CaM integrating action of the nucleus recently has been described (40). Because of the slow relaxation of nucleoplasmic Ca2+ transients compared with those in the cytoplasm, repetitive perinuclear Ca2+ puffs can be integrated into a “staircase” of increasing nucleoplasmic Ca2+ concentration. These Ca2+-integrating events (40) are faster than those described here for CaM, but also contribute to an enhanced and prolonged Ca2+-CaM reaction. There are many Ca2+-CaM-dependent enzymes in the nucleus (41–43), and the integration of nuclear CaM accumulation described here will potentiate the reactions catalyzed by these enzymes and could have consequences for many important events including gene expression (20, 43, 44). In the case of CaM kinase II there is potential for a further level of integration, because isolated enzyme studies show that the autophosphorylated kinase traps CaM in the presence of Ca2+ for a prolonged period. Each successive Ca2+ spike in a train therefore recruits CaM onto holoenzymes that still retain bound CaM (43). Oscillations in the cytosolic-free Ca2+ level maximize gene expression for a given amount of inositol 1,4,5-trisphosphate (45). Dolmetsch et al. (46) also show the relative importance of Ca2+ oscillations in maximizing gene expression. Integration of Ca2+ spike-driven pulses of CaM influx to the nucleoplasm into a sustained CaM elevation (Fig. 6) may optimize the effect of Ca2+ spikes on Ca2+-CaM-dependent reactions.
The translocation of Ca2+ into the SG region also may have important consequences. The main result of a [Ca2+]i elevation in the SG region is exocytosis (47). CaM is essential for Ca2+-regulated exocytosis in Paramecium (48), and Ca2+/CaM may signal the completion of docking and trigger a late step of vacuole fusion (49). In pancreatic acinar cells, the immunosuppressant cyclosporin A inhibits a Ca2+-CaM-dependent protein phosphatase and exocytotic enzyme secretion (50). A rapid, but transient, translocation of CaM into the SG region (Figs. 1–6) could provide an initial boost to the Ca2+-dependent secretion of digestive enzymes across the apical membrane. It may be important that agonist concentrations evoking repetitive [Ca2+]i spikes induce repetitive pulses of CaM movement into the SG region (Fig. 6).
After an agonist-induced global [Ca2+]i elevation in pancreatic acinar cells, there is polarized Ca2+ transport across the plasma membrane with a particularly intense extrusion, mediated by the CaM-regulated plasma membrane Ca2+-ATPase (51), from the apical secretory granule-containing region (26, 52). The translocation of CaM into the SG region (Figs. 1–6) may be an important factor in stimulating Ca2+ extrusion across the apical membrane.
Acknowledgments
This work was supported by a Medical Research Council Programme Grant to O.H.P. and A.V.T. O.H.P. is a Medical Research Council Research Professor. M.C. is a Wellcome Trust Prize Ph.D. student. T.T. was supported by a Fellowship from The University of Hirosaki, Japan.
ABBREVIATIONS
- CaM
calmodulin
- SG
secretory granules
- ER
endoplasmic reticulum
- CCK
cholecystokinin
- ACh
acetylcholine
- [Ca2+]i
cytosolic Ca2+ concentration
- TA-CaM
2-chloro-(ɛ-amino-Lys75)-(6-(4-N,N-diethylamino-phenyl)-1,3,5-triazin-4-yl)-CaM
- DTAF-CaM
5-(4,6-dichlorotriazinyl)-amino-fluorescein-CaM
References
- 1.Means A R, Tash J S, Chafouleas J G. Physiol Rev. 1982;62:1–39. doi: 10.1152/physrev.1982.62.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Cruzalegui F H, Means A R. J Biol Chem. 1993;268:26171–26178. [PubMed] [Google Scholar]
