

Abstract
Phosphorylation and spatial reorganization of the vimentin network have been implicated in mediating smooth muscle contraction, cell migration, and cell mitosis. In this report, stimulation of cultured smooth muscle cells with 5-hydroxytryptamine (5-HT) induced PAK1 phosphorylation at Thr-423 (an indication of PAK activation). Treatment with PAK led to disassembly of wild type vimentin filaments, but not vimentin S56A (alanine substitution at serine-56) mutant filaments, as assessed by an in vitro filament assembly assay. Furthermore, stimulation with 5-HT resulted in the dissociation of Crk-associated substrate (CAS, an adapter protein associated with smooth muscle force development) from cytoskeletal vimentin. Expression of the vimentin S56A mutant in cells inhibited the increase in phosphorylation at Ser-56 and ratios of soluble vimentin to insoluble vimentin (an index of vimentin disassembly), and the dissociation of CAS from cytoskeletal vimentin in response to 5-HT activation as compared to cells expressing wild type vimentin. Because CAS may be involved in PAK activation, PAK phosphorylation was evaluated in cells expressing S56A vimentin mutant. Expression of vimentin S56A mutant depressed PAK phosphorylation at Thr-423 induced by 5-HT. The expression of the vimentin S56A mutant also inhibited the spatial reorientation of vimentin filaments in cells in response to 5-HT stimulation. Our results suggest that vimentin phosphorylation at Ser-56 may inversely regulate PAK activation possibly via the increase in the amount of soluble CAS upon agonist stimulation of smooth muscle cells. Additionally, vimentin phosphorylation at this position is critical for vimentin filament spatial rearrangement elicited by agonists.
The intermediate filament framework is one of the three cytoskeletal systems in mammalian cells. Its well-spread filamentous structure from the nucleus to the plasma membrane is believed to provide protection against various mechanical stresses (1,2). Intermediate filaments also undergo disassembly/assembly and spatial reorganization in cells in response to external stimulation and during mitosis. The dynamic property in the intermediate filament system plays a fundamental role in mediating changes in cell shape, cell division and migration, signaling molecule distribution, and smooth muscle force development (1,3–7).
The dynamic characteristics of the intermediate filament network may be regulated by protein phosphorylation. Vimentin is the most abundant intermediate filament protein in various cell types including smooth muscle cells (2,5,6). Vimentin phosphorylation in association with vimentin disassembly and spatial reorganization occurs during mitosis or in response to extracellular stimulation (8,9). In cultured smooth muscle cells, contractile stimulation triggers vimentin phosphorylation at Ser-56 concurrently with vimentin partial disassembly and spatial reorientation (6).
The disassembly and spatial reorganization of the vimentin network may regulate the translocation of certain molecules (7,10,11). The adapter protein p130 Crk-associated substrate (CAS) has been shown to participate in the signaling processes that regulate smooth muscle contraction and cell migration (12–14). Our recent studies have suggested that vimentin phosphorylation and disassembly are related to CAS redistribution during contractile activation of smooth muscle (10). In addition, external stress initiates Rho kinase redistribution associated with vimentin depolymerization in fibroblasts and the translocation of Ca2+/calmodulin dependent protein kinase II (CamKII) in differentiated smooth muscle cells, which may be an important event for cell signaling (7,11).
p21-activated kinase (PAK) may be an upstream regulator of the vimentin network (6,9). In cultured smooth muscle cells, agonist-mediated vimentin phosphorylation at Ser-56 and spatial reorientation of the vimentin network are inhibited by silencing of PAK1, a dominant isoform in smooth muscle (6,15). Additionally, PAK has been implicated in modulating smooth muscle contraction; introduction of an active PAK isoform into smooth muscle potentiates force development at constant intracellular calcium (16). Expression of an inactive PAK1 mutant attenuates migration of cultured smooth muscle cells in response to platelet-derived growth factor (15).
In response to external stimulation, PAK undergoes autophosphorylation at Thr-423, which increases PAK activity towards substrates (17,18). In addition to the small GTPases Cdc42/Rac1, the activity of PAK may be regulated by PKL (paxillin kinase linker)/PIX (guanine nucleotide exchange factor)(19–21). CAS has been shown to interact with PKL/PIX via CrkII and paxillin (21–23). Thus, CAS could be involved in the regulation of PAK activation.
The aim of this study was to test the hypothesis that vimentin phosphorylation at Ser-56 may play a critical role in regulating PAK activation and the spatial reorientation of the vimentin network. Our results demonstrate that PAK-mediated vimentin phosphorylation at Ser-56 leads to increases in vimentin disassembly and CAS dissociation from the vimentin network during 5-HT stimulation. Expression of a non-phosphorylatable vimentin mutant S56A (alanine substitution at serine-56) attenuates the increase in PAK activation and the structural rearrangement of vimentin filaments upon agonist stimulation.
EXPERIMENTAL PROCEDURES
Smooth muscle cell culture
Primary canine tracheal smooth muscle cells were prepared and cultured according to the methods previously described (6). Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum or growth bovine serum (1:10, v/v) and antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin and 2.5 μg/ml amphotericin-B). Cells within passage four were then serum-deprived 24 hours before biochemical and immunofluorescence or fluorescence experiments. These smooth muscle cells in culture express high levels of smooth muscle specific α-actin as determined by immunoblot analysis.
Assessment of protein phosphorylation
Smooth muscle cells were treated with SDS sample buffer containing 1.5% dithiothreitol, 2% SDS, 80 mM Tris/HCl, pH 6.8, 10% (v/v) glycerol and 0.01% Brompphenol Blue. Cells were boiled in the buffer for 4 min and separated by SDS-PAGE. Proteins were transferred to nitrocellulose membranes, after which the membranes were blocked with 2% bovine serum albumin. To analyze PAK phosphorylation, membranes were reacted with phospho-PAK1 (Thr-423)/PAK2 (Thr-403) antibody (Cell Signaling) followed by horseradish peroxidase-conjugated anti-rabbit Ig (ICN Biomedicals). Proteins were visualized by enhanced chemiluminescence (SuperSignal, Pierce) using Fuji Image System LAS3000. The membranes were stripped and reprobed with PAK1 antibody (Cell Signaling). For determination of vimentin phosphorylation, membranes were probed with the use of a site-specific phosphorylation antibody for Ser-56 (pS56 antibody), stripped and reprobed with monoclonal vimentin antibody (BD Biosciences, clone RV202) followed by the incubation with horseradish peroxidase conjugated anti-mouse Ig (Amersham Life Sciences) (6). Phosphoprotein and total protein were quantified by scanning densitometry of immunoblots (Fuji Multigauge Software). Changes in protein phosphorylation were expressed as a magnitude increase over levels of phosphorylation in unstimulted cells. The luminescent signals from all immunoblots were within the linear range.
Preparation of recombinant proteins
PCR mediated mutagenesis was carried out on pEGFP-vimentin (kindly provided by Dr Robert D. Goldman, Northwestern University, Chicago, IL) to generate the vimentin mutant S56A. DNA sequencing was used to confirm the mutation of vimentin. The PCR product and wild-type vimentin cDNA were subcloned into pGEX-4T at BamHI and EcoRI sites, followed by transformation of E. coli BL21. Recombinant proteins were harvested and purified according to the Amersham’s manual of GST gene fusion system. Thrombin (Amersham Biosciences, Piscataway, NJ) was used to release the recombinant protein from glutathione-sepharose beads, and then was removed by incubation with p-aminobenzamidine-agarose beads (Sigma).
