

Abstract
We found that interleukin (IL)–1β levels were elevated in the lungs of mice infected with Histoplasma capsulatum. Hence, we examined the influence that IL-1β and IL-1 signaling has on host defenses against pulmonary histoplasmosis. In IL-1 receptor 1 knockout (IL-1R−/−) mice challenged intratracheally, fungal recovery on day 7 after infection exceeded that in wild-type (WT) mice. Antibody neutralization of IL-1β also exacerbated infection. For both groups of mice, the absence of bioactive cytokine led to a failure to control infection in a high proportion of mice. The absence of signaling had a modest effect on host resistance in mice with secondary histoplasmosis. Several perturbations in host defense mechanisms were detected in the lungs of IL-1R−/− mice. The number of CD4+ cells was decreased, and transcription of the gene for inducible nitric oxide synthase was depressed transiently. IL-4 and IL-10 levels were elevated in the lungs of IL-1R−/− mice, compared with those in the lungs of WT mice. Conversely, interferon-γ levels were decreased. Thus, IL-1 contributes to host resistance to infection with H. capsulatum.
Infection with Histoplasma capsulatum is common in the midwestern and southeastern parts of the United States. Most infected individuals do not seek medical care, either because they are asymptomatic or because they have only a mild, influenza-like illness. After an individual is exposed by inhalation, the organism undergoes intracellular replication within monocytes and macrophages in the lungs until cellular immunity is activated to arrest growth. The failure of the host to restrict replication of the fungus leads to a progressive infection in which multiple visceral organs and the bone marrow are involved [1]. Progressive infection may develop after either reinfection or reactivation. The continued multiplication of the fungus represents a failure of the integrity of the host immune system. The cells of the adaptive immune system that are largely responsible for protective immunity after initial contact with the fungus are CD4+ cells [2-4]. In reexposure histoplasmosis, either CD4+ cells or CD8+ cells are sufficient to protect against a challenge [5]. The cytokines that promote protective immunity are interleukin (IL)–12, interferon (IFN)–γ, tumor necrosis factor (TNF)–α, and granulocyte-macrophage colony-stimulating factor (GM-CSF) [6-12]. Conversely, IL-4 and IL-10 depress cellular immunity to H. capsulatum [6, 9, 11, 13, 14]. The balance between those cytokines that enhance immunity and those that inhibit immunity largely dictates the outcome of infection.
One of the major issues in understanding the resistance mechanisms against pathogenic microbes is the delineation of the links between the innate and the adaptive immune system. In this regard, IL-1 is a pivotal cytokine that has many functions. This cytokine activates monocytes and macrophages, increases T cell proliferation, and is proinflammatory [15-17]. Because of the central role it plays in immunity, we examined whether this cytokine contributes to the host response to pulmonary histoplasmosis. The data demonstrate that IL-1 is induced in the lungs of mice infected with H. capsulatum and that the inability of mice to signal through the IL-1 receptor (R) and the neutralization of IL-1β are associated with progressive infection.
MATERIALS AND METHODS
Mice
Male C57BL/6 mice and breeding pairs of IL-1R1 knockout (IL-1R−/−) mice were purchased from Jackson Laboratories. The latter were of a C57BL/6 background and were bred at the University of Cincinnati College of Medicine. Only male mice were used in experiments. Mice were housed in isolator cages and were maintained by the Department of Laboratory Animal Medicine, University of Cincinnati, which is accredited by the American Association for Accreditation of Laboratory Animal Medicine. All animal experiments were done in accordance with the Animal Welfare Act guidelines of the National Institutes of Health.
Preparation of H. capsulatum and infection of mice
H. capsulatum yeast cells (strain G217B) were prepared as described elsewhere [9]. To produce infection in naive mice, animals were inoculated intratracheally (int) with varying numbers of H. capsulatum yeast cells in 50 μL of Hanks' balanced salt solution (HBSS). To produce secondary histoplasmosis, mice were inoculated int with 1 × 104 yeast cells in 50 μL of HBSS; 8 weeks later, the mice were rechallenged int with 2 × 106 yeast cells.
Organ culture for H. capsulatum
Recovery of H. capsulatum was performed as described elsewhere [9]. Fungal burden is expressed as the mean ± SE number of colony-forming units (cfu) per whole organ. The limit of detection was 1 × 102 cfu/whole organ.
Isolation of lung leukocytes
Lung leukocytes were isolated as described elsewhere [18]. Homogenates were filtered through a 60-μm nylon mesh (Spectrum Laboratories) and were then washed 3 times with HBSS. Leukocytes were isolated by separation on a 40%–70% Percoll gradient (Pharmacia). Cells were washed 3 times with HBSS before examination.
In vivo neutralization of IL-1β
Groups of mice were given 1 mg of monoclonal antibody (MAb; clone B122 [hamster IgG1]) to IL-1β intraperitoneally 24 h and 1 h before inoculation with 2 × 106 yeast cells int. The MAb, purchased from Pharmingen, has low endotoxicity, is azide free, and neutralizes only IL-1β [19]. Control mice received an equivalent amount of hamster IgG.
Flow-cytometric analysis of cell populations
The phenotype of cells from mouse lungs was determined by incubating lung leukocytes with fluorescein isothiocyanate–conjugated MAbs (BD Biocsciences) to CD4 (clone GK1.5), CD8 (clone 2.43), Ly-6G (Gr-1; clone RB6-8C5), and Mac-3 (clone M3/84). Cells were washed and resuspended in PBS (pH 7.4) containing 2% bovine serum albumin and 0.01% sodium azide, and fluorescence intensity was assessed using a FACScaliber flow cytometer (BD Biosciences). The number of cells was calculated by multiplying the total number of leukocytes by the percentage of cells bearing a specific surface receptor.
RNA isolation and cDNA synthesis
RNA was isolated from mouse lungs by use of Trizol and cDNA that was synthesized as described elsewhere [18].
