

Abstract
We tested the hypothesis that multipotent stromal cells from human bone marrow (hMSCs) can provide a potential therapy for human diabetes mellitus. Severe but nonlethal hyperglycemia was produced in NOD/scid mice with daily low doses of streptozotocin on days 1–4, and hMSCs were delivered via intracardiac infusion on days 10 and 17. The hMSCs lowered blood glucose levels in the diabetic mice on day 32 relative to untreated controls (18.34 mM ± 1.12 SE vs. 27.78 mM ± 2.45 SE, P = 0.0019). ELISAs demonstrated that blood levels of mouse insulin were higher in the hMSC-treated as compared with untreated diabetic mice, but human insulin was not detected. PCR assays detected human Alu sequences in DNA in pancreas and kidney on day 17 or 32 but not in other tissues, except heart, into which the cells were infused. In the hMSC-treated diabetic mice, there was an increase in pancreatic islets and β cells producing mouse insulin. Rare islets contained human cells that colabeled for human insulin or PDX-1. Most of the β cells in the islets were mouse cells that expressed mouse insulin. In kidneys of hMSC-treated diabetic mice, human cells were found in the glomeruli. There was a decrease in mesangial thickening and a decrease in macrophage infiltration. A few of the human cells appeared to differentiate into glomerular endothelial cells. Therefore, the results raised the possibility that hMSCs may be useful in enhancing insulin secretion and perhaps improving the renal lesions that develop in patients with diabetes mellitus.
Keywords: insulin, pancreas, streptozotocin, transplantation
Previous publications presented conflicting observations as to whether cells from bone marrow can provide a potential therapy for diabetes mellitus. One strategy (1–4) was to differentiate plastic adherent marrow cells in culture into insulin-secreting cells. A second strategy was to transplant diabetic mice with genetically labeled marrow and to search for labeled insulin-producing cells in the recipient mice. One study using a CRE-LoxP-GFP system found that 1.7–3% of the cells in islets of the recipient mice were marrow-derived and that GFP-labeled donor cells isolated from the islets expressed insulin, glucose transporter 2, and transcription factors typically found in β cells (5). Three subsequent reports in which mice were transplanted with GFP-expressing bone marrow did not find evidence of marrow cells becoming insulin-producing cells in the pancreas of recipient mice (6–8), but in the reports it was difficult to exclude the possibility that the GFP gene was inactivated or that GFP-labeled cells were destroyed as they engrafted into islets. A third strategy was to determine whether systemically administered marrow cells enhanced regeneration of pancreatic insulin-producing cells in diabetic models. Hess et al. (9) reported that in NOD/scid mice in which diabetes was induced with streptozotocin (STZ), partial marrow ablation followed by transplantation of either GFP-labeled whole-marrow or GFP-labeled c-kit+ cells from murine marrow-enhanced regeneration of islets, lowered blood sugar, and increased blood insulin levels. In related experiments, retro-orbital infusion of large numbers of human umbilical cord cells into obese mice that were genetic models of type 2 diabetes decreased blood sugar (10). There also was attenuation of glomerular hypertrophy and tubular dilatation, but engraftment of the cells was not assayed. In additional experiments, multiple infusions of unfractionated marrow cells into mice with STZ-induced diabetes lowered blood sugar and improved the histomorphology of the pancreas (11). In experiments in which NOD mice were used as a model for type 1 diabetes, transplantation of wild-type bone marrow lowered blood sugar if the transplant was performed before, but not after, the onset of hyperglycemia (12). Also, murine embryonic stem cells differentiated to synthesize insulin and lowered blood sugar in STZ-induced diabetic mice, but the cells produced tumors (13).
If marrow cells are therapeutically useful in diabetes, some of the most attractive candidates are the plastic adherent cells from human marrow referred to variously as fibroblastic colony-forming units, mesenchymal stem cells, or multipotent stromal cells (MSCs) (14–17). MSCs are readily obtained from a patient and rapidly expanded in culture so that it is feasible to administer very large numbers of autologous cells. After systemic infusion, the cells home to injured tissues and repair them by several different mechanisms, including differentiating into multiple cellular phenotypes, providing cytokines and chemokines (17), enhancing the proliferation of tissue-endogenous stem/progenitor cells (18), and perhaps cell fusion (19) or transfer of mitochondria (20). In addition, MSCs suppress some immune reactions (21). A further attractive feature of MSCs is that they were tested in clinical trials and provided promising results without any apparent toxicity in patients (22–25).
Here, the effectiveness of MSCs from human bone marrow (hMSCs) was tested in immunodeficient NOD/scid mice in which an incomplete model for type 2 diabetes was produced with multiple low doses of STZ. The use of human cells in mice made it possible to readily detect and assay the effectiveness of the donor cells without the use of exogenous labels, to avoid the largely unexplained difficulties encountered in expanding murine MSCs in culture (26, 27), and to test the cells that are the most relevant for potential trials in patients.
Results
The Diabetic Model.
