

Abstract
Prolactin (PRL) modulates proliferation in the mammary gland and other tissues, in part through inducing transcription of cyclin D1, a key regulator of G, phase cell cycle progression. We showed previously that PRL, via Jak2, induces binding of Stat5 to a distal GAS site (GAS1) in the cyclin D1 promoter. However, full promoter activity requires additional regions, and in this paper we explored PRL-induced activity at sites other than GAS1. We defined a second PRL-responsive region spanning −254 to −180 that contains a second GAS site (GAS2) and an Oct-1 binding site. Although mutational analysis indicated independence from GAS2, proximal promoter activity remained Stat5-dependent, suggesting alternative mechanisms. EMSA showed that Oct-1 binds the −254 to −180 region and that PRL decreased Oct-1 binding, leading to increased PRL-responsiveness of the proximal cyclin D1 promoter in multiple cell lines. This suggests a role for Oct-1 in PRL-dependent control of cyclin D1 transcription.
Keywords: Prolactin, Cyclin D1, Stat5, Oct-1
1. Introduction
Prolactin (PRL) modulates a wide variety of physiological processes, including well-characterized activities in mammary gland function and reproduction, as well as more recently recognized roles in metabolism, maternal behavior, immune function, hair follicles, lacrimal glands and osteogenesis (for reviews, Freeman et al., 2000; Goffin et al., 2002). PRL exerts trophic effects on many of these targets, promoting proliferation (for review, Buckley, 2001). Although the mechanism that mediates this action of PRL has not been elucidated for many of these systems, PRL has been shown to increase transcription of members of the cyclin D family in some cell types (Brockman et al., 2002; Friedrichsen et al., 2003). These cyclins associate with cyclin-dependent kinases (cdk)4 and 6 in early G1 phase of the cell cycle to initiate cell cycle progression. This key role makes them important sites of integration for multiple hormones and growth factors. Knowledge of PRL signaling to these cell cycle regulators is critical for understanding the role of PRL and its interactions with other factors both in physiologic development as well as diseases of these same tissues.
Activation of the PRL receptor (PRLR) initiates multiple kinase cascades, the most studied of which is the Jak-Stat pathway (for reviews, Bole-Feysot et al., 1998; Clevenger et al., 2003). Jak2 phosphorylation of signal transducers and activators of transcription (Stats) leads to their dimerization, translocation into the nucleus and subsequent activation of GAS enhancer sites in the promoters of target genes. The cyclin D1 promoter contains two consensus GAS sites at −457 and −224, and we have shown that PRL induces binding of Stat5 to the more distal GAS site (GAS1) to enhance promoter activity (Brockman et al., 2002). Mutation of GAS1, but not the more proximal GAS2, abolished the effect of PRL on activity of a 1 kb promoter construct in CHO cells.
However, as reported herein, deletion of the more distal promoter revealed additional sites that modulate PRL-responsiveness. Another Stat5-responsive region was defined within the proximal cyclin D1 promoter, which contained the GAS2 site and a binding site for Oct-1, a ubiquitous transcription factor. PRL modulates the action of constitutively bound Oct-1 at this site, independent of an intact GAS2 site, leading to an increase in PRL-stimulated promoter activity. Understanding the mechanisms for PRL-dependent regulation of cyclin D1 expression may reveal important pathways for maintenance of proper cell cycle progression in PRL target cells, or processes leading to the aberrant proliferation seen in cancer, including breast cancer.
2. Materials and methods
2.1. Materials
Stat antibodies (Stat1, sc-592X; Stat3, sc-7179; Stat5a, sc-1081X; Stat5b, sc-835) and Oct-1 antibody (sc-232X) were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). γ-32P-ATP was purchased from Amersham Pharmacia Biotech (Arlington Heights, IL). Bovine PRL, Lot AFP7170E and human PRL, Lot AFP9042, were obtained through NHPP, NIDDK and Dr. A.F. Parlow.