In vitro kinase assay
Purified wild-type vimentin or the vimentin mutant S56A (0.1 mg/ml) was incubated with 2 μg/ml activated PAK (Upstate) for 10 min or 30 min in a buffer containing 20 mM Hepes (pH7.5), 60 mM NaCl, 2 mM MgCl2, 5 mM EGTA and 100 μM ATP. The reaction mixture was boiled in the SDS sample buffer for 5 min, and proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were probed with the use of the phospho-vimentin antibody for Ser-56 (pS56 antibody), stripped and reprobed with vimentin antibody.
Immunofluorescence molecular imaging
We developed a molecular immunostaining technique to visualize vimentin filament assembly in vitro. Briefly, purified wild-type vimentin and vimentin mutant S56A (0.2 mg/ml) were placed in the kinase buffer (see above) in the presence or absence of PAK for 30 min. NaCl (150 mM) was then added into the reaction mix followed by incubation at 37°C for 1 hr to initiate filament formation. The mixture was treated with 0.1% glutaraldehyde at room temperature for 30 min to crosslink vimentin filaments, and was then plated on slides followed by the fixation with 4% paraformaldehyde for 15 min. The purified molecules were reacted with monoclonal vimentin antibody (BD Biosciences) followed by incubation with anti-mouse IgG conjugated to Alexa 488 (Molecular Probes, Eugene, Oregon). Vimentin filament formation was examined and analyzed under laser scanning confocal microscope (Zeiss).
Analysis of the ratios of soluble protein to insoluble protein in cells
The amounts of soluble and insoluble vimentin were evaluated by the method described previously (4,6). Smooth muscle cells were washed with ice-cold HBSS, and then scraped, collected and centrifuged at 8000 g for 5 min. The resulting pellets were incubated at 37°C for 30 min with a buffer containing 1% Nonidet P-40, 10% (v/v) glycerol, 20 mM Hepes, pH 7.6, 150 mM NaCl, 2 mM sodium orthovanadate, 2 mM molybdate, 2 mM sodium pyrophosphate and protease inhibitors (2 mM benzamidine, 0.5 mM aprotinin and 1 mM PMSF). The soluble (disassembled) and insoluble (assembled) fractions were collected after centrifugation at 2,100 g at 4°C for 30 min, and assessed by immunoblot analysis using vimentin antibody. The ratios of soluble vimentin to insoluble vimentin were determined after scanning densitometry of the immunoblots. To assess ratios of soluble CAS to insoluble (vimentin-associated) CAS and ratios of insoluble CAS to cytoskeletal vimentin, the membranes were stripped and reprobed with CAS antibody (BD Biosciences, clone 24).
Cell transfection and assessment of protein expression
Cultured smooth muscle cells were transfected with plasmids encoding EGFP-tagged wild-type vimentin and vimentin mutant S56A using transfection kits (Lipofectamine™ and Plus™, Invitrogen, CA). After 48 hrs, the cells were incubated in serum free medium for 1 day. Cells were treated with 1 × SDS sample buffer to extract and denature proteins, after which the proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes. To assess the expression of recombinant proteins, the membranes were detected with the use of EGFP antibody (Lab Vision, clone GFP02) or vimentin antibody.
Cell fluorescence analysis
After transfection with plasmids containing EGFP-labeled proteins, cells on coverslips were fixed with paraformaldehyde and were mounted on slides using Fluoromount-G (Fishers). Cells were also stained with 4′,6-diamidino-2-phenylindole (DAPI) to visualize the nucleus. Cell images were viewed under confocal fluorescence microscope. Cells with reorganized vimentin filaments were determined and quantitatively analyzed by the method previously described (6).
Statistical analysis
All statistical analysis was performed using Prism 4 software (GraphPad Software, San Diego, CA). Comparison among multiple groups was performed by one-way analysis of variance followed by post test (Tukey’s multiple comparison test). Differences between pairs of groups were analyzed by Student-Newman-Keuls test or Dunn’s method. Values of n refer to the number of experiments used to obtain each value. P < 0.05 was considered to be significant.
RESULTS
Activation with 5-HT induces PAK phosphorylation in smooth muscle cells
PAK has been implicated in the modulation of smooth muscle functions (6,15,16). To evaluate whether agonist stimulation activates PAK, cultured tracheal smooth muscle cells were treated with 10 μm 5-HT for 5–15 min, or left unstimulated. Blots of protein extracts from these cells were probed using phospho-PAK1 (Thr-423) antibody, stripped and reprobed by use of PAK1 antibody.
The activation of smooth muscle cells with 5-HT led to the enhancement of PAK phosphorylation at Thr-423. As shown in Figure 1A, PAK1 phosphorylation at Thr-423 was increased 5 min after stimulation with 5-HT, and was sustained for the 15-min duration. The phosphorylation levels in smooth muscle cells were increased by 1.5-fold 10–15 min after activation with 5-HT (Figure 1B, n = 4).
Figure 1. Stimulation of smooth muscle cells by 5-HT induces PAK1 phosphorylation at Thr-423.
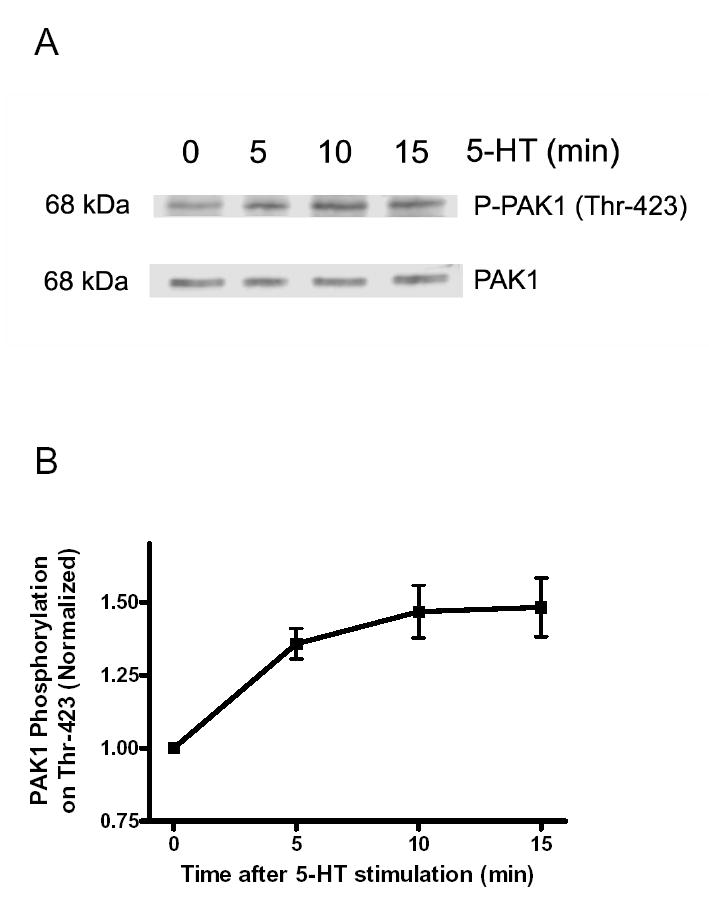
(A) Immunoblots illustrating PAK phosphorylation in smooth muscle cells in response to 5-HT stimulation. Smooth muscle cells were exposed to 10 μM 5-HT for 5–15 min, or unstimulated. Blots of these cells were probed by use of phospho-PAK1 (Thr-423), stripped and reprobed with PAK1 antibody. (B) PAK phosphorylation is normalized to the levels obtained from unstimulated smooth muscle cells. Values shown are mean ± SE (n = 4).
PAK catalyzes vimentin phosphorylation at Ser-56 in vitro
To determine whether PAK directly catalyzes vimentin phosphorylation, we evaluated the phosphorylation state of vimentin phosphorylation at Ser-56 mediated by PAK in vitro. Recombinant wild type vimentin was incubated with active PAK for 10 min or 30 min in the kinase buffer. Vimentin phosphorylation on Ser-56 was assessed by immunoblot analysis using the site-specific vimentin phosphorylation antibody (6). Incubation of the purified wild type vimentin with PAK resulted in the enhancement of vimentin phosphorylation at Ser-56; the levels of vimentin phosphorylation were dramatically increased 30 min after the treatment with PAK (Fig. 2, A and B, n = 4).