STZ was used to produce diabetes in NOD/scid mice. The mice do not spontaneously develop diabetes but lack functional B and T cells and have lymphopenia and hypogammaglobulinemia together with a normal hematopoietic microenvironment (28). Multiple low doses of STZ were administered to the mice (Scheme 1) under conditions that tend to minimize nephrotoxicity from the drug (29). In initial experiments, we administered 35 mg/kg STZ daily for 5 days following the protocol of Hess et al. (9), but the mice either died or had to be killed after 3–5 weeks because of severe weight loss and cachexia. Therefore, we administered 35 mg/kg for 4 days only. With the 4-day regimen, blood glucose levels increased from normal levels (5.92 mM ± 0.98 SE) to severe hyperglycemic levels (29.70 mM ± 2.42 SE) that reached the renal threshold (Fig. 1A). The mice survived for over 1 month without administration of insulin. The diabetic mice weighed less on days 32 than controls (24.03 g ± 3.13 SD vs. 27.83 g ± 1.65 SD; n = 5, P = 0.02). Also, the diabetic mice had a marked increase in urinary volume at days 39–45 (5.04 ml ± 3.18 SD vs. 0.44 ml ± 0.3 SD; n = 7, P = 0.005). None of the mice, however, developed albuminuria (data not shown).
Scheme. 1.

Experimental design.
Fig. 1.

Effects of hMSCs on blood glucose and mouse insulin levels in STZ-induced diabetic NOD/scid mice. (A) Blood glucose levels in untreated diabetic mice (STZ) and in hMSC-treated diabetic mice (STZ + hMSCs). Values are mean ± SE from three experiments. (B) Blood glucose levels in untreated diabetic mice and diabetic mice infused with human fibroblasts (STZ + hFibroblasts). Differences on day 10 reflect variations in untreated mice before fibroblasts were infused. Values are mean ± SD. (C) Blood levels of mouse insulin on day 32 in diabetic mice (STZ), hMSC-treated diabetic mice (STZ + hMSCs), and normal mice. Values are mean ± SD. ∗, Values that differ from each other with P = 0.0018.
Infusion of hMSCs Lowered Blood Sugar and Increased Blood Insulin.
Approximately 2.5 × 106 hMSCs were infused into the diabetic mice on day 10 and again on day 17. To avoid aggregation of the hMSCs and to ensure reproducible delivery, the hMSCs were suspended in a large volume of buffer (150 μl) at a concentration of ≈17,000 cells per μl and injected through the chest wall into the left cardiac ventricle. The blood glucose levels in the hMSC-treated diabetic mice (Fig. 1A) decreased significantly by days 24 and 32 (P = 0.0003 and 0.0019, respectively). There was no difference between untreated diabetic mice and hMSC-treated diabetic mice on day 32 in body weight (23.7 g ± 2.37 SD; n = 15), but there was a reduction on days 39–42 in urinary volume (2.20 ml ± 3.3 SD vs. 5.04 ml ± 3.18 SD; n = 7, P = 0.029). Human skin fibroblasts infused into the diabetic mice under the same conditions had no effect on blood glucose levels (Fig. 1B).
ELISAs on blood demonstrated that the administration of the hMSCs to the diabetic mice increased the levels of circulating mouse insulin (0.70 μg/liter ± 0.11 SD vs. 0.30 μg/liter ± 0.04 SD; n = 5 or 9; P = 0.0018; Fig. 1C). ELISAs on the same samples for human insulin were negative (data not shown).
Detection of Human DNA from hMSCs in Pancreas and Kidney of Diabetic NOD/scid Mice.
Tissues from the hMSC-treated diabetic mice were assayed for engraftment by real-time PCR for human Alu sequences (30). In 9 of 13 mice, from 0.11% to 2.9% of the human DNA infused as hMSCs was detected in the pancreas on days 17 or 32 (Table 1). In 4 of the 13 mice, no human genomic DNA was detected on day 32, perhaps because of the technical difficulty in consistently injecting cells into the left ventricle. In 6 mice in which human DNA was detected in the pancreas, the kidneys and other organs also were assayed. Human DNA was detected in the kidneys of all 6 mice (Table 1). In 4 of these 6 mice, the recovery of human DNA in kidney was unusually high and accounted for 6.7–11.6% of the human DNA infused as hMSCs. Variable amounts of human DNA (equivalent to 0–0.22% of the infused DNA) also were detected in the hearts of mice into which the hMSCs were infused (data not shown). Human Alu sequences were not detected in lung, liver, and spleen. Also, human Alu sequences were not detected in any of the same tissues 22 days after infusion of cultured human fibroblasts (Table 1).
Table 1.