Real-time quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) for the inducbile NO synthase (iNOS) gene
qRT-PCR for iNOS (also called “NOS2”) was performed using a TaqMan kit and primers from Applied Bio-systems. Samples were analyzed using an ABI Prism 7500 from Applied Biosystems. Transcript quantification was standardized to expression in an uninfected lung. In each experiment, expression of the gene for hypoxanthine phosphoribosyl transferase, a housekeeping gene, was analyzed. These data are expressed as the log10 of relative quantification. The conditions for amplification were as follows: 50°C for 2 min; 95°C for 10 min, followed by 40 cycles of 95°C for 15 s; and 60°C for 1 min.
Measurement of NO levels
The quantity of NO released by lung leukocytes was assessed as described elsewhere [6].
Measurement of cytokine levels
Lung homogenates were prepared as described elsewhere [11], and levels of IL-1β, IL-4, IL-10, IFN-γ, and TNF-α were quantified by ELISA. All reagents were purchased from R&D Systems. The amount of protein in homogenates of lungs from various groups of mice did not differ; therefore, these data are expressed as the quantity of cytokine per milliliter of homogenate.
Assessment of eosinophil number
Lung leukocytes were subjected to cytospin, and slides were stained with Diff-Quik (Fisher Scientific). Five hundred cells were counted for each slide.
Histopathological analysis
Mice were killed, and their lungs were inflated with 10% buffered formalin. Five-micron-thick tissue sections were prepared and stained with hematoxylin-eosin. Histopathological analysis was performed by a comparative pathologist, and slides were read in a blinded manner.
Statistical analysis
Differences between groups were analyzed by analysis of variance (ANOVA). Differences in survival were analyzed by the log rank test.
RESULTS
Induction of IL-1β in the lungs of mice infected with H. capsulatum
We infected C57BL/6 mice with 2 × 106 H. capsulatum yeast cells int and, at serial intervals of infection, measured the quantity of IL-1β in lung homogenates. Infection with this fungus induced a prompt release of IL-1β on day 1 of infection. The quantity of this cytokine peaked on day 7 and then decreased on days 14 and 21 (figure 1). However, the amount of IL-1β in the lungs on day 21 did not decrease to preinfection levels; in fact, the amount in the lungs remained relatively constant on days 14 and 21. The amount of IL-1β in the lungs on each day of infection was significantly higher than that on day 0 (P < .01, ANOVA), but the levels during infection did not differ significantly (P ≥ .05, ANOVA) between each other.
Figure 1.
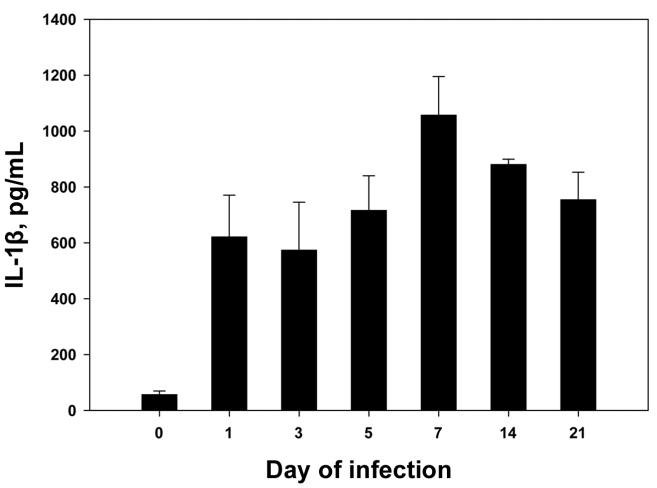
Interleukin (IL)–1β levels in homogenates of lungs from uninfected mice and from mice infected with Histoplasma capsulatum. Mice (n = 5–6/group) were infected with 2 × 106 yeast cells intratracheally, and, at serial intervals of infection, lung homogenates were prepared and analyzed by ELISA for levels of IL-1β. Data are mean ± SE values.
Fungal burden and course of infection in IL-1R−/− and IL-1β–neutralized mice
The finding that levels of IL-1β were elevated in mice after a nonlethal challenge with H. capsulatum stimulated us to investigate whether this cytokine contributes to host defenses against this fungus. Hence, we challenged wild-type (WT) mice and IL-1R−/− mice [20] with 2 × 106 yeast cells int and quantified fungal burden in lungs and spleens on day 7 of infection. The numbers of colony-forming units in the lungs, but not in the spleens, of IL-1R−/− mice were moderately but significantly higher (P < .01, ANOVA) than those in WT mice (figure 2A and 2B). Subsequently, we determined whether the impaired immunity could be observed with smaller inoculum sizes. We challenged WT and IL-1R−/− mice with either 2 × 105 or 1 × 104 yeast cells int and assessed fungal burden in lungs and spleens on day 7 of infection. IL-1R−/− mice challenged with 2 × 105 yeast cells int manifested a pronounced elevation (P < .01, ANOVA) in the numbers of colony-forming units in both lungs and spleens, compared with those in WT mice (figure 2C and 2D). Likewise, IL-1R−/− mice exposed to 1 × 104 yeast cells int also exhibited a statistically significant increase in the numbers of colony-forming units in lungs (P < .05, ANOVA) and spleens (P < .01, ANOVA) (figure 2E and 2F).
Figure 2.