2.2. Plasmid constructs
The D1Δ-944 construct was generously provided by Dr. Rolf Müller at the IMT in Marburg, Germany (Herber et al., 1994). Truncations of D1Δ-944 (D1Δ-304, D1Δ-254, D1Δ-180 and D1Δ-71) were constructed by restriction enzyme digest, filling in the cut ends and ligation of the resulting plasmid. Mutation of the Oct site within D1Δ-304 was accomplished by PCR with complementary mutant oligos corresponding to the sequence 5′-CGCGGATCCAAGCTTGTCGACCC-3′. Mutations were confirmed by sequence analysis. The long form of the bovine PRLR was expressed in pcDNA3 (Invitrogen, Carlsbad, CA) (Scott et al., 1992), and the human PRLR construct was provided by Dr. C. Clevenger (Kline et al., 1999). Stat1 and 3 constructs were provided by Dr. J. Darnell (Zhong et al., 1994; Wen et al., 1995), Stat5a and 5b wild-type constructs were provided by Dr. J. Rosen (Kabotyanski and Rosen, 2003), Stat5a dominant-negative construct was from Dr. P. Bertics, University of Wisconsin, and Stat5-Δ53C and Stat5-VVV constructs were from Dr. R. Ilaria (Ilaria, Jr. et al., 1999). The cytomegalovirus-β-galactosidase construct was obtained from Dr. C. Caskey (MacGregor and Caskey, 1989).
2.3. Cell culture and transient transfection
Chinese hamster ovary (CHO-K1) cells (ATCC no. CCL-61) were maintained in DMEM/F12 containing 5% FBS and penicillin/streptomycin (Life Technologies, Inc. Gaithersburg, MD). MCF-10A human mammary epithelial cells were maintained in DMEM/F12 containing 5% horse serum, penicillin/streptomycin, 100 ng/ml cholera toxin, 20 ng/ml EGF, 500 ng/ml hydrocortisone and 0.01 ng/ml insulin. Cells were transfected using SuperFect (QIAGEN Inc., Valencia, CA) as described previously (Brockman et al., 2002), except that some transfections employed human PRLR and were consequently treated with human PRL, instead of the bovine hormone. Results obtained were indistinguishable between the two systems.
2.4. Reporter gene assays
Luciferase activity of cell lysates was determined by adding 25 μl lysate to 100 μl of luciferase substrate in a Turner Designs Model 20/20 luminometer (Turner Designs, Sunnyvale, CA). (β-galactosidase activity was measured by the Galacto Light Plus kit (Tropix Inc., Bedford, MA). Luciferase values were corrected for transfection efficiency by determining the ratio of luciferase activity/μl to β-galactosidase activity/μl and expressed as relative luciferase units. Statistical analyses were performed using Prism v.3.02 (GraphPad Software Inc., San Diego, CA, USA).
2.5. Preparation of nuclear extracts
Chinese hamster ovary cells that were stably transfected with PRLR (CHO-D6) (Gao et al., 1996) were plated at a density of 2.5 × 106 cells/10 cm plate in DMEM/F12 media with 5% FBS and 0.5 mg/ml geneticin, and incubated at 37 °C overnight. Cells were placed in serum-free DMEM/F12 for approximately 18 h at 37 °C, and then stimulated with 10 nM PRL for various times before harvesting nuclear extracts as described previously (Tseng and Schuler, 1998). Protein concentrations of nuclear extracts were determined by the BCA protein assay (Pierce Chemical Co., Rockford, IL).
2.6. EMSA
EMSA was performed as described previously (Tseng and Schuler, 1998). Dried gels were visualized and quantitated on a Storm Phosphoimaging System. Overlapping EMSA probes consisted of synthetic double-stranded oligonucleotides obtained from Integrated DNA Technologies (Coralville, IA). Probe sequences are described in Table 1. To assay for competition of probe binding, a double-stranded Oct-1 oligonucleotide (5′-AGAGGATCCATGCAAATGGACGTACG-3′) was used. For supershift assays, 1 μg of antibody was incubated with nuclear extracts for 45 min at 25 °C before addition of radiolabeled probe.
Table 1.