Engraftment assayed by real-time PCR for Alu
| Animal/cells | Days | Pancreas | Kidney |
|---|---|---|---|
| 1/hMSC | 17 | 2.95 ± 0.06 | 6.70 ± 0.06 |
| 2/hMSC | 32 | 1.02 ± 0.31 | 0.05 ± 0.004 |
| 3/hMSC | 32 | 0.78 ± 0.05 | 11.58 ± 2.16 |
| 4/hMSC | 32 | 0.22 ± 0.03 | 0.03 ± 0.05 |
| 5/hMSC | 32 | 0.07 ± 0.01 | 10.62 ± 0.715 |
| 6/hMSC | 32 | 0.04 ± 0.02 | 9.82 ± 1.23 |
| 7/hMSC | 32 | 0.36 ± 0.02 | NA |
| 8/hMSC | 32 | 0.19 ± 0.09 | NA |
| 9/hMSC | 32 | 0.11 ± 0.01 | NA |
| 10–13/hMSC | 32 | ND | ND |
| 14–18/hFibro | 22 | ND | ND |
Values are percentage of human DNA infused as cells. NA, not assayed; ND, not detected; hFibro, human skin fibroblasts. Tissues were assayed either 17 or 32 days (Scheme 1) or 22 days after hFibro.
Increased Pancreatic Islets in hMSC-Treated Diabetic Mice.
Tissues from mice with high levels of human Alu sequences were selected for histological processing. Pancreases from the STZ-diabetic mice contained smaller islets (Fig. 2A), less mouse insulin immunoreactivity (Fig. 2 B and C), and a decreased number of islets per section (Fig. 2D). In pancreases from hMSC-treated diabetic mice, the islets appeared larger compared with islets from untreated diabetic mice (Fig. 2A). Also, the islets had an increase in mouse insulin immunoreactivity (Fig. 2 B and C), and there was an increase in number of islets per section (Fig. 2D). Many of the islets in the hMSC-treated diabetic mice appeared to bud off of the pancreatic ducts (Figs. 2A and 3).
Fig. 2.

Histology of pancreas from diabetic mice (STZ-treated), hMSC-treated diabetic mice (STZ + hMSCs), and control mice (Normal) at day 32. (A) Morphology of islets stained with hematoxylin and eosin. Sections (5-μm) are magnified ×400. (B) Islets labeled antibodies for mouse insulin. Nuclei labeled with DAPI. Sections (5-μm) are magnified ×400. (C) Insulin pixels per islet. Values are mean ± SD. ∗, Values that differ from each other with P = 0.0079. (D) Islets per section. Values are mean ± SD. ∗, Values that differ from each other with P = 0.002. n = 4 or 5.
Fig. 3.

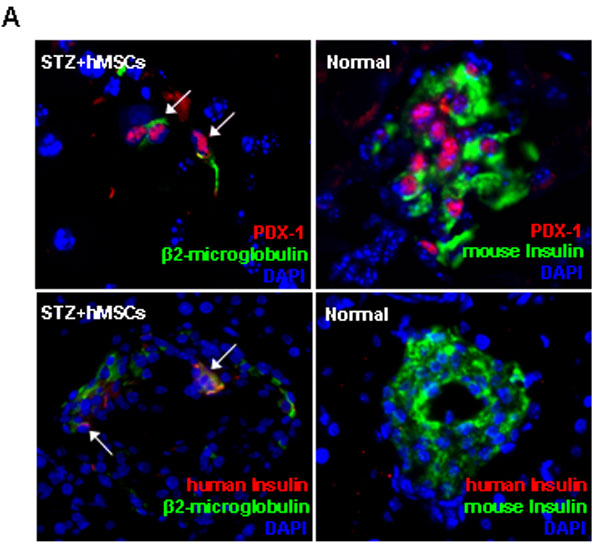
IHC of pancreas from hMSC-treated diabetic NOD/scid mice on day 32. Sections were colabeled with antibodies for human cells (β2-microglobulin) and mouse insulin. Nuclei were stained with DAPI. Sections (5-μm) are magnified ×400. Dotted line, outlines of ducts; arrows, human cells; arrowheads, human cells colabeled for mouse insulin.
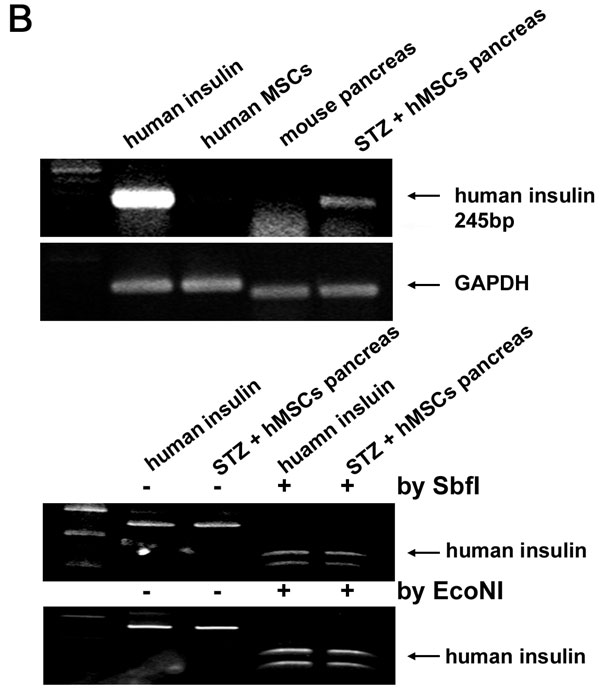
Small numbers of human cells were detected in islets of the hMSC-treated diabetic mice by labeling sections with antibodies to human β2-microglobulin and mouse insulin (Fig. 3). A few of the cells labeled for human β2-microglobulin colabeled with a human-specific antibodies both to PDX-1 and human insulin (Fig. 6A, which is published as supporting information on the PNAS web site). Qualitative RT-PCR assays of RNA from the pancreas of one hMSC-treated diabetic mouse detected mRNA for human insulin (Fig. 6B). However, samples from 11 additional hMSC-treated diabetic mice were negative both by immunolabeling and RT-PCR assays for human insulin.