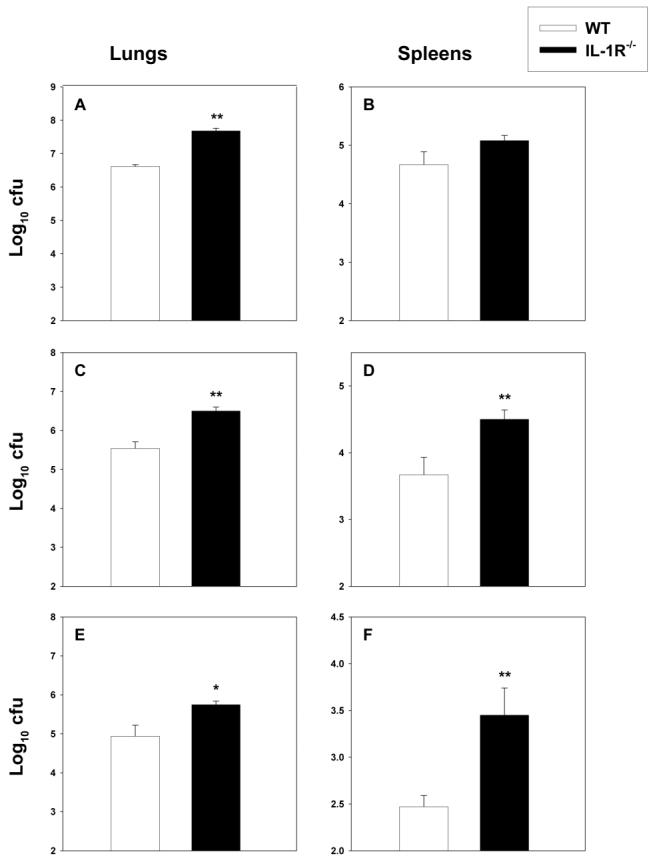
Fungal burden in wild-type (WT) and interleukin-1 receptor 1 knockout (IL-1R−/−) mice infected with Histoplasma capsulatum. Mice (n = 6–8/group) were inoculated with 2 × 106 (A and B), 2 × 105 (C and D), or 1 × 104 (E and F ) yeast cells intratracheally, and fungal recovery in lungs and spleens was assessed on day 7 of infection. Data are mean ± SE values for each group. Results from 1 of 3–4 experiments are shown. *P < .05 and **P < .01 (analysis of variance).
Because the absence of IL-1 signaling impaired the efficiency of fungal clearance, we determined whether the course of infection was altered. As shown in figure 3, only IL-1R−/− mice that received 2 × 106 yeast cells demonstrated an alteration in survival (P < .01, log rank test). Approximately 80% of the IL-1R−/− mice that received this inoculum size died by day 20. In contrast, WT mice and those IL-1R−/− mice that received either 2 × 105 or 1 × 104 yeast cells survived. The WT mice were killed after 40 days of infection, and no H. capsulatum organisms were detected in their lungs or spleens (limit of detection, 1 × 102 cfu/whole organ).
Figure 3.
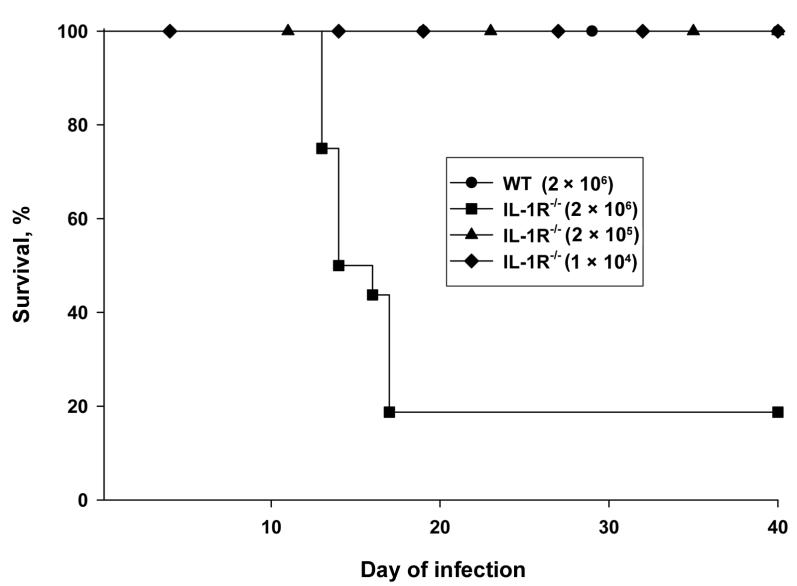
Survival of interleukin-1 receptor 1 knockout (IL-1R−/−) mice infected with Histoplasma capsulatum. IL-1R−/− mice (n = 8–16/group) were inoculated with 2 × 106, 2 × 105, or 1 × 104 yeast cells and monitored for survival. A group of wild-type (WT) mice (n = 8) were inoculated concomitantly with 2 × 106 yeast cells and monitored for survival. Data are pooled from 3 experiments.
To validate the above findings in primary infection, we treated C57BL/6 mice with MAb to IL-1β or with an equal amount of hamster IgG and challenged them with 2 × 106 yeast cells int. Fungal burden and survival were assessed. Neutralization of IL-1β was associated with a higher fungal burden (P < .01, ANOVA) and a marked increase in mortality (P < .01, log rank test) (figure 4A-4C).
Figure 4.