- 3.Schulman H. Curr Opin Cell Biol. 1993;5:247–253. doi: 10.1016/0955-0674(93)90111-3. [DOI] [PubMed] [Google Scholar]
- 4.Means A R. FEBS Lett. 1994;347:1–4. doi: 10.1016/0014-5793(94)00492-7. [DOI] [PubMed] [Google Scholar]
- 5.Niki I, Yokokura H, Sudo T, Kato M, Hidaka H. J Biochem. 1996;120:685–698. doi: 10.1093/oxfordjournals.jbchem.a021466. [DOI] [PubMed] [Google Scholar]
- 6.Babu Y S, Sack J S, Greenhough T J, Bugg C E, Means A R, Cook W J. Nature (London) 1985;315:37–40. doi: 10.1038/315037a0. [DOI] [PubMed] [Google Scholar]
- 7.Babu Y S, Bugg C E, Cook W J. J Mol Biol. 1988;204:191–204. doi: 10.1016/0022-2836(88)90608-0. [DOI] [PubMed] [Google Scholar]
- 8.Ikura M. Trends Biochem Sci. 1996;21:14–17. [PubMed] [Google Scholar]
- 9.Dash P K, Karl K A, Colicos M A, Prymes R, Kandel E R. Proc Natl Acad Sci USA. 1991;88:5061–5065. doi: 10.1073/pnas.88.11.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frank D A, Greenberg M E. Cell. 1994;79:5–8. doi: 10.1016/0092-8674(94)90394-8. [DOI] [PubMed] [Google Scholar]
- 11.Matthews R P, Guthrie C R, Wailes L M, Zhao X, Means A R, McKnight G S. Mol Cell Biol. 1994;14:6107–6116. doi: 10.1128/mcb.14.9.6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayford M, Wang J, Kandel E R, O’Dell T J. Cell. 1995;81:891–904. doi: 10.1016/0092-8674(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 13.Bito H, Deisseroth K, Tsien R W. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- 14.Bito H, Deisseroth K, Tsien R W. Curr Opin Neurobiol. 1997;7:419–429. doi: 10.1016/s0959-4388(97)80072-4. [DOI] [PubMed] [Google Scholar]
- 15.Finkbeiner S, Greenberg M E. BioEssays. 1997;19:657–660. doi: 10.1002/bies.950190803. [DOI] [PubMed] [Google Scholar]
- 16.Simmen R C M, Dunbar B S, Guerreiro V, Chafouleas J G, Clark J H, Means A R. J Cell Biol. 1984;99:588–593. doi: 10.1083/jcb.99.2.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serratosa J, Pujol M J, Bachs O, Carafoli E. Biochem Biophys Res Commun. 1988;150:1162–1169. doi: 10.1016/0006-291x(88)90751-6. [DOI] [PubMed] [Google Scholar]
- 18.Pruschy M, Ju Y, Spitz L, Carafoli E, Goldfarb D S. J Cell Biol. 1994;127(6):1527–1536. doi: 10.1083/jcb.127.6.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luby-Phelps K, Hori M, Phelps J M, Won D. J Biol Chem. 1995;270:21532–21538. doi: 10.1074/jbc.270.37.21532. [DOI] [PubMed] [Google Scholar]
- 20.Deisseroth K, Heist E K, Tsien R W. Nature (London) 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- 21.Palade G E. Science. 1975;189:347–358. doi: 10.1126/science.1096303. [DOI] [PubMed] [Google Scholar]
- 22.Thorn P, Lawrie A M, Smith P M, Gallacher D V, Petersen O H. Cell. 1993;74:661–668. doi: 10.1016/0092-8674(93)90513-p. [DOI] [PubMed] [Google Scholar]
- 23.Petersen O H, Petersen C C H, Kasai H. Annu Rev Physiol. 1994;56:297–319. doi: 10.1146/annurev.ph.56.030194.001501. [DOI] [PubMed] [Google Scholar]
- 24.Mogami H, Nakano K, Tepikin A V, Petersen O H. Cell. 1997;88:49–55. doi: 10.1016/s0092-8674(00)81857-7. [DOI] [PubMed] [Google Scholar]
- 25.Mogami H, Tepikin A V, Petersen O H. EMBO J. 1998;17:435–442. doi: 10.1093/emboj/17.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belan P, Gerasimenko O, Petersen O H, Tepikin A V. Cell Calcium. 1997;22:5–10. doi: 10.1016/s0143-4160(97)90084-1. [DOI] [PubMed] [Google Scholar]
- 27.Török K, Trentham D R. Biochemistry. 1994;33:12807–12820. doi: 10.1021/bi00209a012. [DOI] [PubMed] [Google Scholar]