Glomerular Morphology in hMSC-Treated Diabetic Mice.
Kidneys from untreated diabetic mice at day 32 contained many abnormal glomeruli with increased deposits of extracellular matrix protein in the mesangium (Fig. 4A). In kidneys from hMSC-treated diabetic mice that had high levels of human Alu sequences, glomeruli were more normal in appearance. The differences were accentuated by labeling kidney sections with antibodies to mouse macrophages/monocytes (Fig. 4 B and C). In the untreated diabetic mice, there was a marked increase in macrophages in the glomeruli; few were seen in the glomeruli from the hMSC-treated diabetic mice.
Fig. 4.

Renal glomeruli from diabetic mice (STZ), hMSC-treated diabetic mice (STZ + hMSCs), and control (Normal) mice on day 32. (A) Glomeruli stained with periodic acid-Schiff. Sections (8-μm) are magnified ×400. (B) Glomeruli labeled with antibodies to mouse macrophages/monocytes. Sections (8-μm) are magnified ×400. (C) Pixels per glomerulus in sections labeled with antibodies to mouse macrophages/monocytes. Values are mean ± SD; n ≥ 20 sections per mouse. ∗, Values that differ from each other with P < 0.005.
Kidneys that showed high levels of engraftment of human Alu sequences (Table 1) also were assayed for human cells with antibody to human nuclei antigen. Frozen sections labeled with antibodies to human nuclei antigen demonstrated that human cells were present in the glomeruli of hMSC-treated diabetic mice (Fig. 5 and Figs. 7 and 8 and Movie 1, which are published as supporting information on the PNAS web site). In some sections, human cells were present in about one-fifth of the glomeruli (Fig. 7), an observation consistent with the PCR assays for human Alu sequences (Table 1). Human cells were not found in tubules. Most positive glomeruli had one human cell. Glomeruli with two or more human cells were rare, and, in such glomeruli, the human cells usually were dispersed widely. Therefore, the results suggested that the human cells had not propagated after engrafting in kidney.
Fig. 5.

Renal glomeruli from hMSC-treated diabetic mice on day 32. Sections (5-μm) are magnified ×400. (A–D) Glomeruli labeled with antibodies for human nuclei antigen and mouse/human fibronectin. Some human cells that are colabeled have rounded morphology of mesangial cells. (E--H) Glomeruli labeled with antibodies for human nuclei antigen and mouse/human podocalyxin. No colabeling is detected. (J–L) Three-dimensional views of cells in glomeruli labeled for human nuclei antigen and for mouse/human endothelial cells (CD31). Some human cells appear to be colabeled and have the elongated morphology of endothelial cells. As indicated in the three-dimensional views, the CD31 epitope is present on several aspects of the human cells. Arrows, human cells; dotted arrows, planes for deconvolution; dotted lines, outlines of glomeruli. For additional three-dimensional deconvolved images, see Figs. 7–9 and Movie 1, which are published as supporting information on the PNAS web site.
Double immunohistochemistry (IHC) suggested that some of the human cells colabeled with a monoclonal antibody to CD31 (platelet-endothelial cell adhesion molecule 1, or PECAM-1), an epitope irregularly distributed on membranes of endothelial cell (Figs. 5 I–L and 7–9). CD31 was not expressed in cultured hMSCs (data not shown). Also, in some sections in which the cells were in the appropriate orientation, the human cells that colabeled with CD31 had the elongated morphology of endothelial cells (Fig. 5L and 8). Therefore, the results suggested that some of the human cells had differentiated into endothelial cells. Some of the human cells colabeled for fibronectin (Fig. 5 A–L), a protein expressed in mesangial cells. The colabeled cells had the rounded morphology of mesangial cells. However, fibronectin was expressed in cultured hMSC, and, therefore, it was not clear whether the cells had differentiated into mesangial cells. No cells were found that colabeled with antibodies to human nuclei antigen and podocalyxin, a protein expressed in podocytes (Fig. 5 E–H).
Discussion
Two aspects of the observations made here are remarkable: (i) the selective homing of hMSCs to both pancreatic islets and renal glomeruli of the diabetic mice and (ii) the ability of the cells to repair the tissues.