Figure 2. Vimentin phosphorylation at Ser-56 is catalyzed by PAK in vitro.

(A) Immunoblots illustrating the phosphorylation of wild type vimentin and vimentin S56A mutant (alanine substitution at Ser-56) by PAK. PAK-mediated protein phosphorylation was determined by the method described in the Experimental Procedures. PAK catalyzed Ser-56 phosphorylation of wild type vimentin while little phosphorylation was detected in the vimentin S56A mutant. (B) The phosphorylation levels of various treatments are normalized to the phosphorylation level of wild type vimentin in the absence of PAK. * Significantly higher phosphorylation levels in the presence of PAK as compared to the level of wild type vimentin obtained without PAK treatment (P < 0.05). All values are means ± SE (n = 6). pS56 Ab, phospho-vimentin (Ser-56) antibody.
To assess whether Ser-56 is a major phosphorylation site on vimentin mediated by PAK, phosphorylation of the vimentin mutant S56A was also evaluated. Phosphorylation levels of the vimentin S56A mutant were relatively lower as compared to wild type vimentin and they were not significantly increased after the addition of PAK (Fig. 2, n = 4, P < 0.05).
PAK induces disassembly of wild type vimentin filaments, but not vimentin S56A mutant filaments, in vitro
Vimentin phosphorylation is associated with vimentin disassembly in smooth muscle cells upon agonist activation (6). To assess whether vimentin phosphorylation on Ser-56 by PAK modulates its assembly/disassembly, we developed an in vitro filament assembly assay. Soluble wild type vimentin and its mutant S56A were treated with or without activated PAK, after which 150 mM NaCl was added to initiate vimentin filament formation. These proteins were plated on slides and were immunostained with vimentin antibody.
In the absence of PAK, wild-type vimentin was filamentous and part of vimentin filaments was assembled into a super-coiled structure (Fig. 3, panel a). The formation of super-coiled vimentin filaments might be due to lack of tension imposed on the filaments. Treatment with PAK led to the disassembly of wild type vimentin intermediate filaments (Fig. 3, panel b). Likewise, vimentin mutant S56A displayed a filamentous structure when it was not treated with PAK (Fig. 3, panel c). However, incubation of the mutant with PAK did not trigger the disassembly of the vimentin S56A mutant filaments (Fig. 3, panel d). The results suggest that vimentin phosphorylation on Ser-56 by PAK directly leads to vimentin filament disassembly.
Figure 3. PAK treatment inhibits assembly of wild type vimentin filaments, but not vimentin S56A mutant filaments.
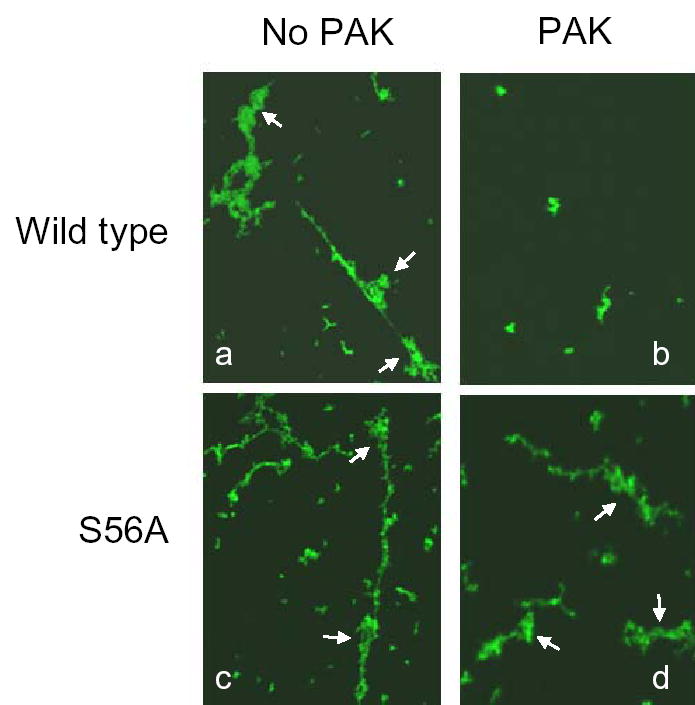
Purified wild-type vimentin and vimentin mutant S56A were placed in the kinase buffer in the presence or absence of PAK for 30 min. NaCl (150 mM) was added to initiate filament formation followed by crosslink with 0.1% glutaraldehyde. The samples were plated on slides and then immunostained for vimentin. PAK treatment prevented the filament assembly of wild type vimentin (a and b). In contrast, the vimentin S56A mutant was able to form a filamentous structure in the presence of PAK (c and d). Images are representatives of three identical experiments. Arrows point to super-coiled structures of vimentin filaments.
The amount of CAS in insoluble vimentin fraction is reduced in smooth muscle cells elicited by 5-HT
The vimentin framework binds to certain signaling partners such as CamKII and Rho kinase (7,11). The adapter protein CAS and the small GTPase Cdc42 have been shown to regulate active force development in smooth muscle (12,14,24,25). To determine whether CAS and Cdc42 associate with the vimentin network, soluble and insoluble (cytoskeletal) vimentin from unstimulated smooth muscle cells was separated by SDS-PAGE, and blots of the fractions were probed with antibodies against CAS, Cdc42 and vimentin. CAS, but not Cdc42, was found in the fractions of both insoluble vimentin and soluble vimentin (Fig. 4A). Quantification analysis showed that 25% of total CAS was associated with insoluble vimentin fraction (Fig. 4B, n = 8).
Figure 4. Crk-associated substrate (CAS) is dissociated from cytoskeletal vimentin upon 5-HT stimulation.
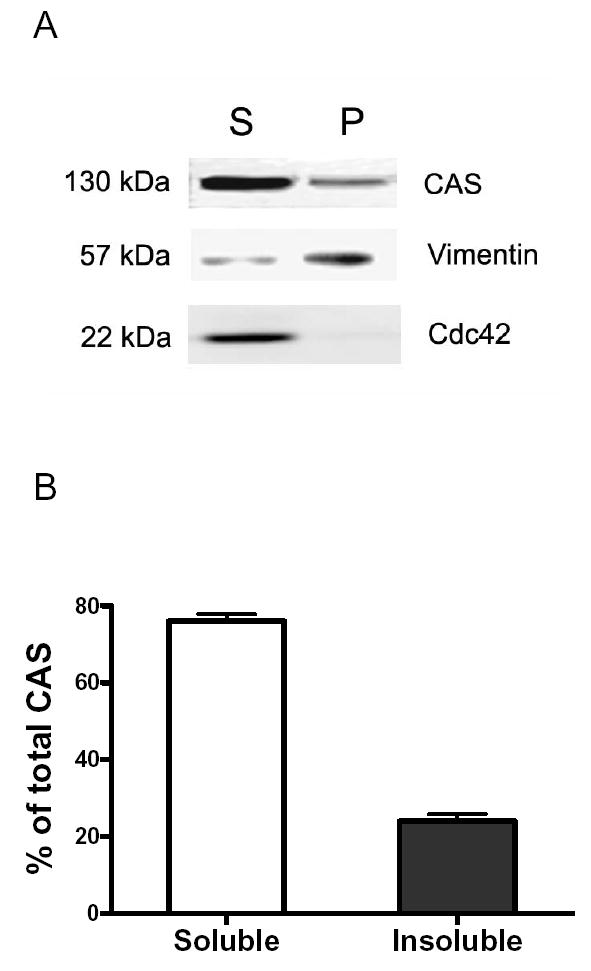

(A) CAS is associated with cytoskeletal vimentin. Blots of vimentin supernatant (S) and pellet (P) fractions from cultured tracheal smooth muscle cells were probed for CAS, Cdc42 and vimentin. CAS, but not Cdc42, was found in the cytoskeletal (pellet) vimentin fraction. (B) The amount of CAS on the immunoblots from each fraction is quantified after scanning densitometry analysis. The relative portion of soluble CAS is expressed as soluble CAS/total CAS (soluble CAS + insoluble CAS) × 100; the relative portion of insoluble CAS, insoluble CAS/total CAS × 100 (n = 8). (C) Blots of equal amount of vimentin from unstimulated and 5-HT stimulated cells (10 μM, 15 min) were probed with use of CAS antibody and vimentin antibody. The inset represents an immunoblot showing the effects of 5-HT stimulation on the amount of CAS in cytoskeletal vimentin. The ratios of CAS to vimentin in stimulated cells are normalized to that in unstimulted cells. *Significantly lower ratios of CAS/vimentin in stimulated cells as compared to unstimulated cells (P < 0.05, n = 5). (D) Insoluble CAS/soluble CAS ratios in cells elicited by 5-HT activation are normalized to those in unstimulated cells. *Significantly lower ratios in stimulated cells when compared with unstimulated cells (P < 0.05, n = 5). All values are means ± SE.