- 28.Zimprich F, Török K, Bolsover S R. Cell Calcium. 1995;17:233–238. doi: 10.1016/0143-4160(95)90038-1. [DOI] [PubMed] [Google Scholar]
- 29.Vandermeers A, Vandermeers-Piret M-C, Rathe J, Kutzner R, Delforge A, Christophe J A. Eur J Biochem. 1977;81:379–386. doi: 10.1111/j.1432-1033.1977.tb11962.x. [DOI] [PubMed] [Google Scholar]
- 30.Gerasimenko O V, Gerasimenko J V, Petersen O H, Tepikin A V. Pflügers Arch. 1996;432:1055–1061. doi: 10.1007/s004240050234. [DOI] [PubMed] [Google Scholar]
- 31.Torok K, Wilding M, Groigno L, Patel R, Whitaker M. Current Biol. 1998;8:692–699. doi: 10.1016/s0960-9822(98)70275-1. [DOI] [PubMed] [Google Scholar]
- 32.Kasai H, Augustine G J. Nature (London) 1990;349:735–738. doi: 10.1038/348735a0. [DOI] [PubMed] [Google Scholar]
- 33.Toescu E C, Lawrie A M, Petersen O H, Gallacher D V. EMBO J. 1992;11:1623–1629. doi: 10.1002/j.1460-2075.1992.tb05208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petersen C C H, Toescu E C, Petersen O H. EMBO J. 1991;10:527–533. doi: 10.1002/j.1460-2075.1991.tb07979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hofer A M, Landolfi B, Debellis L, Pozzan T, Curci S. EMBO J. 1998;17:1986–1995. doi: 10.1093/emboj/17.7.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malviya A N, Rogue P, Vincendon G. Proc Natl Acad Sci USA. 1990;87:9270–9274. doi: 10.1073/pnas.87.23.9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerasimenko O V, Gerasimenko J V, Tepikin A V, Petersen O H. Cell. 1995;80:439–444. doi: 10.1016/0092-8674(95)90494-8. [DOI] [PubMed] [Google Scholar]
- 38.Lee M A, Dunn R C, Clapham D E, Stehno-Bittel L. Cell Calcium. 1998;23:91–101. doi: 10.1016/s0143-4160(98)90107-5. [DOI] [PubMed] [Google Scholar]
- 39.Sweitzer T D, Hanover J A. Proc Natl Acad Sci USA. 1996;93:14574–14579. doi: 10.1073/pnas.93.25.14574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lipp P, Thomas D, Berridge M J, Bootman M D. EMBO J. 1997;16:7166–7173. doi: 10.1093/emboj/16.23.7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Agell N, Aligue R, Alemany V, Castro A, Jaime M, Jesus-Pojol M, Rius E, Serratosa J, Taules M, Bachs O. Cell Calcium. 1998;23:115–121. doi: 10.1016/s0143-4160(98)90109-9. [DOI] [PubMed] [Google Scholar]
- 42.Bito H. Cell Calcium. 1998;23:143–150. doi: 10.1016/s0143-4160(98)90113-0. [DOI] [PubMed] [Google Scholar]
- 43.Heist E K, Schulman H. Cell Calcium. 1998;23:103–114. doi: 10.1016/s0143-4160(98)90108-7. [DOI] [PubMed] [Google Scholar]
- 44.Malviya A N, Rogue P J. Cell. 1998;92:17–23. doi: 10.1016/s0092-8674(00)80895-8. [DOI] [PubMed] [Google Scholar]
- 45.Li W, Llopis J, Whitney M, Zlokarnik G, Tsien R Y. Nature (London) 1998;392:936–941. doi: 10.1038/31965. [DOI] [PubMed] [Google Scholar]
- 46.Dolmetsch R E, Xu K, Lewis R S. Nature (London) 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- 47.Maruyama Y, Inooka G, Li Y X, Miyashita Y, Kasai H. EMBO J. 1993;12:3017–3022. doi: 10.1002/j.1460-2075.1993.tb05970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kerboeuf D, LeBerre A, Dedieu J-C, Cohen J. EMBO J. 1993;12:3385–3390. doi: 10.1002/j.1460-2075.1993.tb06012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peters C, Mayer A. Nature (London) 1998;396:575–580. doi: 10.1038/25133. [DOI] [PubMed] [Google Scholar]
- 50.Groblewski G E, Wagner A C C, Williams J A. J Biol Chem. 1994;269:15111–15117. [PubMed] [Google Scholar]
- 51.Carafoli E. J Biol Chem. 1992;267:2115–2118. [PubMed] [Google Scholar]
- 52.Belan P V, Gerasimenko O V, Tepikin A V, Petersen O H. J Biol Chem. 1996;271:7615–7619. doi: 10.1074/jbc.271.13.7615. [DOI] [PubMed] [Google Scholar]