Previous reports demonstrated only very low levels of engraftment after systemic infusion MSCs into uninjured adult rodents (see ref. 31). In contrast, the results obtained here indicated that up to 3% of the infused hMSCs engrafted into pancreas and up to 11% of the infused cells engrafted into kidney in the diabetic mice (Table 1). Intracardiac infusion instead of i.v. infusion of the cells probably decreased trapping of the cells in the capillary beds of the lung, but it was apparent that the highest levels of engraftment were seen in the two organs damaged in the diabetic model, and no cells were detected in lung, liver, or spleen. The cells in the renal glomeruli were single cells, an observation suggesting that they engrafted immediately after systemic infusion into the mice, probably in response to specific signals from the injured tissues.
The infused hMSCs improved the hyperglycemia and increased blood levels of mouse insulin in the diabetic mice. A few of the human cells that engrafted into the pancreas differentiated so as to express both PDX-1 and human insulin. However, the major effect of the hMSCs treatment was to increase the number of mouse islets and mouse insulin-producing cells. In the treated diabetic mice, new islets appeared to bud off pancreatic ducts that are the source of islets during early development of the pancreas (32). Therefore, the effects of the hMSCs may be similar to the recent observations that hMSCs implanted into the dentate gyrus of the hippocampus of immunodeficient mice enhanced proliferation, migration, and neural differentiation of the nearby endogenous mouse neural stem cells (18).
The engraftment of the hMSCs into kidney was associated with improvements in glomerular morphology, a decrease in mesangial thickening, and a decrease in macrophage infiltration. STZ is a DNA-alkylating reagent, and single large doses produce tubular necrosis, but repeated lower doses and the resulting hyperglycemia produce glomerular changes more typical of, but not identical to, diabetic nephropathy (29). The observations here do not rigorously rule out the possibility that the improvements in the glomeruli were secondary to the lower blood glucose levels in the treated diabetic mice. However, it was striking that the human cells were found exclusively in the glomeruli and that some the cells apparently differentiated into endothelial cells. Therefore, the simplest interpretation of the data are that the engrafted hMSCs either prevented the pathological changes in the glomeruli or enhanced their regeneration.
The observations made here raise the possibility that hMSCs may be useful to treat both the hyperglycemia and the accompanying renal damage seen in diabetic patients. Autologous hMSCs are readily generated in a few weeks from patients (33), and the risk from administration of autologous hMSCs to patients should be minimal. In contrast to embryonic stem cells and some immortal cell lines, malignant transformations have not been observed with MSCs unless the cells are expanded extensively in culture under stressful conditions (34). In mice, MSCs enhanced the growth of cancers (35), and therefore there is a small risk that administration of autologous hMSCs to a patient will enhance to growth of an unsuspected tumor. Also, there is a risk of pulmonary emboli if the cells are allowed to aggregate in suspension before i.v. infusion. In addition, culture of hMSCs usually is performed in medium containing FCS that the cells internalize (36) and that can produce immune reactions with repeated administrations of the cells (37). However, the calf proteins can be removed metabolically by short-term culture with human serum (36) or avoided by culture in medium containing platelet lysates in place of the FCS (38, 39). In effect, the apparent risks from administration of autologous hMSCs seem small compared with the potential therapeutic benefits. MSCs or related cells from bone marrow have been shown to produce beneficial effects in animal models for a variety of diseases and in several clinical trials, including, apparently, clinical trials in heart disease that are now being conducted at multiple medical centers (17, 40, 41). Therefore, it is difficult to exclude the possibility that systemic infusion of autologous hMSCs in patients with diabetes could have beneficial effects in several of the many tissues damaged by the disease.
Materials and Methods
STZ-Induced Diabetes in Mice.
Male immunodeficient NOD/scid mice (NOD.CB17-Prkdcscid/J; The Jackson Laboratory, Bar Harbor, ME) at 7–8 weeks of age were injected i.p. with 35 mg/kg STZ (Sigma-Aldrich, St. Louis, MO) daily on days 1–4. STZ was solubilized in sodium citrate buffer, pH 4.5, and injected within 15 min of preparation. The mice were maintained under sterile conditions, and all animal work was carried out under protocols approved by the Institutional Animal Care and Utilization Committees of Tulane University Health Sciences Center and the Ochsner Clinic Foundation.
Preparation and Infusion of Cells.
Extensively characterized preparations of hMSCs (42) from normal healthy donors were obtained from the Tulane Center for the Preparation and Distribution of Adult Stem Cells (www.som.tulane.edu/gene_therapy/distribute.shtml). Frozen vials of ≈106 passage 1 hMSCs were plated at high density in complete culture medium containing 17% FCS for 24 h, replated at 100 cells per cm2, incubated for 7–9 days until they were ≈70% confluent, and harvested at passage 2 with 0.25% trypsin and 1 mM EDTA at 37°C for ≈5 min (42). For transplantation, the cells were washed with PBS by centrifugation, suspended in Hank's balanced salt solution at a concentration of ≈17,000 cells per μl, and maintained at 4°C. Mice were anesthetized i.p. with a mixture of ketamine (91 mg/kg) and xylazine (9 mg/kg), and 150 μl of cell suspension was injected through the chest wall into the left ventricle. Human skin fibroblasts (Hs 68; American Type Culture Collection, Rockville, MD) were expanded from frozen vials through two passages as recommended by the supplier, lifted with trypsin/EDTA, and processed as the hMSCs before infusion into mice.