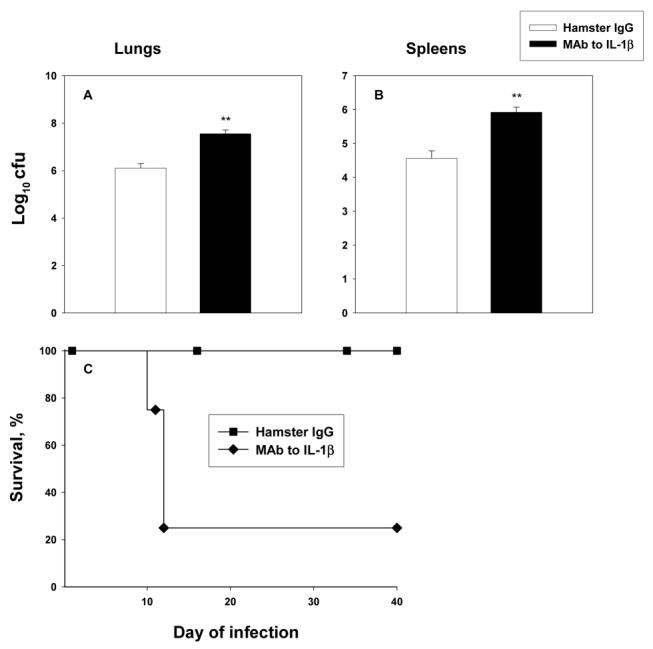
Effect of interleukin (IL)–1β neutralization. Mice (n = 6/group) were given 2 mg of monoclonal antibody (MAb) to IL-1b or an equal amount of hamster IgG and then were inoculated with 2 × 106 yeast cells intratracheally. On day 7 of infection, mice were killed, and fungal burden was determined in lungs (A) and spleens (B). Data are mean ± SE values for each group. Panel C shows survival rates for groups of IL-1β–neutralized mice and control mice (n = 8) monitored for 40 days. **P < .01 (analysis of variance).
We next sought to determine whether IL-1R−/− mice can control secondary infection as well as WT mice. WT and IL-1R−/− mice were inoculated with 1 × 104 yeast cells int and were rechallenged 8 weeks later with 2 × 106 yeast cells. On day 7 of rechallenge, some of the mice were killed, and fungal burden was assessed in lungs and spleens. The numbers of colony-forming units did not differ between the 2 groups of mice on day 7 (figure 5). On day 14, the numbers of colony-forming units in the lungs and spleens of IL-1R−/− mice were demonstrably higher (P < .01, ANOVA) than those in WT mice, but both sets of mice had eliminated the fungus by day 21. Observation of IL-1R−/− mice with secondary histoplasmosis for 40 days did not reveal any evidence of progressive infection—a set of mice killed on day 40 of infection revealed <1 × 102 cfu/whole organ.
Figure 5.
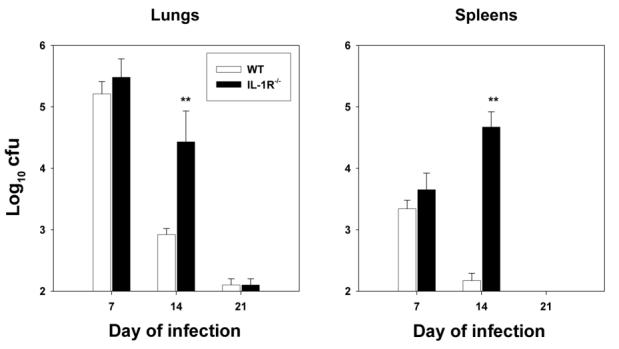
Fungal burden in wild-type (WT) and interleukin-1 receptor 1 knockout (IL-1R−/−) mice with secondary histoplasmosis. Mice (n = 5–6/group) were inoculated with 1 × 104 Histoplasma capsulatum yeast cells intratracheally (int) and were rechallenged 8 weeks later with 2 × 106 yeast cells int. Fungal burden was assessed on days 7, 14, and 21 of infection. Data are mean ± SE values for each group. Results from 1 of 2 experiments are shown. **P < .01 (analysis of variance).
Inflammatory response in IL-1R−/− mice
Because IL-1 is a proinflammatory cytokine, we reasoned that its inability to signal through its receptor might modulate the inflammatory response to this fungus. We inoculated WT and IL-1R−/− mice with 2 × 106 yeast cells and analyzed the numbers of CD4+, CD8+, Gr-1+ (neutrophils), and Mac-3+ (macrophages) cells in lungs and spleen on days 1, 3, and 7 of infection. On day 1, the number of Gr-1+ cells in the lungs, but not in the spleens, of IL-1R−/− mice was significantly less than that in WT mice (P < .01, ANOVA), but the numbers of T cells and Gr-1+ cells were similar in both organs (table 1). By day 3, the number of Gr-1+ cells in the lungs of IL-R−/− mice was still less than that in WT mice, but the difference was insignificant. No differences between the numbers of cells in the spleens of the 2 strains were noted. A moderate but significant decrease (P < .05, ANOVA) in the number of CD4+ cells in the lungs of IL-1R−/− mice was observed on day 7 (table 1). Otherwise, the numbers of CD8+, Gr-1+, and Mac-3+ cells in lungs did not differ between the 2 groups. There were no significant differences (P ≥ .05, ANOVA) in the sizes of these cell populations in spleens between WT and IL-1R−/− mice. The alteration in the size of the lung CD4+ population on day 7 was not observed in IL-1R−/− mice challenged with 2 × 105 yeast cells. The number of CD4+ cells in the lungs of WT mice (mean ± SE, 1.8 × 105 ± 0.3 × 105 cells; n = 4) did not differ from that in IL-1R−/− mice (mean ± SE, 1.6 × 105 ± 0.3 × 105).
Table 1.
Inflammatory response in the lungs of wild-type (WT) and interleukin-1 receptor 1 knockout (IL-1R−/−) mice (n = 6–10/group) infected with Histoplasma capsulatum.