Vimentin filaments undergo partial disassembly upon agonist stimulation of smooth muscle cells (6). Vimentin disassembly has been implicated in the redistribution of Rho kinase in fibroblasts (11). We also evaluated the effects of 5-HT stimulation on the association of CAS with cytoskeletal vimentin in smooth muscle cells. Treatment with 5-HT led to the decrease in the amount of CAS associated with cytoskeletal vimentin. The ratio of CAS to cytoskeletal vimentin was statistically lower in smooth muscle tissues in response to 5-HT stimulation as compared to unstimulated smooth muscle cells (Fig. 4C, n = 5, P < 0.05).
The distribution of CAS in different fractions was also evaluated in response to agonist stimulation. Activation with 5-HT led to a decrease in the amount of CAS in insoluble fractions. The ratios of insoluble CAS/soluble CAS elicited by 5-HT stimulation were lower when compared to unstimulated smooth muscle cells (Fig. 4D, n = 5, P < 0.05).
Vimentin phosphorylation at Ser-56 in response to 5-HT stimulation is diminished in smooth muscle cells expressing vimentin S56A mutant
Thus far, our results suggested that the PAK-catalyzed vimentin phosphorylation and disassembly are associated with the redistribution of CAS upon agonist stimulation of smooth muscle cells. To determine whether vimentin phosphorylation and disassembly influence the CAS redistribution, we attempted to express the vimentin S56A in smooth muscle cells to block vimentin phosphorylation and disassembly, and then evaluated the CAS redistribution as compared to cells expressing wild type vimentin.
First, we assessed whether vimentin phosphorylation in response to 5-HT stimulation is depressed in cells expressing the vimentin S56A mutant. pEGFP plasmids alone or plasmids encoding wild type vimentin or vimentin mutant S56A were transfected into smooth muscle cells. Immunoblot analysis showed that EGFP-labeled recombinant vimentin was highly expressed; approximately 90% of total vimentin was recombinant vimentin in the transfected cells (Fig. 5, A and B). Smooth muscle cells expressing wild type vimentin or vimentin mutant S56A were stimulated with 10 μm 5-HT for 5–15 min, or left unstimulated. Blots of protein extracts from these cells were probed using phospho-vimentin (Ser-56) antibody, stripped and reprobed by use of EGFP antibody.
Figure 5. Ser-56 is a major phosphorylation site on vimentin in smooth muscle cells stimulated by 5-HT.


(A) Smooth muscle cells were transfected with pEGFP plasmids and the plasmids encoding wild type (WT) vimentin or S56A vimentin mutant, or they were untransfected. Protein extracts of these cells were immunoblotted (IB) with EGFP antibody. EGFP-tagged proteins with 85-kDa molecular weight were detected in the extracts of cells treated with the plasmids for WT and S56A vimentin, but not in untransfected cells or in cells transfected with empty vector, indicating effective expression of the fusion proteins in these cells. Blots are representative of four identical experiments. (B) Protein extracts of untransfected cells and cells transfected with the empty vector or the plasmids for WT or S56A vimentin were immunoblotted with monoclonal vimentin antibody. In cells transfected with constructs of WT vimentin or S56A vimentin mutant, approximately 90% of total vimentin was the tagged recombinant proteins. Blots are representative of four identical experiments. (C) Smooth muscle cells expressing WT vimentin or S56A vimentin were stimulated with 10 μm 5-HT for 5–15 min, or left unstimulated. Blots of these cells were probed with use of phospho-vimentin antibody (pS56 Ab), stripped and reprobed with EGFP antibody. Vimentin phosphorylation in response to 5-HT stimulation is reduced in cells expressing S56A vimentin mutant as compared to in cells expressing WT vimentin. (D) Vimentin phosphorylation is normalized to the level of unstimulated cells expressing WT vimentin. *Significantly lower values for each time point in cells expressing S56A vimentin mutant than in cells expressing wild type vimentin (P < 0.05). The values represent means ± SE (n = 4).
Stimulation with 5-HT resulted in increases in vimentin phosphorylation at Ser-56 in the cells transfected with plasmids encoding wild type vimentin (Fig. 5C). The phosphorylation levels in the cells expressing wild type vimentin were increased by 1.7 fold after stimulation with 5-HT for 10–15 min (Fig. 5D), which is similar to our previous results from untransfected cells (6). However, the phosphorylation levels in response to 5-HT stimulation in the cells expressing vimentin mutant S56A was significantly lower as compared with cells expressing wild type vimentin (Fig. 5, C and D, n = 4, P < 0.05).
Stimulation with 5-HT does not lead to the increase in the ratios of soluble vimentin to insoluble vimentin in cells expressing vimentin mutant S56A
To assess whether vimentin phosphorylation at Ser-56 affects vimentin disassembly in vivo, smooth muscle cells expressing wild type vimentin or vimentin mutant S56A were stimulated with 10 μM 5-HT for 5–15 min, or left unstimulated. The ratios of soluble vimentin to insoluble vimentin were then determined.
Activation with 5-HT increased the amount of soluble vimentin in cells expressing wild type vimentin; however, the 5-HT mediated increase in the level of soluble vimentin was attenuated in cells expressing vimentin mutant S56A (Fig. 6A). The ratios of soluble vimentin to cytoskeletal vimentin in response to 5-HT stimulation were lower in cell expressing vimentin S56A mutant than in cells expressing wild type vimentin (Fig. 6B, n = 4, P < 0.05).
Figure 6. Increases in soluble vimentin/insoluble vimentin ratios in response to 5-HT stimulation are attenuated in cells expressing vimentin S56A mutant.