Assays for Blood Glucose and Insulin.
Blood glucose was assayed in tail-vein blood with a glucometer (Elite Diabetes Care System; Bayer, Leverkusen, Germany) after a 4-h morning fast. Blood insulin was assayed on blood obtained by intracardiac puncture of anesthetized mice before they were killed on day 32 by using both a mouse-specific ELISA kit and a human-specific ELISA kit (Ultrasensitive Mouse Insulin ELISA and Insulin Ultrasensitive ELISA; Mercodia, Uppsala, Sweden). Details for RT-PCR assays for insulin can be found in Supporting Text, which is published as supporting information on the PNAS web site.
Preparation of Tissue Samples.
Mice were injected i.p. with ketamine/xylazine and perfused through the left ventricle with 20 ml of PBS and then through the right ventricle with 5 ml of PBS before tissues were isolated by dissection. The distal half of the pancreas, one kidney, and other organs were quick-frozen at −80°C for DNA and RNA assays. The proximal half of the pancreas was fixed overnight in 10% buffered formalin and incubated overnight at 4°C in 30% sucrose/PBS. The tissues were then embedded (Tissue-Tek OCT Compound; Sakura Finetek, Torrance, CA), frozen in dry-ice-cooled isopentane, and sectioned at 5–8 μm on a cryostat. Samples of kidney for histology were fixed with the same protocol, embedded in paraffin, and cut at 8 μm. Samples of kidney for IHC were frozen as above and sectioned at 8–30 μm.
Real-Time PCR Assays.
Frozen tissues were homogenized, DNA was extracted with phenol/chloroform (Phase Lock Gel; Eppendorf/Brinkmann Instruments, Inc., Westbury, NY) and precipitated with ethanol, and total DNA was assayed by UV absorbance. Real-time PCR assay was performed with 200 ng of target DNA, Alu-specific primers, and a fluorescent probe (30) by using an automated instrument (Model 7700; Applied Biosystems, Foster City, CA). Values for the amount of target DNA in each sample were corrected by assays for the single-copy mouse albumin gene (31).
Histology and IHC.
For histology of pancreas, sections were stained with hematoxylin and eosin (Richard Allan Scientific, Kalamazoo, MI). For histology of kidney, sections were stained with periodic acid-Schiff (PAS; Richard Allan Scientific). For IHC, frozen sections were incubated for 18 h at 4°C with primary antibodies to an anti-human β2-microglobulin (1:200; Roche, Basel, Switzerland), anti-human nuclei antigen (1:200; clone 235–1; Chemicon, Temecula, CA), anti-human insulin (1:40; clone E2E3C2; Calbiochem, San Diego, CA), anti-mouse insulin (1:50; clone 182410; R & D Systems, Minneapolis, MN), anti-mouse/human PDX-1 (1:50; clone 267712; R & D Systems), anti-mouse/human podocalyxin (1:100; clone 222328; R & D Systems), anti-mouse macrophages/monocytes (1:25; clone MOMA-2; Chemicon), anti-mouse/human fibronectin (1:80; Chemicon), or anti-mouse/human CD31 (1:500; clone MEC 13.3; BD Biosciences, San Jose, CA). The slides were washed three times for 5 min with PBS and incubated for 45 min at room temperature with species-specific secondary antibodies (1:1,000; Alexa-594 or Alexa-488; Molecular Probes, Eugene, OR). Controls included omitting the primary antibody. Slides were evaluated by epifluorescence microscopy (Eclipse E800; Nikon, Melville, NY). Pixels were counted with a software program (ImageJ, NIH Image). Islets labeled for mouse insulin per section were counted manually. For deconvolution microscopy, images were acquired at 0.4-μm intervals on epifluorescence microscope Leica DMRXA equipped with an automated x, y, z stage and CCD camera (Sensicam; Intelligent Imaging Innovations, Denver, CO). Deconvolution and 3D reconstructions then were performed by using commercial software (Slidebook Software; Intelligent Imaging Innovations).
Urine Assays.
Mice on days 39–45 were placed in individual metabolic cages (NALGENE Labware, Rochester, NY), and 18-h urine samples were assayed for albumin (QuantiChrom bacillus Calmette–Guérin Albumin Assay Kit; Bioassay Systems, Haywood, CA).
Statistical Analyses.
Student's t test was used for P values.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health Grant P40 RR 17447 and grants from the W. M. Keck Foundation, HCA the Healthcare Company, and the Louisiana Gene Therapy Research Consortium.