| Day of infection, organ, mouse type | Cell type |
|||
|---|---|---|---|---|
| CD4+ | CD8+ | Gr-1+ | Mac-3+ | |
| Day 1 | ||||
| Lung | ||||
| WT | 0.6 ± 0.2 | 0.4 ± 0.1 | 6.2 ± 1.8 | 1.4 ± 0.2 |
| IL-1R−/− | 0.4 ± 0.2 | 0.3 ± 0.1 | 2.6 ± 0.3a | 1.3 ± 0.1 |
| Spleen | ||||
| WT | 28.4 ± 2.3 | 13.6 ± 0.9 | 8.4 ± 0.5 | 5.9 ± 0.4 |
| IL-1R−/− | 24.6 ± 2.3 | 13.1 ± 1.0 | 8.5 ± 0.2 | 5.4 ± 0.2 |
| Day 3 | ||||
| Lung | ||||
| WT | 2.1 ± 0.4 | 1.7 ± 0.3 | 4.6 ± 0.9 | 0.6 ± 0.1 |
| IL-1R−/− | 2.7 ± 0.6 | 2.1 ± 0.6 | 2.4 ± 0.5 | 0.4 ± 0.1 |
| Spleen | ||||
| WT | 20.9 ± 3.3 | 14.6 ± 1.3 | 12.4 ± 2.3 | 3.3 ± 0.5 |
| IL-1R−/− | 33.6 ± 3.8 | 17.3 ± 1.4 | 14.2 ± 0.6 | 2.6 ± 0.5 |
| Day 7 | ||||
| Lung | ||||
| WT | 7.8 ± 1.1 | 3.5 ± 0.9 | 13.1 ± 2.0 | 1.7 ± 0.4 |
| IL-1R−/− | 3.9 ± 0.8a | 2.5 ± 0.6 | 13.0 ± 2.5 | 2.8 ± 0.8 |
| Spleen | ||||
| WT | 36.0 ± 1.9 | 11.7 ± 1.0 | 13.8 ± 1.6 | 0.7 ± 0.1 |
| IL-1R−/− | 42.7 ± 3.6 | 16.0 ± 1.4 | 16.5 ± 1.8 | 0.3 ± 0.1 |
NOTE. Data are mean ± SE no. (105) of inflammatory cells and are pooled from 2 experiments.
P<.01, compared with the response in WT mice (analysis of variance).
We examined the histopathological appearance of lungs on day 7 of infection in both groups of mice. The lungs of WT mice demonstrated a modest alveolitis that was composed of an admixture of neutrophils and lymphocytes (figure 6A), whereas the lungs of IL-1R−/− mice contained a moderate alveolitis that was composed principally of neutrophils (figure 6B).
Figure 6.

Histopathological analysis of the lungs of Histoplasma capsulatum–infected mice. Lungs from mice were inflated, fixed, and stained with hematoxylin-eosin. Panel A shows a lung from an infected control mouse, and panel B shows a lung from an interleukin-1 receptor 1 knockout (IL-1R−/−) mouse. Original magnification, × 100.
Cytokine profile in the lungs of WT and IL-1R−/− mice
Because IL-1R−/− mice that received 2 × 106 yeast cells failed to control infection, we analyzed lung cytokine levels during the first 7 days of infection, seeking to determine whether the poor immunity was associated with changes in cytokine levels. WT and IL-1R−/− mice were inoculated with 2 × 106 yeast cells and, on days 3, 5, and 7 of infection, the levels of IFN-γ, IL-4, IL-10, and TNF-α were measured. IFN-γ levels did not differ on days 3 and 5 but were decreased (P < .01, ANOVA) in the lungs of IL-1R−/− mice on day 7 (figure 7). IL-4, IL-10, and TNF-α levels were significantly increased (P < .05 to P < .01, ANOVA) in the lungs of IL-1R−/− mice on days 3, 5, and 7 (figure 7). Because the cytokine response was biased to Th2 in IL-1R−/− mice, we examined lung leukocytes for the presence of eosinophils. The lungs of infected WT mice (n = 4) contained <0.2% eosinophils, whereas the lungs of IL-1R−/− mice (n = 4) contained a mean ± SE of 1.4% ± 0.2% (P < .05, ANOVA).
Figure 7.
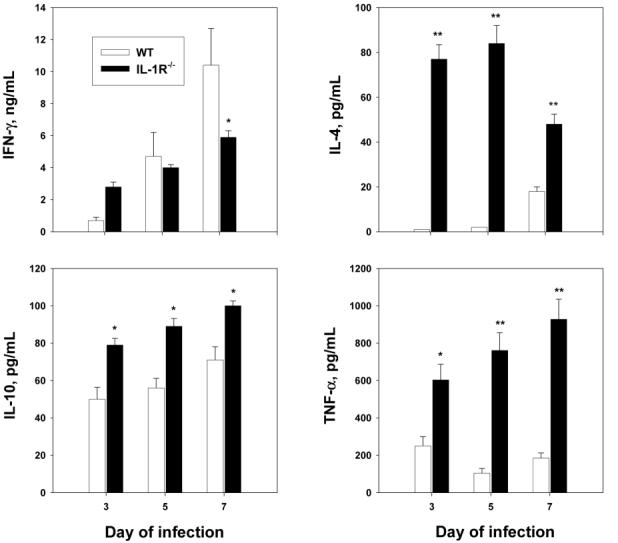
Cytokine levels in homogenates of lungs from wild-type (WT) and interleukin-1 receptor 1 knockout (IL-1R−/−) mice infected with Histoplasma capsulatum. Mice (n = 4–6/group) were infected with 2 × 106 yeast cells, and levels of IL-4, IL-10, interferon (IFN)–γ, and tumor necrosis factor (TNF)–α were assessed in lung homogenates on days 3, 5, and 7 of infection. Data are mean ±SE values for each group. Results from 1 of 2 experiments are shown. *P<.05 and **P<.01 (analysis of variance).
We assessed iNOS induction in both WT and IL-1R−/− mice by qRT-PCR. Synthesis was significantly decreased (P < .05, ANOVA) on days 1 and 3 of infection in IL-1R−/− mice but was similar in both groups of mice on day 5 (table 2). The alterations in iNOS transcription were also apparent in the production of NO by lung leukocytes ex vivo (table 3).
Table 2.