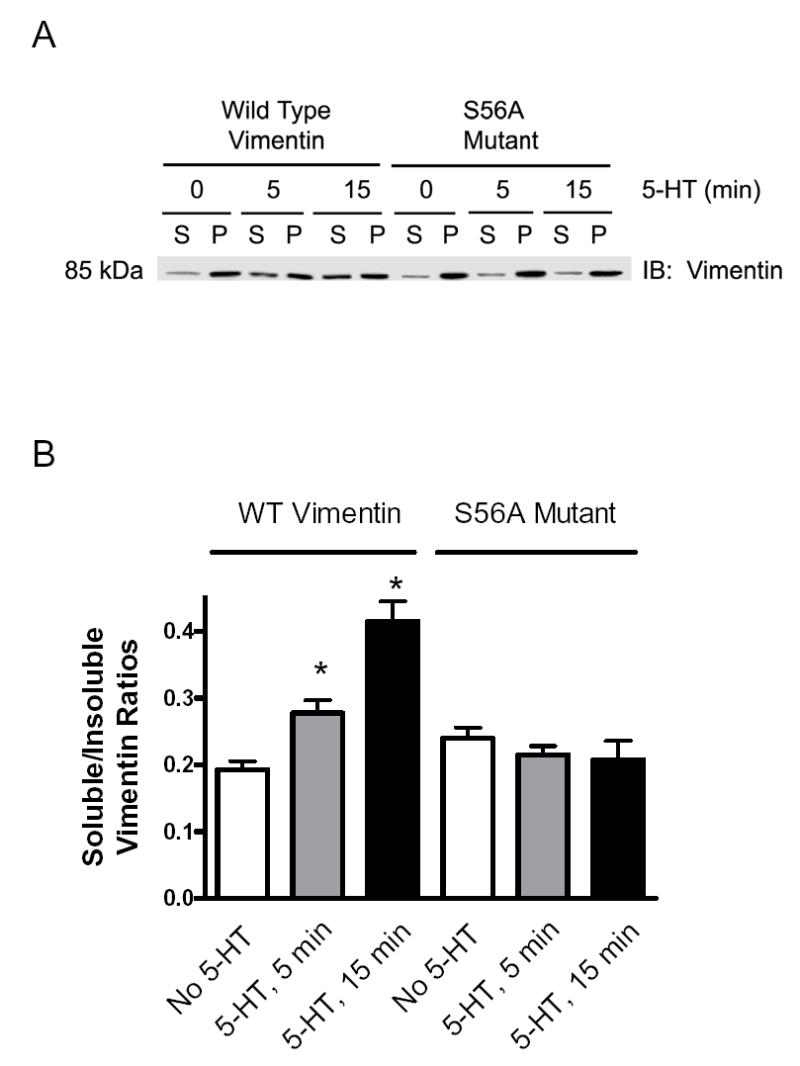
(A) Immunoblot illustrating the effects of the expression of S56A vimentin mutant on the amount of soluble (supernatant) and insoluble (pellet) vimentin elicited by 5-HT. Smooth muscle cells expressing wild type vimentin or S56A vimentin mutant were stimulated with 10 μM 5-HT for 5–15 min, or they were not stimulated. Supernatant (S) and pellet (P) vimentin was assessed by fractionation. (B) The ratios of soluble vimentin over insoluble vimentin were determined scanning densitometry of immunoblots for fractions. * Significantly greater ratios of soluble vimentin/insoluble vimentin in stimulated cells than in corresponding unstimulated cells (P < 0.05). All values are means ± SE (n = 4–5).
The redistribution of CAS elicited by 5-HT is depressed in cells expressing S56A vimentin mutant
To evaluate whether vimentin phosphorylation at Ser-56 is required for CAS redistribution, we assessed the ratios of vimentin-associated (insoluble) CAS to cytoskeletal vimentin in unstimulated or stimulated (10 μM 5-HT, 15 min) smooth muscle cells expressing wild type vimentin or vimentin mutant S56A.
Expression of S56A vimentin mutant suppressed the decrease in the CAS/vimentin ratios in response to 5-HT stimulation. In cells expressing wild type vimentin, the ratios of insoluble CAS to cytoskeletal vimentin elicited by 5-HT was significantly decreased as compared to unstimulated cells. In contrast, the ratios of insoluble CAS over cytoskeletal vimentin were not significantly different in unstimulated or 5-HT stimulated cells expressing the vimentin S56A mutant (Fig. 7 A, P < 0.05, n = 7).
Figure 7. Expression of the non-phosphorylatable vimentin mutant attenuates the redistribution of CAS stimulated by 5-HT.
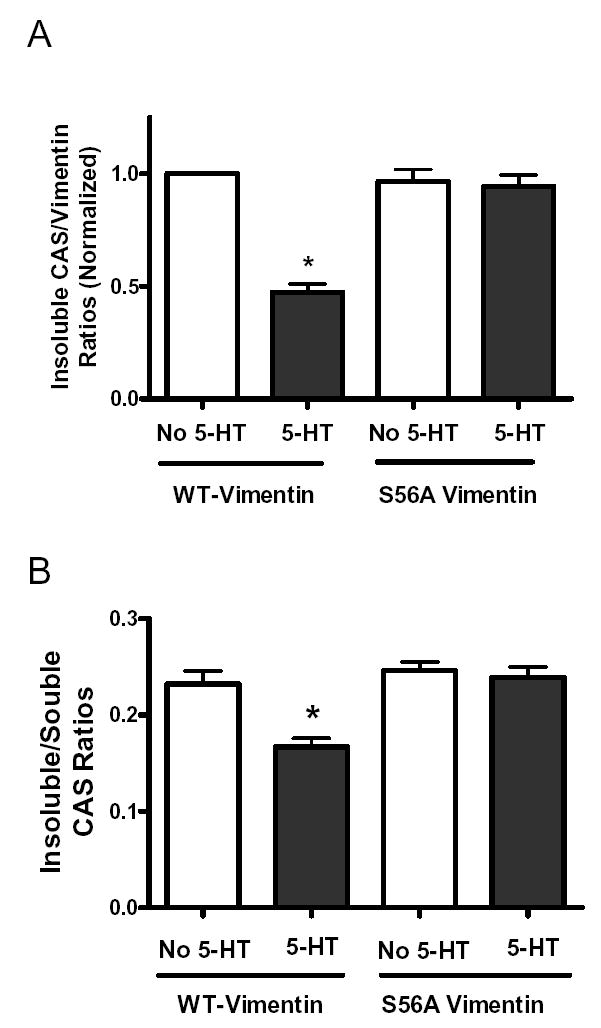
(A) Smooth muscle cells expressing recombinant proteins were stimulated with 10 μM 5-HT for 15 min, or left unstimulated. The ratios of insoluble CAS over cytoskeletal vimentin were then determined. Ratios are normalized to the level in unstimulated cells transfected with plasmids for wild type vimentin. The decrease in the CAS/vimentin ratios stimulated by 5-HT was inhibited in cells expressing S56A vimentin mutant as compared to cells expressing wild type vimentin (n = 7). (B) Insoluble CAS/soluble CAS ratios were assessed for unstimulated and 5-HT stimulated cells expressing recombinant proteins. The decrease in ratios of insoluble CAS/soluble CAS elicited by 5-HT was suppressed in cells expressing vimentin S56A mutant when compared with cells expressing wild type vimentin (n = 10). * Significantly lower ratios of soluble CAS/insoluble CAS in stimulated cells than in corresponding unstimulated cells (P < 0.05). All values are means ± SE.
Moreover, we assessed the effects of the vimentin S56A mutant on ratios of vimentin-associated CAS to soluble CAS. The insoluble CAS/soluble CAS ratios were decreased in 5-HT stimulated cells expressing wild type vimentin when compared with the cells not treated with 5-HT. However, the ratios of insoluble CAS to soluble CAS in response to 5-HT stimulation were not reduced in cells expressing the S56A vimentin mutant as compared to unstimulated cells (Fig. 7B, n = 10, P < 0.05).
Activation of PAK1 elicited by 5-HT stimulation is suppressed in cells expressing vimentin S56A mutant
Since the dissociation of CAS from the vimentin network is regulated by vimentin phosphorylation and CAS may affect PAK activity (19,22), we determined whether vimentin phosphorylation at Ser-56 reciprocally regulates PAK1. Smooth muscle cells expressing wild type vimentin or vimentin mutant S56A were stimulated with 10 μM 5-HT for 15 min or left unstimulated. PAK1 phosphorylation from these cells was evaluated by immunoblot analysis.