EMSA probe sequences
| Probe | 5′-Sequence-3′ |
|---|---|
| EMSA probe 1 | GGAGAAAGGCTGCAGCGGGGCGAT |
| EMSA probe 2 | CGATTTGCATTTCTATGAAAACCG |
| EMSA probe 3 | CCGGACTACAGGGGCAACTCCGCC |
| EMSA probe 4 | GCCGCAGGGCAGGCGCGGCGCCTC |
| OCT-GAS2 | GCTGCAGCGGGGCGATTTGCATTTCTATGAAAACCGGAC |
| OCTmut | GCTGCAGCGGGGCGAGTCGAAGTTCTATGAAAACCGGAC |
3. Results
3.1. The proximal cyclin D1 promoter retains PRL-induced activity in the absence of GAS2
The cyclin D1 promoter, as shown in Fig. 1A, is a complex promoter containing consensus binding sites for a number of transcription factors, including two GAS sites whose sequences match the consensus for regions of PRL-induced Stat binding in other systems (Schmitt-Ney et al., 1992). We have previously shown that mutation of the GAS1 site, but not GAS2, in the context of the cyclin D1 promoter, results in a loss of PRL-induced cyclin D1 promoter activity (Brockman et al., 2002). Therefore, we surmised that truncation of the promoter at −304 (D1Δ-304), which removes the GAS1 site but leaves GAS2 intact, would also abolish PRL-responsiveness. To test this, the D1Δ-944 and D1Δ-304 constructs were transiently transfected into Chinese hamster ovary cells, since these cells have proven to be a robust model for PRL activation of the Jak/Stat pathway (Gao et al., 1996; Brockman et al., 2002; Novaro et al., 2003) and in contrast to many PRL target cells, including breast cancer cells, produce negligible levels of endogenous PRL (for review, Clevenger et al., 2003). Surprisingly, the activity of D1Δ-304 was also stimulated by PRL to a similar extent as the D1Δ-944 promoter construct (Fig. 1B). To determine whether D1Δ-304 activity was due to PRL-induced activity at GAS2, this GAS site was mutated within the truncated construct and the responsiveness of this construct (D1Δ-304G2M) to PRL was assessed. As shown in Fig. 1B, mutation of the GAS2 site resulted in no loss of PRL-induced activity, suggesting that GAS2 was not important for PRL action.
Fig. 1.

PRL responsiveness of the proximal cyclin D1 promoter is not dependent on GAS2. (A) Diagram of the cyclin D1 promoter showing the position of selected transcription factor binding sites. (B) CHO cells were transiently transfected with PRLR, β-galactosidase and either pXP2 empty vector, D1Δ-944, D1Δ-304 or D1Δ-304 with a mutated GAS2 site (Δ304G2M), as described in Section 2. Transfected cells were cultured in serum-free media with (solid bar) or without (open bar) 10 nM PRL. After 24 h, samples were assayed for luciferase activity. β-Galactosidase activity was used to correct for transfection efficiencies, and activity was presented relative to the untreated pXP2-transfected cells. Data represent the mean of at least three separate experiments ± S.E.M. Numbers above solid bars denote the fold change compared to untreated control and asterisks indicate a statistically significant increase in PRL-treated promoter activity compared with non-PRL-treated control (*P<0.05, **P<0.005) using ANOVA with Bonferroni post-test.
3.2. Stat5 is important for PRL-induced proximal promoter activity
To determine whether PRL-induced proximal cyclin D1 promoter activity is mediated by Stats, the activity of the truncated promoter construct was assayed in the presence of Stat5 wild-type and dominant-negative constructs, since Stat5 has been shown to be important for signaling to the GAS1 site in the cyclin D1 promoter, as well as in other PRL-induced systems (for reviews, Bole-Feysot et al., 1998; Clevenger et al., 2003). As shown in Fig. 2A, transfection of a Stat5a wild-type construct further increased the PRL-induced promoter activity (approximately two- to five-fold), while the Stat5 dominant-negative construct, which displays dominant-negative activity for both Stat5a and 5b, completely abolished induction of promoter activity. Similar experiments were done using Stat5 dominant negative constructs that are reportedly defective in either transcriptional activation (Stat5a/Δ53C) or DNA binding (Stat5a/VVV) (Ilaria, Jr. et al., 1999). As shown in Fig. 2B, both of these constructs abolished PRL-induced proximal promoter activity, as well. Constructs expressing wild-type and dominant negative Stats 1 and 3 did not alter PRL action (data not shown).
Fig. 2.