Inducbile NO synthase transcription in wild-type (WT) and interleukin-1 receptor 1 knockout (IL-1R−/−) mice (n = 4/group) infected with Histoplasma capsulatum.
| Day of infection |
||||
|---|---|---|---|---|
| Mouse type | 1 | 3 | 5 | 7 |
| WT | 0.40 ± 0.01 | 1.04 ± 0.09 | 1.90 ± 0.12 | 2.29 ± 0.16 |
| IL-1R−/− | 0.03 ± 0.01a | 0.37 ± 0.02a | 2.37 ± 0.21 | 2.22 ± 0.16 |
NOTE. Data are mean ± SE values and are expressed as the log10 of relative quantification.
P<.05, compared with the response in WT mice (analysis of variance).
Table 3.
NO release in wild-type (WT) and interleukin-1 receptor 1 knockout (IL-1R−/−) mice (n = 5/group) infected with Histoplasma capsulatum.
| Day of infection |
||||
|---|---|---|---|---|
| Mouse type | 1 | 3 | 5 | 7 |
| WT | 20.2 ± 2.6 | 45.6 ± 4.2 | 60.3 ± 5.1 | 68.3 ± 6.2 |
| IL-1R−/− | 7.1 ± 1.1a | 15.3 ± 1.3a | 67.8 ± 5.1 | 69.4 ± 2.5 |
NOTE. Data are mean ± SE NO level and are expressed in micromoles per milliliter.
P<.05, compared with the response in WT mice (analysis of variance).
DISCUSSION
Here, we have demonstrated that IL-1 signaling and IL-1β contribute to the generation of protective immunity against infection with H. capsulatum. This cytokine is produced by many types of cells, including monocytes, macrophages, dendritic cells, and neutrophils [16]. Analysis of the role played by IL-1 in host resistance was prompted by the finding that IL-1β levels were elevated in infected mice. To determine whether this finding had a functional correlate, we examined whether the absence of signaling or in vivo neutralization altered clearance. Impaired fungal elimination was observed in IL-1R−/− mice that received inoculum sizes ranging from 1 × 104 to 2 × 106 yeast cells. However, an inability to control infection among IL-1R−/− mice was observed only in those receiving the highest inoculum size. These data indicate that the susceptibility of IL-1R−/− mice is dependent on the size of the challenge. Thus, a threshold likely exists at which mice can tolerate an infectious inoculum.
Despite the alteration in protective immunity observed during primary infection, the same inoculum size (2 × 106yeast cells) induced only a modest alteration in fungal recovery during secondary histoplasmosis. The alteration in the numbers of colony-forming units was detected only transiently. Hence, in IL-1R−/− mice, the adaptive immune response was acquired and maintained.
The elevated levels of IL-1 observed after pulmonary challenge contrast with those found in mice injected intravenously with yeast cells [21]. In that model, the IL-1 activity of splenocytes was sharply decreased during the acute phase of infection, and the decrement corresponded with an increase in fungal burden. After clearance, the levels were restored to those in control mice. These diametrically opposed findings may have resulted from the differing routes of infection (intravenous vs. intrapulmonary). Alternatively, the opposed findings raise the possibility that the IL-1–producing cells in the lungs may respond to yeast cells differently than do splenocytes.
The influence of endogenous IL-1 as a mediator in innate immunity has been well documented. This cytokine possesses proinflammatory properties and acts as an endogenous pyrogen. Knowledge of the contribution of this cytokine to protective immunity, rather than to just inflammation, has begun to expand. Both the inability to signal through IL-1R and the lack of IL-1 lead to exacerbation of infection with several intracellular pathogens, including Mycobacterium tuberculosis, Leishmania major, and Listeria monocytogenes [22-24]. The pivotal role for IL-1 is not restricted to intracellular pathogens, because recombinant IL-1 has been used to decrease the burden of infection with Klebsiella pneumoniae, Pseudomonas aeruginosa, and Staphylococcus aureus [25, 26]. Thus, the accumulated evidence indicates that either IL-1 or IL-1 signaling is a critical component of host mechanisms of resistance to intracellular as well as extracellular pathogens.
Because IL-1 plays a prominent role in systemic inflammation, we examined the possibility that the failure to control infection was a consequence of impaired migration of cells into the lungs of IL-1R−/− mice. In this regard, only the number of CD4+ cells was significantly decreased in the lungs of mice. Given the ability of IL-1 to enhance inflammation, there was only a selective decrement in the number of CD4+ cells, rather than a global change. The CD4+ cell population is critically important in host resistance to primary infection with H. capsulatum. It is the principal generator of IFN-γ, which is necessary for the resolution of primary infection [2]. A profound deficiency in this cell subset leads to uncontrolled histoplasmosis in mice [2, 5]. In humans, a low CD4+ cell count enhances the risk of reactivation of disease [27]. The moderate reduction in the size of this cell population possibly contributed to the more aggressive nature of the infection in mice that lacked the capacity to signal through IL-1R. It is unlikely, however, that the decrement was sufficiently profound to be the major cause of the inimical course of infection, because infection with 2 × 105 yeast cells was not accompanied by a change in the size of the CD4+ cell population. Another potential contributing factor to the Th2 shift was the decrease in the number of neutrophils, because these cells can be important in promoting a Th1 phenotype [28]. Our findings with our highest inoculum size differ slightly from findings in IL-1R−/− mice infected with M. tuberculosis—in the lungs of these mice, the lack of IL-1 signaling was associated with an increased number of colony-forming units but was not associated with an alteration in the number of CD4+ cells [22]. These results demonstrate that host responses to different pathogens vary, despite a common immune defect.