Phosphorylation of PAK1 at Thr-423 in response to 5-HT stimulation was enhanced in cells expressing wild type vimentin. Nevertheless, the agonist-induced PAK1 phosphorylation at this site was significantly suppressed in cells expressing vimentin S56A mutant as compared to cells expressing wild type vimentin (Fig. 8, n = 5, P < 0.05).
Figure 8. The increase in PAK phosphorylation elicited by 5-HT is diminished in cells expressing vimentin S56A mutant.
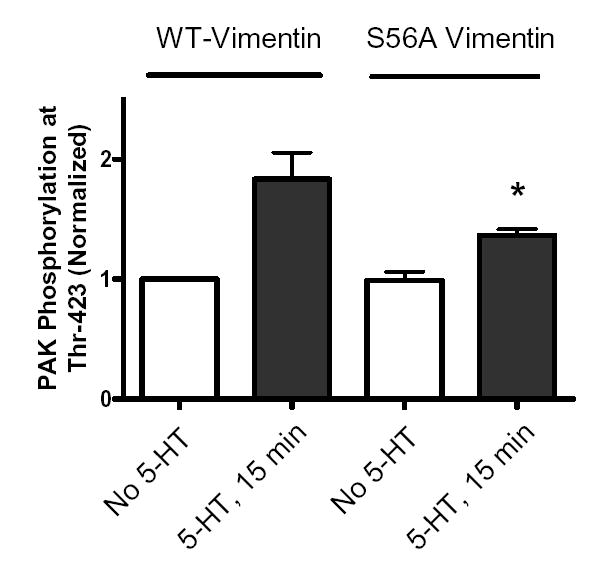
Smooth muscle cells expressing wild type vimentin or S56A vimentin mutant were exposed to 10 μM 5-HT for 15 min, or unstimulated. Blots of these cells were probed by use of phospho-PAK1 (Thr-423), stripped and reprobed with PAK1 antibody. PAK phosphorylation is normalized to the levels obtained from unstimulated cells transfected with wild type vimentin. *Significantly lower phosphorylation levels in stimulated cells containing S56A vimentin mutant than in stimulated cells expressing wild type vimentin (P < 0.05). Values shown are mean ± SE (n = 5).
Spatial reorientation of vimentin filaments in response to 5-HT activation is suppressed in cells expressing vimentin mutant S56A
Phosphorylation of vimentin has been shown to be associated with spatial restructuring of the vimentin network (6,26). To assess whether vimentin phosphorylation at Ser-56 is required for its filamentous reorganization in smooth muscle, cells transfected with wild type vimentin or vimentin mutant S56A were stimulated with 10 μM 5-HT for 15 min or left unstimulated after transfection for 48 hrs. These cells were then fixed and analyzed under fluorescence microscope.
Vimentin phosphorylation at Ser-56 is crucial for its spatial reorganization in smooth muscle cells upon 5-HT stimulation. In unstimulated cells transfected with wild type vimentin (Fig. 9A, panel a), vimentin exhibited curved filamentous structures. Exposure of cells transfected with wild type vimentin to 5-HT induced the straightness of vimentin filaments (Fig. 9A, panel b). The 5-HT induced spatial reorientation of the GFP tagged vimentin fibers is similar to that of native vimentin filaments in smooth muscle cells (6). In unstimulated cells expressing vimentin mutant S56A, vimentin fibers also showed the curvature (Fig. 9A, panel c). However, stimulation with 5-HT did not induce the spatial rearrangement; the intermediate filaments remained curved in the stimulated cells transfected with vimentin mutant S56A (Fig. 9A, panel d). Stimulation with 5-HT resulted in the increase in the numbers of cells displaying the reorganized intermediate filaments in cells expressing wild type vimentin, but not in cells expressing vimentin S56A mutant (Fig. 9B).
Figure 9. Activation of smooth muscle cells with 5-HT does not induce structural rearrangement of vimentin S56A mutant filaments.
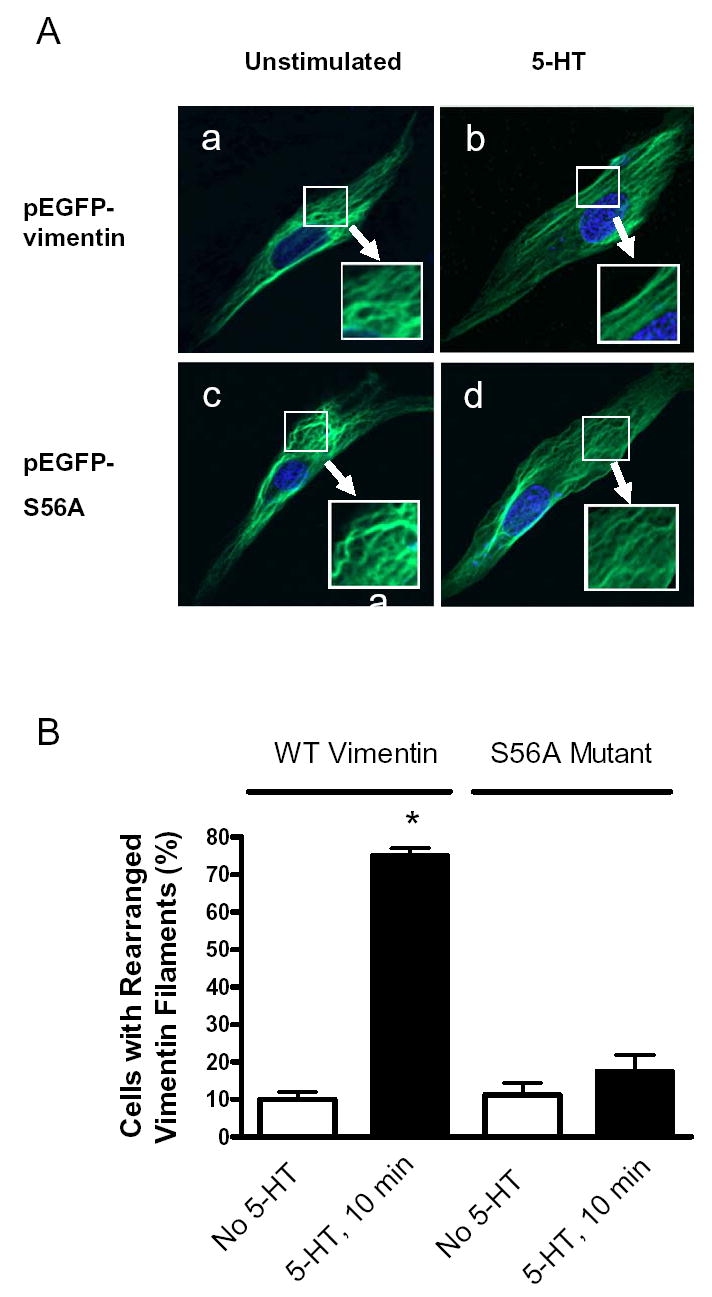
(A) Smooth muscle cells expressing EGFP-tagged wild type vimentin or vimentin S56A mutant were stimulated with 10 μM 5-HT for 15 min, or left not stimulated. Images of vimentin or its mutant filaments in these cells were viewed under confocal fluorescence microscope. In cells expressing wild type vimentin, stimulation with 5-HT induced the reorientation of vimentin filaments from a curved structure (panel a) to a straight structure (panel b). In contrast, activation with 5-HT did not trigger the structural rearrangement of vimentin S56A mutant filaments in cells (panel c and d). The DAPI-stained nuclei were shown in blue. (B) Vimentin filaments displaying straightness longer than one-quarter of cell length are considered as straight filaments. Cells with more than four straight vimentin or its mutant filaments are counted as cells with rearranged filaments. Quantitative analysis is expressed as (numbers of cells with rearranged vimentin/mutant filaments)/(numbers of total cells observed) × 100 (6). *Significantly higher percentage of cells with rearranged vimentin filaments in 5-HT stimulated cells expressing wild type vimentin compared to corresponding unstimulated cells (P < 0.05). The values represent means ± SE (n = 4).