PRL-induced proximal promoter activity is Stat5-dependent. (A) CHO cells were transiently transfected with PRLR, β-galactosidase and either D1Δ-304 alone, or with D1Δ-304 in the presence of Stat5a WT or Stat5a DN as indicated. (B) CHO cells were transiently transfected with PRLR, β-galactosidase, D1Δ-304 and either Stat5a WT, Stat5a/Δ53C or Stat5a/VVV. For both sets of experiments, transfected cells were cultured in serum-free media with (solid bar) or without (open bar) 10 nM PRL. After 24 h, samples were assayed for luciferase activity. β-Galactosidase activity was used to correct for transfection efficiencies, and activity was presented relative to untreated D1Δ-304-transfected cells (A) or untreated Stat5a WT (B). Data represent the mean of at least three separate experiments ± S.E.M. Numbers above solid bars denote the fold change compared to untreated control, and asterisks indicate a statistically significant increase in PRL-treated promoter activity compared with non-PRL-treated control (*P<0.05, ***P<0.001) using ANOVA with Bonferroni post-test.
3.3. Mutational analysis of the proximal cyclin D1 promoter
To define a region within the proximal cyclin D1 promoter that is responsible for PRL-induced activity, further truncations of D1Δ-304 were constructed at −254, −180 and −71 as shown in Fig. 3A. Activity assays of these truncated promoters revealed that PRL-induced activity is lost as the promoter is truncated from −254 to −180 (Fig. 3B). Computer analysis (TFBIND, Human Genome Center, University of Tokyo) revealed the GAS2 site, and a binding site for Oct-1, as well as several degenerate sites, all within the −243 to −220 regions. Although mutation of GAS2 had no influence on proximal promoter activity (Fig. 1B), it was still unclear which proteins were binding to the promoter within the −254 to −180 regions.
Fig. 3.

PRL responsiveness localizes to the −254 to −180 promoter region. (A) Diagram of the cyclin D1 promoter showing the length of promoter truncations and the relative position of selected transcription factor binding sites. (B) CHO cells were transiently transfected with PRLR, β-galactosidase and truncated cyclin D1 promoter reporter constructs D1Δ-944, D1Δ-304, D1Δ-254, D1Δ-180 or D1Δ-71. Transfected cells were cultured in serum-free media with (solid bar) or without (open bar) 10 nM PRL. After 24 h, samples were assayed for luciferase activity. β-Galactosidase activity was used to correct for transfection efficiencies, and activity was presented relative to the untreated D1Δ-944-transfected cells. Data represent the mean of at least three separate experiments ± S.E.M. Numbers above solid bars denote the fold change compared to untreated control and asterisks indicate a statistically significant increase in PRL-treated promoter activity compared with non-PRL-treated control (*P<0.05, ***P<0.001) using ANOVA with Bonferroni post-test.
3.4. Multiple proteins bind to the proximal promoter
EMSA was used to visualize protein complexes that bind to the −254 to −180 region of the promoter, with overlapping primers that span this region, as shown in Fig. 4A. These studies showed a major protein complex binding to the −243 to −220 regions (Fig. 4B). Minor complexes of lower mobility bound to the −263 to −240 and −222 to −199 regions as well; the former complex appears to be non-specific since its binding was not competed away with unlabeled oligonuclcotide. Additional EMSAs using different overlapping primers did not reveal any other complexes (data not shown). Since consensus binding sites for Oct-1 and Stats are contained within the −243 to −220 promoter region, antibodies specific for these transcription factors were used in EMSA supershift experiments to determine whether the −243 to −220 binding complex contained either of these proteins. As shown in Fig. 4C, antibodies to Stat1, 3, 5a or 5b that are capable of supershifting Stat-containing complexes (Brockman et al., 2002; Mynard et al., 2002; Mackey and Darlington, 2004), did not change the mobility or intensity of the EMSA complex, indicating that Stats were not present, assuming that the Stat epitopes were not masked from the antibody. However, antibody to Oct-1 did reduce the mobility of most of the protein complex, and furthermore, an unlabeled Oct-1 oligonucleotide competed with labeled probe for protein binding. These results indicated that Oct-1 was present in the −243 to −220 binding complex, and suggested that Oct-1 is binding to its consensus binding site within this region.
Fig. 4.