Abbreviations
- MSC
multipotent stromal cell
- hMSC
MSC from human bone marrow
- STZ
streptozotocin
- IHC
immunohistochemistry.
Footnotes
The authors declare no conflict of interest.
References
- 1.Tang DQ, Cao LZ, Burkhardt BR, Xia CQ, Litherland SA, Atkinson MA, Yang LJ. Diabetes. 2004;53:1721–1732. doi: 10.2337/diabetes.53.7.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oh SH, Muzzonigro TM, Bae SH, LaPlante JM, Hatch HM, Petersen BE. Lab Invest. 2004;84:607–617. doi: 10.1038/labinvest.3700074. [DOI] [PubMed] [Google Scholar]
- 3.Choi KS, Shin JS, Lee JJ, Kim YS, Kim SB, Kim CW. Biochem Biophys Res Commun. 2005;330:1299–1305. doi: 10.1016/j.bbrc.2005.03.111. [DOI] [PubMed] [Google Scholar]
- 4.Chen LB, Jiang XB, Yang L. World J Gastroenterol. 2004;10:3016–3020. doi: 10.3748/wjg.v10.i20.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ianus A, Holz GG, Theise ND, Hussain MA. J Clin Invest. 2003;111:843–850. doi: 10.1172/JCI16502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi JB, Uchino H, Azuma K, Iwashita N, Tanaka Y, Mochizuki H, Migita M, Shimada T, Kawamori R, Watada H. Diabetologia. 2003;46:1366–1374. doi: 10.1007/s00125-003-1182-9. [DOI] [PubMed] [Google Scholar]
- 7.Lechner A, Yang YG, Blacken RA, Wang L, Nolan AL, Habener JF. Diabetes. 2004;53:616–623. doi: 10.2337/diabetes.53.3.616. [DOI] [PubMed] [Google Scholar]
- 8.Taneera J, Rosengren A, Renstrom E, Nygren JM, Serup P, Rorsman P, Jacobsen SE. Diabetes. 2006;55:290–296. doi: 10.2337/diabetes.55.02.06.db05-1212. [DOI] [PubMed] [Google Scholar]
- 9.Hess D, Li L, Martin M, Sakano S, Hill D, Strutt B, Thyssen S, Gray DA, Bhatia M. Nat Biotechnol. 2003;21:763–770. doi: 10.1038/nbt841. [DOI] [PubMed] [Google Scholar]
- 10.Ende N, Chen R, Reddi AS. Biochem Biophys Res Commun. 2004;321:168–171. doi: 10.1016/j.bbrc.2004.06.121. [DOI] [PubMed] [Google Scholar]
- 11.Banerjee M, Kumar A, Bhonde RR. Biochem Biophys Res Commun. 2005;328:318–325. doi: 10.1016/j.bbrc.2004.12.176. [DOI] [PubMed] [Google Scholar]
- 12.Kang EM, Zickler PP, Burns S, Langemeijer SM, Brenner S, Phang OA, Patterson N, Harlan D, Tisdale JF. Exp Hematol. 2005;33:699–705. doi: 10.1016/j.exphem.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 13.Fujikawa T, Oh SH, Pi L, Hatch HM, Shupe T, Petersen BE. Am J Pathol. 2005;166:1781–1791. doi: 10.1016/S0002-9440(10)62488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Owen M, Friedenstein AJ. Ciba Found Symp. 1988;136:42–60. doi: 10.1002/9780470513637.ch4. [DOI] [PubMed] [Google Scholar]
- 15.Caplan AI. J Orthop Res. 1991;9:641–650. doi: 10.1002/jor.1100090504. [DOI] [PubMed] [Google Scholar]
- 16.Prockop DJ. Science. 1997;276:71–74. doi: 10.1126/science.276.5309.71. [DOI] [PubMed] [Google Scholar]
- 17.Prockop DJ, Gregory CA, Spees JL. Proc Natl Acad Sci USA. 2003;100(Suppl 1):11917–11923. doi: 10.1073/pnas.1834138100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munoz JR, Stoutenger BR, Robinson AP, Spees JL, Prockop DJ. Proc Natl Acad Sci USA. 2005;102:18171–18176. doi: 10.1073/pnas.0508945102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spees JL, Olson SD, Ylostalo J, Lynch PJ, Smith J, Perry A, Peister A, Wang MY, Prockop DJ. Proc Natl Acad Sci USA. 2003;100:2397–2402. doi: 10.1073/pnas.0437997100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spees JL, Olson SD, Whitney MJ, Prockop DJ. Proc Natl Acad Sci USA. 2006;103:1283–1288. doi: 10.1073/pnas.0510511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le Blanc K, Tammik L, Sundberg B, Haynesworth SE, Ringden O. Scand J Immunol. 2003;57:11–20. doi: 10.1046/j.1365-3083.2003.01176.x. [DOI] [PubMed] [Google Scholar]