DISCUSSION
The intermediate filament protein vimentin undergoes phosphorylation at Ser-56 and partial filament disassembly in cultured smooth muscle cells upon stimulation with 5-HT (6). In this report, PAK is able to directly initiate vimentin disassembly associated with Ser-56 phosphorylation. Second, the adapter protein CAS is dissociated from cytoskeletal vimentin during 5-HT activation. The expression of the non-phosphorylatable vimentin mutant S56A attenuates the dissociation of CAS from cytoskeletal vimentin. Third, the expression of vimentin S56A mutant suppresses PAK activation and the spatial reorganization of vimentin filament framework upon agonist stimulation. These results suggest that vimentin phosphorylation at Ser-56 and its disassembly may play a critical role in regulating PAK activation and the vimentin cytoskeleton.
In cultured smooth muscle cells, PAK silencing attenuates vimentin phosphorylation upon contractile activation (6). In this report, stimulation with 5-HT induced PAK1 autophosphorylation at Thr-423 in smooth muscle cells. In inactive status, the amino-terminal domain of PAK interacts with the carboxyl-terminal catalytic motif, forming an intramolecular inhibitory conformation and diminishing enzyme activity. Autophosphorylation at Thr-423 has been proposed to induce a conformational change for the activation of PAK (18). Thus, the results suggest that stimulation with 5-HT may increase PAK1 activity in smooth muscle cells.
Although the intermediate filament framework in the cell is resistant to the extraction with detergents, its assembly/disassembly may be regulated by phosphorylation (1,2). Contractile stimulation of smooth muscle cells induces phosphorylation at Ser-56, which is located at the globular head domain of vimentin molecule (2,6). In in vitro studies of this report, treatment of wild type vimentin with PAK initiated the almost complete filament disassembly while substitution of alanine at Ser-56 impaired its disassembly. These results suggest that PAK mediated Ser-56 phosphorylation profoundly influences the assembly status of vimentin filaments. Because the effects of serine phosphorylation of a protein can be mimicked by introduction of a negative charge at the corresponding serine position (27), we speculate that phosphorylation at Ser-56 creates a negative charge in the side chain, which may facilitate changes in vimentin conformation associated with disassembly.
There is evidence that the vimentin filament framework harbors Rho kinase and CamKII, which may be dissociated from the network when vimentin partial disassembly or structural rearrangement occurs (7,11). The adapter protein CAS may participate in the signaling process that mediates smooth muscle force development (12,14). In the present study, CAS associates with the vimentin cytoskeleton and, agonist activation triggers the dissociation of CAS from cytoskeletal vimentin. The amount of CAS dissociated from cytoskeletal vimentin is approximately 50% of total CAS associated with vimentin filaments. Furthermore, the expression of the vimentin S56A mutant in smooth muscle cells attenuates the CAS redistribution during agonist stimulation. Since vimentin phosphorylation at Ser-56 and partial disassembly are also inhibited in cells expressing the vimentin S56A mutant, we propose that contractile stimulation of smooth muscle cells leads to vimentin phosphorylation at this position, inducing vimentin disassembly and CAS release from the vimentin filament network.
Changes in physiological status of a substrate may conversely serve as a regulator of its upstream enzyme, forming a feedback mechanism. As mentioned above, PAK is able to mediate vimentin phosphorylation both in vitro and in vivo. In the present study, PAK activation in response to 5-HT stimulation is inhibited in cells expressing S56A vimentin mutant, suggesting that vimentin phosphorylation at Ser-56 reciprocally regulates PAK activity. The positive feedback mechanism may render PAK in an active status during agonist stimulation. The mechanisms by which vimentin phosphorylation at this residue regulates PAK activity are unknown. Because CAS can be dissociated from the vimentin network during contractile activation, it is possible that the increase in soluble CAS mediated by vimentin phosphorylation may facilitate the activation of PAK. When activated, CAS may be able to form a protein complex containing CrkII, paxillin, p95-PKL and PAK, which may mediate the activation of PAK (21,22).
In addition to the regulation of signaling molecule redistribution, vimentin phosphorylation may be associated with spatial restructuring of the vimentin network (6). In the present study, 5-HT stimulated vimentin phosphorylation at position-56 was reduced in cells expressing the vimentin S56A mutant. Moreover, spatial reorganization of the vimentin cytoskeleton in response to stimulation with 5-HT was also attenuated in cells expressing the S56A vimentin mutant. These results lead us to suggest that phosphorylation at Ser-56 is required for vimentin structural reorganization during agonist activation of smooth muscle cells.
As mentioned above, PAK treatment of vimentin protein in vitro triggered almost complete filament disassembly while vimentin filaments underwent partial disassembly and spatial rearrangement in the cells in response to agonist activation. The difference between the in vitro study and the in situ study may stem from the lower extent of vimentin phosphorylation in the cells. Our prior investigation has shown that approximately 65% of soluble vimentin gets phosphorylation at Ser-56 in smooth muscle cells elicited by 5-HT, which is associated with the partial vimentin disassembly as estimated by a cell fractionation assay, and is regulated by PAK (6). Thus, we postulate that smooth muscle cells may possess limited amount of PAK, and/or, PAK has limited access to vimentin. Upon agonist stimulation, vimentin in the cells undergoes partial phosphorylation and partial disassembly that may facilitate the spatial reorientation of vimentin filaments. In contrast, in the in vitro study, sufficient amount of PAK is added into the reaction system, and/or PAK has full access to vimentin. PAK treatment is able to induce comprehensive filament disassembly.
The biological significance of vimentin filament rearrangement in smooth muscle cells is not fully understood. The spatial reorganization might facilitate contractile element reorganization and/or the linkage of intermediate filaments to desmosomes, which may be a part of cellular processes that affect force development in smooth muscle (5,6,28–30). The structural rearrangement of vimentin filaments might also be associated with the mitogenic processes in smooth muscle cells induced by 5-HT (6,31).
Thus, we propose a unique vimentin cytoskeletal signaling mechanism in smooth muscle cells. Activation of PAK by 5-HT stimulation may catalyze vimentin phosphorylation at Ser-56, leading to vimentin disassembly and the dissociation of the adapter protein CAS from the vimentin network. The released CAS may facilitate the formation of a multi-protein complex including CAS and PAK, maintaining PAK in an active state. In addition, vimentin phosphorylation at this position may induce the spatial reorientation of vimentin filaments, which may be associated with contractile element reorganization in response to agonist stimulation (Fig. 10).
Figure 10. Vimentin cytoskeleton signaling in smooth muscle cells.
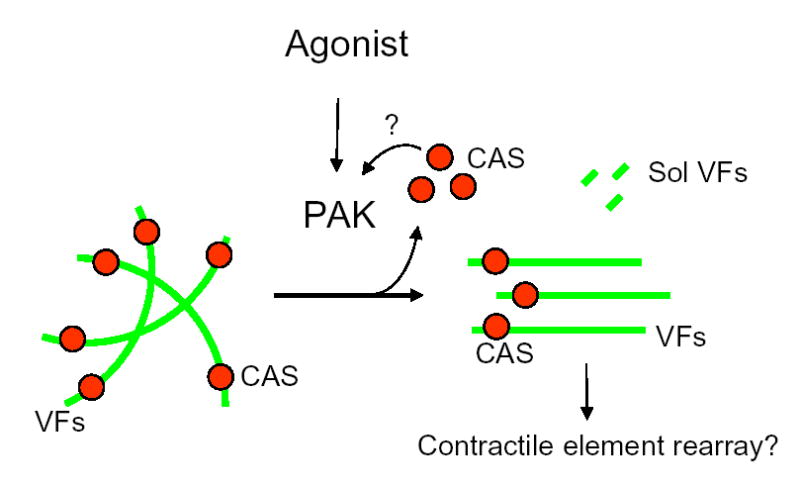
Agonist stimulation of smooth muscle cells may activate PAK, leading to vimentin phosphorylation at Ser-56 and vimentin disassembly, which may initiate the partial dissociation of CAS (red balls) from vimentin filaments (VFs, green lines). The increase in soluble (Sol) CAS may maintain PAK in an active state possibly via the formation of a multi-protein complex (See Discussion). In addition, vimentin phosphorylation may induce the spatial reorientation of vimentin filaments, facilitating contractile element reorganization in response to agonist stimulation.