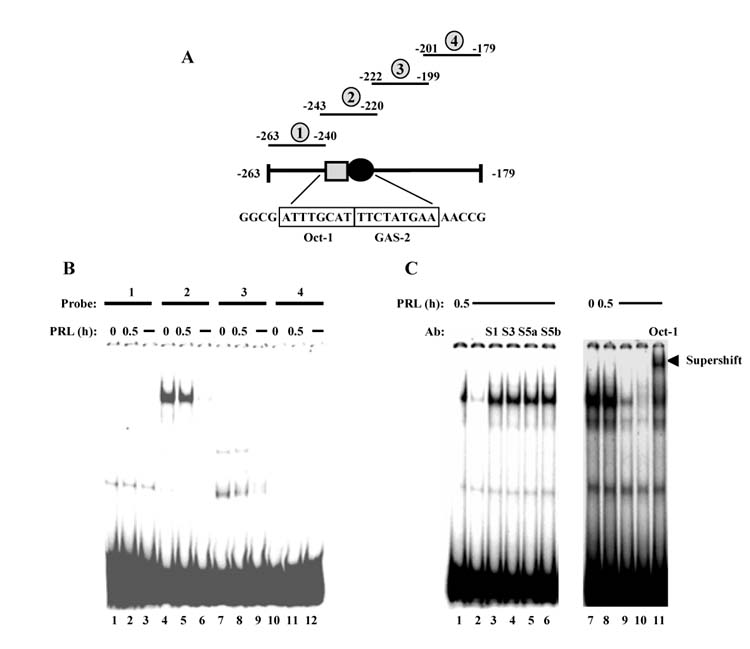
EMSA visualizes proteins bound to the −254 to −180 promoter region. (A) Diagram of the −263 to − 179 region of the cyclin D1 promoter showing selected transcription factor binding sites and relative positions of EMSA probes 1–4. (B) CHO cells stably transfected with PRLR were treated for 0.5 h ± 10 nM PRL, nuclear extracts were prepared, then subjected to EMSA with labeled probes that span the −263 to −179 region of the cyclin D1 promoter, described in (A). Lanes 3, 6, 9 and 12 contain nuclear extracts pretreated with unlabeled probe to compete for specific binding. (C) EMSA done with nuclear extracts prepared from CHO cells stably transfected with PRLR and treated for 0.5 h ± 10 nM PRL as indicated, then incubated with labeled probe 2 described in (A). Lanes 2 and 9 contain nuclear extracts pretreated with unlabeled probe 2, and lane 10 contains nuclear extract pretreated with an unlabeled Oct-1 oligonucleotide to compete for specific binding. To supershift protein complexes, lanes 3–6 contain nuclear extracts pretreated with antibodies to Stat1 (S1), Stat3 (S3), Stat5a (S5a) and Stat5b (S5b), respectively, while lane 11 contains nuclear extract pretreated with 8 μg Oct-1 antibody.
3.5. Oct-1 binding to the proximal promoter
Transcriptional activity of Oct-1 depends on the promoter context, with repression of activity occurring when the Oct-1 site overlaps the site for an activator protein (for review, Phillips and Luisi, 2000; Sytina and Pankratova, 2003). Due to the close proximity of the Oct-1 site to the GAS2 site, we theorized that Oct-1 binding may inhibit binding of Stats to GAS2. In order to test this, we used an EMSA probe that contained a mutated Oct-1 site (OCTmut), and compared the binding ability of this probe to a wild-type OctGAS probe (OCT-GAS2). As shown in Fig. 5A, the wild-type OCT-GAS2 probe detected specific binding of a protein complex that supershifted with Oct-1 antibody, but not Stat5 antibody in both untreated and PRL-treated cells. However, the same experiment done with the OCTmut probe did not detect any specific protein complex, indicating that the Oct-1 site is necessary for complex formation. Since Stat5 did not bind to the GAS2 site in the OCTmut probe, this suggests that steric hindrance by Oct-1 does not explain the lack of Stat5 binding observed with the wild-type OCT-GAS2 probe.
Fig. 5.

PRL modulates Oct-1 binding at the Oct-GAS2 site. (A) EMSA done with labeled wild-type OCT-GAS2 or OCTmut probes, described in Table 1, using nuclear extract prepared from CHO cells stably transfected with PRLR and treated for 0.5h ± 10 nM PRL as indicated. Lanes 2, 6, 10 and 14 contain nuclear extract pretreated with unlabeled probe to compete for specific binding. For supershifts, nuclear extracts in lanes 3, 7, 11 and 15 were pretreated with Oct-1 antibody (O), and lanes 4, 8, 12 and 16 were pretreated with Stat 5a antibody (S5). (B) EMSA done with labeled OCT-GAS2 probe, using nuclear extract prepared from CHO cells stably transfected with PRLR that were treated with 10 nM PRL for a time course of 0.5–4 h. Lane 6 contains nuclear extract pretreated with unlabeled OCT-GAS2 to compete for specific binding.