In the present study, deviation to a Th2 response was manifest in the lungs of IL-1R−/− mice during the first 7 days of infection, despite a modest change in the inflammatory response. Although the sources of IL-4 and IL-10 in H. capsulatum–infected IL-1R−/− mice are not known, the decrease in the number of CD4+ cells suggests that cells other than T lymphocytes may be important in the release of these cytokines. In immunocompetent mice, H. capsulatum is a potent inducer of Th1 responses after challenge with nonlethal inoculum sizes [18]. Th2 responses dominate when the challenge is lethal or when the mice are deficient in IL-12, IFN-γ, GM-CSF, or TNF-α [6, 8, 9]. IL-1 does contribute to the polarization of Th1 and Th2 responses [29]. In IL-1R−/− mice infected with M. tuberculosis, IFN-γ production by splenocytes is sharply decreased in response to engagement of the T cell receptor complex [22]. IL-1R−/− mice infected with L. major manifest a deviation in the cytokine phenotype [22]. Splenocytes from infected IL-1R−/− mice produce increased amounts of IL-4 and IL-10 in response to antigen stimulation [30]. Thus, IL-1 signaling appears to be necessary to induce a Th1 response to several intracellular pathogens.
IL-1 has been reported to be a key factor in the polarization of T cell–mediated immunity. T cells from IL-1α– or IL-1β– deficient mice do not polarize in the presence of IL-4 alone but do polarize when exogenous IL-1 is added [30]. The failure of IL-1–deficient mice to develop a Th2 response impairs the expulsion of the worm Trichuris muris, a parasite whose elimination is largely dependent on a Th2 response. These findings contrast with the response observed here in H. capsulatum–infected mice. The inability of bioactive IL-1 to signal in H. capsulatum–infected mice led to a marked shift from a Th1 response to a Th2 response, as was evidenced by a reversal of the balance between IFN-γ and IL-4. IL-10 levels were also elevated in relation to those in WT mice. Because a Th1 response is crucial for host survival during histoplasmosis, the decreased levels of IFN-γ, in combination with the elevated levels of IL-4 and IL-10, are the most likely explanation for the impairment of immunity observed in IL-1R−/− mice.
TNF-α is necessary for protection [6, 12, 31]. The lungs of IL-1R−/− mice infected with H. capsulatum exhibited elevated levels of TNF-α during the first 7 days of infection. The increase was insufficient to compensate for an ineffective protective immune response. These results are similar to those from experiments in IL-12– or IFN-γ–deficient mice infected with this fungus [9]. In the absence of IFN-γ, TNF-α levels are elevated but do not overcome the defective protective immune response.
In mice, NO is an important mediator of host resistance to H. capsulatum [11]. Impaired generation of this reactive nitrogen intermediate inhibits protective immunity. Analysis of iNOS transcription and NO generation revealed that they were decreased during the early stages of infection (days 1 and 3) but then rebounded. This transient decrease may have established a permissive environment for growth of the fungus.
In summary, signaling through IL-1R is pivotal for the optimal generation of protective immunity to infection with H. capsulatum. The inability of IL-1 to engage its receptor induces a Th2 profile in association with decreased IFN-γ levels. The shift to a Th2 response, coupled with decreases in the number of CD4+ cells and in NO production, likely impair the protective immune response. These results raise the possibility that drugs that interfere with IL-1 signaling—such as anakinra, the recombinant human IL-1R antagonist—may increase the risk of a more aggressive histoplasmosis for those who reside in an endemic area.
Footnotes
Potential conflicts of interest: G.S.D. has been a consultant for and has received a grant from Amgen Corporation (although the monies did not support or influence the present work).
Financial support: Department of Veterans Affairs (Merit Review grant); National Institute of Allergy and Infectious Diseases (grants AI-42747, AI-34361, and AI-61298).
References
- 1.Deepe GS., Jr . Histoplasma capsulatum. In: Mandell GL, Bennet JE, Dolin R, editors. Principles and practices of infectious diseases. 6th ed. Vol. 2. Elsevier Churchill Livingstone; Philadelphia: 2004. pp. 3012–26. [Google Scholar]
- 2.Gomez AM, Bullock WE, Taylor CL, Deepe GS., Jr Role of L3T4+ T cells in host defense against Histoplasma capsulatum. Infect Immun. 1988;56:1685–91. doi: 10.1128/iai.56.7.1685-1691.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin JS, Yang CW, Wang DW, Wu-Hsieh BA. Dendritic cells cross-present exogenous fungal antigens to stimulate a protective CD8 T cell response in infection by Histoplasma capsulatum. J Immunol. 2005;174:6282–91. doi: 10.4049/jimmunol.174.10.6282. [DOI] [PubMed] [Google Scholar]
- 4.Zhou P, Seder RA. CD40 ligand is not essential for induction of type 1 cytokine responses or protective immunity after primary or secondary infection with Histoplasma capsulatum. J Exp Med. 1998;187:1315–24. doi: 10.1084/jem.187.8.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allendorfer R, Brunner GD, Deepe GS., Jr Complex requirements for nascent and memory immunity in pulmonary histoplasmosis. J Immunol. 1999;162:7389–96. [PubMed] [Google Scholar]
- 6.Allendoerfer R, Deepe GS., Jr Blockade of endogenous TNF-α exacerbates primary and secondary pulmonary histoplasmosis by differential mechanisms. J Immunol. 1998;160:607–82. [PubMed] [Google Scholar]