- 22.Horwitz EM, Prockop DJ, Gordon PL, Koo WW, Fitzpatrick LA, Neel MD, McCarville ME, Orchard PJ, Pyeritz RE, Brenner MK. Blood. 2001;97:1227–1231. doi: 10.1182/blood.v97.5.1227. [DOI] [PubMed] [Google Scholar]
- 23.Koc ON, Day J, Nieder M, Gerson SL, Lazarus HM, Krivit W. Bone Marrow Transplant. 2002;30:215–222. doi: 10.1038/sj.bmt.1703650. [DOI] [PubMed] [Google Scholar]
- 24.Le Blanc K, Rasmusson I, Sundberg B, Gotherstrom C, Hassan M, Uzunel M, Ringden O. Lancet. 2004;363:1439–1441. doi: 10.1016/S0140-6736(04)16104-7. [DOI] [PubMed] [Google Scholar]
- 25.Lazarus HM, Koc ON, Devine SM, Curtin P, Maziarz RT, Holland HK, Shpall EJ, McCarthy P, Atkinson K, Cooper BW, et al. Biol Blood Marrow Transplant. 2005;11:389–398. doi: 10.1016/j.bbmt.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 26.Baddoo M, Hill K, Wilkinson R, Gaupp D, Hughes C, Kopen GC, Phinney DG. J Cell Biochem. 2003;89:1235–1249. doi: 10.1002/jcb.10594. [DOI] [PubMed] [Google Scholar]
- 27.Peister A, Mellad JA, Larson BL, Hall BM, Gibson LF, Prockop DJ. Blood. 2004;103:1662–1668. doi: 10.1182/blood-2003-09-3070. [DOI] [PubMed] [Google Scholar]
- 28.Serreze DV, Leiter EH, Hanson MS, Christianson SW, Shultz LD, Hesselton RM, Greiner DL. Diabetes. 1995;44:1392–1398. doi: 10.2337/diab.44.12.1392. [DOI] [PubMed] [Google Scholar]
- 29.Tay YC, Wang Y, Kairaitis L, Rangan GK, Zhang C, Harris DC. Kidney Int. 2005;68:391–398. doi: 10.1111/j.1523-1755.2005.00405.x. [DOI] [PubMed] [Google Scholar]
- 30.McBride C, Gaupp D, Phinney DG. Cytotherapy. 2003;5:7–18. doi: 10.1080/14653240310000038. [DOI] [PubMed] [Google Scholar]
- 31.Lee RH, Hsu SC, Munoz J, Jung JS, Lee NR, Pochampally R, Prockop DJ. Blood. 2006;107:2153–2161. doi: 10.1182/blood-2005-07-2701. [DOI] [PubMed] [Google Scholar]
- 32.Hardikar AA. Trends Endocrinol Metab. 2004;15:198–203. doi: 10.1016/j.tem.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 33.Sekiya I, Larson BL, Smith JR, Pochampally R, Cui JG, Prockop DJ. Stem Cells. 2002;20:530–541. doi: 10.1634/stemcells.20-6-530. [DOI] [PubMed] [Google Scholar]
- 34.Rubio D, Garcia-Castro J, Martin MC, de la Fuente R, Cigudosa JC, Lloyd AC, Bernad A. Cancer Res. 2005;65:3035–3039. doi: 10.1158/0008-5472.CAN-04-4194. [DOI] [PubMed] [Google Scholar]
- 35.Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Cancer Res. 2002;62:3603–3608. [PubMed] [Google Scholar]
- 36.Spees JL, Gregory CA, Singh H, Tucker HA, Peister A, Lynch PJ, Hsu SC, Smith J, Prockop DJ. Mol Ther. 2004;9:747–756. doi: 10.1016/j.ymthe.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 37.Horwitz EM, Gordon PL, Koo WK, Marx JC, Neel MD, McNall RY, Muul L, Hofmann T. Proc Natl Acad Sci USA. 2002;99:8932–8937. doi: 10.1073/pnas.132252399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamada Y, Ueda M, Hibi H, Nagasaka T. Cell Transplant. 2004;13:343–355. doi: 10.3727/000000004783983909. [DOI] [PubMed] [Google Scholar]
- 39.Doucet C, Ernou I, Zhang Y, Llense JR, Begot L, Holy X, Lataillade JJ. J Cell Physiol. 2005;205:228–236. doi: 10.1002/jcp.20391. [DOI] [PubMed] [Google Scholar]
- 40.Ye L, Haider HK, Sim EK. Exp Biol Med (Maywood) 2006;231:8–19. doi: 10.1177/153537020623100102. [DOI] [PubMed] [Google Scholar]
- 41.Fazel S, Tang GH, Angoulvant D, Cimini M, Weisel RD, Li RK, Yau TM. Ann Thorac Surg. 2005;79:S2238–S2247. doi: 10.1016/j.athoracsur.2005.02.085. [DOI] [PubMed] [Google Scholar]
- 42.Sekiya I, Vuoristo JT, Larson BL, Prockop DJ. Proc Natl Acad Sci USA. 2002;99:4397–4402. doi: 10.1073/pnas.052716199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}