Interestingly, treatment of the cells with PRL appeared to decrease the binding of the Oct-containing protein complex (compare Fig. 4, panel B, lanes 4 and 5), with an average 26.2 ± 6.8% decrease in binding (mean ± S.D., n = 4) after 30min treatment with PRL. Prolonged PRL treatment further decreased the amount of protein complex that bound to the OCT-GAS2 probe (Fig. 5B), resulting in an approximate 60% decrease after 4 h PRL treatment. Western analysis of CHO nuclear proteins showed no difference in Oct-1 protein between untreated and PRL-treated cells (data not shown), consistent with the stability of this protein seen in other systems (for review, Phillips and Luisi, 2000; Sytina and Pankratova, 2003).
3.6. Importance of the Oct-1 site for cyclin D1 promoter activity
Since the EMSA suggested that PRL decreased Oct-1 binding to the proximal cyclin D1 promoter, the importance of an intact Oct-1 site for cyclin D1 promoter activity was determined. For these studies, a mutation at the Oct-1 binding site was introduced into the D1Δ-304 construct, the mutant (Δ304OM) and wild-type constructs were transiently transfected into CHO cells, and PRL responsiveness was assayed. As shown in Fig. 6A, the Oct mutation reduced unstimulated activity of the promoter, but increased PRL responsiveness, from approximately four- to six-fold (Fig. 6B). Similar results were found in MCF-10A cells, a human mammary epithelial cell line derived from non-tumorigenic cells (Fig. 6B). This may suggest a general mechanism for PRL action at this Oct-1 site and may have implications for PRL signaling to cyclin D1 in multiple target cells.
Fig. 6.
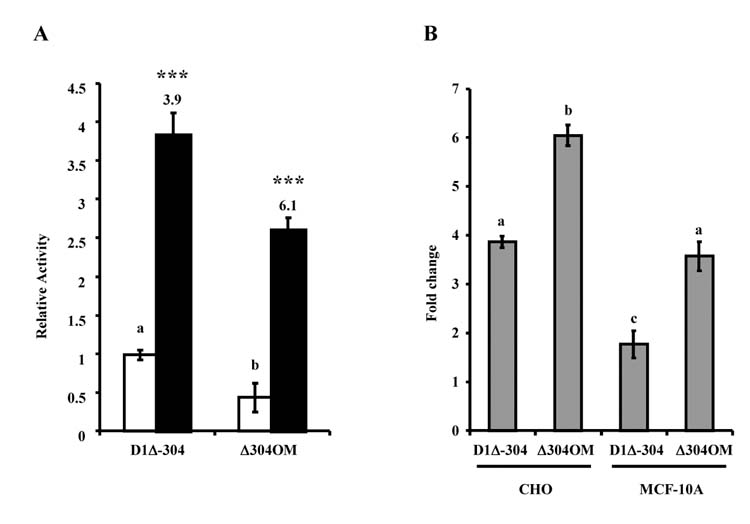
Mutation of the Oct-1 site leads to increased PRL-responsiveness of the promoter. (A) CHO cells were transiently transfected with PRLR, β-galactosidase, Stat5A WT and either D1Δ-304 or Δ304OM reporter constructs. Transfected cells were cultured in serum-free media with (solid bar) or without (open bar) 10 nM PRL. After 24h, samples were assayed for luciferase activity. β-Galactosidase activity was used to correct for transfection efficiencies, and activity was presented relative to untreated D1Δ-304-transfected cells. Data represent the mean of at least three separate experiments ± S.E.M. Numbers above solid bars denote the fold change compared to untreated control and asterisks indicate a statistically significant increase in PRL-treated promoter activity compared with non-PRL-treated control (***P<0.001) using ANOVA with Bonferroni post-test. Different letters denote significant differences in unstimulated promoter activity. (B) Data from CHO cells shown in (A), and from the same experiment done in MCF-10A cells, presented as fold change between untreated and PRL-treated samples. Data represent the mean of at least three separate experiments ± S.E.M., and different letters denote significant differences in fold change.