- 7.Allendoerfer R, Deepe GS., Jr Intrapulmonary response to Histoplasma capsulatum in gamma interferon knockout mice. Infect Immun. 1997;65:2564–9. doi: 10.1128/iai.65.7.2564-2569.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deepe GS, Jr, Gibbons R, Woodward E. Neutralization of endogenous granulocyte-macrophage colony-stimulating factor subverts the protective immune response to Histoplasma capsulatum. J Immunol. 1999;163:4985–93. [PubMed] [Google Scholar]
- 9.Allendoerfer R, Biovin GP, Deepe GS., Jr Modulation of immune responses in murine pulmonary histoplasmosis. J Infect Dis. 1997;175:905–14. doi: 10.1086/513989. [DOI] [PubMed] [Google Scholar]
- 10.Wu-Hsieh B, Zlotnik A, Howard DH. T-cell hybridoma-produced lymphokine that activates macrophages to suppress intracellular growth of Histoplasma capsulatum. Infect Immun. 1984;43:380–5. doi: 10.1128/iai.43.1.380-385.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou P, Sieve MC, Bennett J, et al. IL-12 prevents mortality in mice infected with Histoplasma capsulatum through induction of IFN-γ. J Immunol. 1995;155:785–95. [PubMed] [Google Scholar]
- 12.Zhou P, Miller G, Seder RA. Factors involved in regulating primary and secondary immunity to infection with Histoplasma capsulatum: TNF-α plays a critical role in maintaining secondary immunity in the absence of IFN-γ. J Immunol. 1998;160:1359–68. [PubMed] [Google Scholar]
- 13.Deepe GS, Jr, Gibbons RS. Protective and memory immunity to Histoplasma capsulatum in the absence of IL-10. J Immunol. 2003;171:5353–62. doi: 10.4049/jimmunol.171.10.5353. [DOI] [PubMed] [Google Scholar]
- 14.Peng JK, Lin JS, Kung JT, Finkelman FD, Wu-Hsieh BA. The combined effect of IL-4 and IL-10 suppresses the generation of, but does not change the polarity of, type-1 T cells in Histoplasma infection. Int Immunol. 2005;17:193–205. doi: 10.1093/intimm/dxh200. [DOI] [PubMed] [Google Scholar]
- 15.Dinarello CA. The biological properties of interleukin-1. Eur Cytokine Netw. 1994;5:517–31. [PubMed] [Google Scholar]
- 16.Dinarello CA. Interleukin-1. Cytokine Growth Factor Rev. 1997;8:253–65. doi: 10.1016/s1359-6101(97)00023-3. [DOI] [PubMed] [Google Scholar]
- 17.Dinarello CA. Blocking IL-1 in systemic inflammation. J Exp Med. 2005;201:1355–9. doi: 10.1084/jem.20050640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cain JA, Deepe GS., Jr Evolution of the primary immune response to Histoplasma capsulatum in murine lung. Infect Immun. 1998;66:1473–81. doi: 10.1128/iai.66.4.1473-1481.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hogquist KA, Nett MA, Sheehan KC, Pendleton KD, Schreiber RD, Chaplin DD. Generation of monoclonal antibodies to murine IL-1 beta and demonstration of IL-1 in vivo. J Immunol. 1991;146:1534–40. [PubMed] [Google Scholar]
- 20.Lewthwaite JC, Coates AR, Tormay P, et al. Mycobacterium tuberculosis chaperonin 60.1 is a more potent cytokine stimulator than chaperonin 60.2 (Hsp 65) and contains a CD14-binding domain. Infect Immun. 2001;69:7349–55. doi: 10.1128/IAI.69.12.7349-7355.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watson SR, Schmitt SK, Hendricks DE, Bullock WE. Immunoregulation in disseminated murine histoplasmosis: disturbances in the production of interleukins 1 and 2. J Immunol. 1985;135:3487–93. [PubMed] [Google Scholar]
- 22.Juffermans NP, Florquin S, Camoglio L, et al. Interleukin-1 signaling is essential for host defense during murine pulmonary tuberculosis. J Infect Dis. 2000;182:902–8. doi: 10.1086/315771. [DOI] [PubMed] [Google Scholar]
- 23.Satoskar AR, Okano M, Connaughton S, Raisanen-Sokolwski A, David JR, Labow M. Enhanced Th2-like responses in IL-1 type 1 receptor-deficient mice. Eur J Immunol. 1998;28:2066–74. doi: 10.1002/(SICI)1521-4141(199807)28:07<2066::AID-IMMU2066>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 24.Havell EA, Moldawer LL, Helfgott D, Kilian PL, Sehgal PB. Type I IL-1 receptor blockade exacerbates murine listeriosis. J Immunol. 1992;148:1486–92. [PubMed] [Google Scholar]
- 25.Hultgren OH, Svensson L, Tarkowski A. Critical role of signaling through IL-1 receptor for development of arthritis and sepsis during Staphylococcus aureus infection. J Immunol. 2002;168:5207–12. doi: 10.4049/jimmunol.168.10.5207. [DOI] [PubMed] [Google Scholar]
- 26.Ozaki Y, Ohashi T, Minami A, Nakamura S. Enhanced resistance of mice to bacterial infection induced by recombinant human interleukin-1a. Infect Immun. 1987;55:1436–40. doi: 10.1128/iai.55.6.1436-1440.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wheat LJ, Connolly-Stringfield PA, Baker RL, et al. Disseminated histoplasmosis in the acquired immune deficiency syndrome: clinical findings, diagnosis and treatment, and review of the literature. Medicine (Baltimore) 1990;69:361–74. doi: 10.1097/00005792-199011000-00004. [DOI] [PubMed] [Google Scholar]
- 28.Romani L, Mencacci A, Cenci E, et al. Neutrophil production of IL-12 and IL-10 in candidiasis and efficacy of IL-12 therapy in neutropenic mice. J Immunol. 1997;158:5349–56. [PubMed] [Google Scholar]
- 29.Allendoerfer R, Deepe GS., Jr Regulation of infection with Histoplasma capsulatum by TNFR1 and -2. J Immunol. 2000;165:2657–64. doi: 10.4049/jimmunol.165.5.2657. [DOI] [PubMed] [Google Scholar]
- 30.Helmby H, Grencis RK. Interleukin 1 plays a major role in the development of Th2-mediated immunity. Eur J Immunol. 2004;34:3674–81. doi: 10.1002/eji.200425452. [DOI] [PubMed] [Google Scholar]
- 31.Wu-Hsieh BA, Lee GS, Franco M, Hofman FM. Early activation of splenic macrophages by tumor necrosis factor alpha is important in determining the outcome of experimental histoplasmosis in mice. Infect Immun. 1992;60:4230–8. doi: 10.1128/iai.60.10.4230-4238.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]