Footnotes
The abbreviations used are: PAK, p21-activated kinase; 5-HT, 5-hydroxytryptamine (serotonin); CAS, Crk-associated substrate; CamKII, Ca2+/calmodulin dependent protein kinase II; EGFP, enhanced green fluorescence protein; GST, glutathione S-transferase.
This work was supported by National Heart, Lung, and Blood Institute Grant HL-75388 and an American Heart Association Scientist Development Grant (to D.D.T.).
References
- 1.Eriksson JE, He T, Trejo-Skalli AV, Harmala-Brasken AS, Hellman J, Chou YH, Goldman RD. J Cell Sci. 2004;117:919–932. doi: 10.1242/jcs.00906. [DOI] [PubMed] [Google Scholar]
- 2.Chang L, Goldman RD. Nat Rev Mol Cell Biol. 2004;5:601–613. doi: 10.1038/nrm1438. [DOI] [PubMed] [Google Scholar]
- 3.Bruel A, Paschke S, Jainta S, Zhang Y, Vassy J, Rigaut JP, Beil M. Anticancer Res. 2001;21:3973–3980. [PubMed] [Google Scholar]
- 4.Meriane M, Mary S, Comunale F, Vignal E, Fort P, Gauthier-Rouviere C. J Biol Chem. 2000;275:33046–33052. doi: 10.1074/jbc.M001566200. [DOI] [PubMed] [Google Scholar]
- 5.Wang R, Li Q, Tang DD. Am J Physiol Cell Physiol. 2006;291:C483–C489. doi: 10.1152/ajpcell.00097.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang DD, Bai Y, Gunst SJ. Biochem J. 2005;388:773–783. doi: 10.1042/BJ20050065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marganski WA, Gangopadhyay SS, Je HD, Gallant C, Morgan KG. Circ Res. 2005;97:541–549. doi: 10.1161/01.RES.0000182630.29093.0d. [DOI] [PubMed] [Google Scholar]
- 8.Goto H, Kosako H, Tanabe K, Yanagida M, Sakurai M, Amano M, Kaibuchi K, Inagaki M. J Biol Chem. 1998;273:11728–11736. doi: 10.1074/jbc.273.19.11728. [DOI] [PubMed] [Google Scholar]
- 9.Goto H, Tanabe K, Manser E, Lim L, Yasui Y, Inagaki M. Genes Cells. 2002;7:91–97. doi: 10.1046/j.1356-9597.2001.00504.x. [DOI] [PubMed] [Google Scholar]
- 10.Li QF, Wang R, Tang DD. FASEB J. 2006;20:A1243. [Google Scholar]
- 11.Sin WC, Chen XQ, Leung T, Lim L. Mol Cell Biol. 1998;18:6325–6339. doi: 10.1128/mcb.18.11.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang DD, Tan J. Hypertension. 2003;42:858–863. doi: 10.1161/01.HYP.0000085333.76141.33. [DOI] [PubMed] [Google Scholar]
- 13.Defilippi P, Di SP, Cabodi S. Trends Cell Biol. 2006 doi: 10.1016/j.tcb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 14.Ogden K, Thompson JM, Hickner Z, Huang T, Tang DD, Watts SW. Am J Physiol Heart Circ Physiol. 2006 doi: 10.1152/ajpheart.00229.2006. published on 7/21/2006. [DOI] [PubMed] [Google Scholar]
- 15.Dechert MA, Holder JM, Gerthoffer WT. Am J Physiol Cell Physiol. 2001;281:C123–C132. doi: 10.1152/ajpcell.2001.281.1.C123. [DOI] [PubMed] [Google Scholar]
- 16.McFawn PK, Shen L, Vincent SG, Mak A, Van Eyk JE, Fisher JT. Am J Physiol Lung Cell Mol Physiol. 2003;284:L863–L870. doi: 10.1152/ajplung.00068.2002. [DOI] [PubMed] [Google Scholar]
- 17.Knaus UG, Bokoch GM. Int J Biochem Cell Biol. 1998;30:857–862. doi: 10.1016/s1357-2725(98)00059-4. [DOI] [PubMed] [Google Scholar]
- 18.Jaffer ZM, Chernoff J. Int J Biochem Cell Biol. 2002;34:713–717. doi: 10.1016/s1357-2725(01)00158-3. [DOI] [PubMed] [Google Scholar]
- 19.Bokoch GM. Annu Rev Biochem. 2003;72:743–781. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- 20.Woolfolk EA, Eguchi S, Ohtsu H, Nakashima H, Ueno H, Gerthoffer WT, Motley ED. Am J Physiol Cell Physiol. 2005;289:C1286–C1294. doi: 10.1152/ajpcell.00448.2004. [DOI] [PubMed] [Google Scholar]
- 21.Turner CE, Brown MC, Perrotta JA, Riedy MC, Nikolopoulos SN, McDonald AR, Bagrodia S, Thomas S, Leventhal PS. J Cell Biol. 1999;145:851–863. doi: 10.1083/jcb.145.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown MC, Turner CE. Physiol Rev. 2004;84:1315–1339. doi: 10.1152/physrev.00002.2004. [DOI] [PubMed] [Google Scholar]
- 23.West KA, Zhang H, Brown MC, Nikolopoulos SN, Riedy MC, Horwitz AF, Turner CE. J Cell Biol. 2001;154:161–176. doi: 10.1083/jcb.200101039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang DD, Gunst SJ. J Biol Chem. 2004;279:51722–51728. doi: 10.1074/jbc.M408351200. [DOI] [PubMed] [Google Scholar]
- 25.Tang DD, Zhang W, Gunst SJ. J Biol Chem. 2005;280:23380–23389. doi: 10.1074/jbc.M413390200. [DOI] [PubMed] [Google Scholar]
- 26.Chan W, Kozma R, Yasui Y, Inagaki M, Leung T, Manser E, Lim L. Eur J Cell Biol. 2002;81:692–701. doi: 10.1078/0171-9335-00281. [DOI] [PubMed] [Google Scholar]
- 27.Finley NL, Rosevear PR. J Biol Chem. 2004;279:54833–54840. doi: 10.1074/jbc.M408304200. [DOI] [PubMed] [Google Scholar]
- 28.Kowalczyk AP, Bornslaeger EA, Norvell SM, Palka HL, Green KJ. Int Rev Cytol. 1999;185:237–302. doi: 10.1016/s0074-7696(08)60153-9. [DOI] [PubMed] [Google Scholar]
- 29.Small JV, Gimona M. Acta Physiol Scand. 1998;164:341–348. doi: 10.1046/j.1365-201X.1998.00441.x. [DOI] [PubMed] [Google Scholar]
- 30.Small JV. Bioessays. 1995;17:785–792. doi: 10.1002/bies.950170908. [DOI] [PubMed] [Google Scholar]
- 31.Parrott DP, Lockey PM, Bright CP. Atherosclerosis. 1991;88:213–218. doi: 10.1016/0021-9150(91)90083-f. [DOI] [PubMed] [Google Scholar]