4. Discussion
The classic pathway for PRL activation of transcription occurs through stimulation of Jak2 and consequent activation of Stat5 that then bind to GAS consensus sites in promoter targets. Indeed, our previous studies demonstrated the importance of the Jak2/Stat5 pathway for PRL-induced activity of the distal GAS site in the cyclin D1 promoter (Brockman et al., 2002). However, here we show that following truncation of GAS1, the proximal cyclin D1 promoter retained Stat5-mediated PRL-induced activity, which we localized to a region containing a second GAS site and an immediately adjacent Oct-1 binding site. The proximity of GAS2 to the Oct-1 binding site suggested some level of interaction between these two transcription factors. However, PRL-induced activity of the proximal cyclin D1 promoter, while dependent upon Stat5, did not appear to involve direct Stat5 binding at the GAS2 site. Moreover, PRL decreased constitutively bound Oct-1 at this region. Mutagenesis of this Oct-1 site resulted in an increase in PRL-stimulated promoter activity in both CHO and MCF-10A cells, indicating that Oct-1 plays a negative regulatory role for PRL action in this region of the cyclin D1 promoter.
Stat5 does not mediate PRL action via the GAS2 site, since mutation of GAS2 had no effect on proximal promoter activity. Stat5 is nevertheless important for both unstimulated and PRL-induced proximal promoter activity, as shown by the loss of activity in the presence of either of two Stat5 dominant negative constructs with different modes of action (Ilaria, Jr. et al., 1999). Although both of these mutants can be tyrosine-phosphorylated by Jak2 and dimerize, the Stat5a/Δ53C mutant is truncated prior to the 53 carboxy-terminal amino acids that constitute most of the Stat5 transactivation domain. Conversely, the Stat5a/VVV mutant (V466VV→AAA) contains point mutations in the putative Stat5 DNA binding domain, and so fails to bind DNA, although it retains the transactivation domain. Together, our data suggest that Stat5 must bind DNA to exert its effect. Since Stat5 apparently does not bind GAS2, and we have shown that Stat5-mediated activity resides in the −254 to −180 region, this implies that either Stat5 may bind to an as yet undefined imperfect GAS site in this region, or may bind to the promoter of another protein that then mediates cyclin D1 promoter activity. However, examination of the −254 to −180 promoter sequence does not reveal an obvious GAS site or half-site, and EMSA of this region does not show a complex that supershifts with Stat5 antibody (Fig. 4 and data not shown). This interpretation may be complicated by a report suggesting that in COS-1 cells, homodimers of a Stat5b VVVI mutant, which is analogous to the Stat5a/VVV mutant, do not effectively accumulate in the nucleus upon PRL stimulation (Luo and Yu-Lee, 2000). It is unclear whether heterodimers of Stat5a/VVV and wild-type Stat5 would act similarly in our system; such an activity would open the possibility that Stat5 may act on the cyclin D1 promoter through protein–protein interactions.
Although EMSA did not reveal the capability of Stat5 to bind to the GAS2 site, Oct-1 appears to be bound to its consensus binding site at −234 in the cyclin D1 promoter in unstimulated cells, while this binding is reduced in PRL-treated cells. This is a common mechanism for gene regulation, where some transcription factors, including Oct-1, bind DNA but are inactive until a later signal is received, permitting rapid regulation of transcription (Brivanlou and Darnell, 2002). Depending on the promoter context, Oct-1 can either activate or suppress transcription. Transcriptional activation frequently occurs at promoter sites that allow cooperative binding between Oct-1 and another transcription factor, while repression occurs at promoter sites where the Oct-1 binding site overlaps that of an activator protein. Since the Oct-1 site is immediately adjacent to GAS2 in the proximal cyclin D1 promoter, we had initially postulated that constitutive Oct-1 binding hinders Stat5 interaction with GAS2. In this model, a PRL-dependent decrease in Oct-1 binding would then allow Stat5 to associate with GAS2. However, mutation of the Oct-1 site did not permit binding of nuclear proteins in either unstimulated or PRL-treated cells, refuting this model.
Functionally, mutation of the Oct-1 site in the proximal cyclin D1 promoter decreased unstimulated promoter activity, suggesting that Oct-1 is important for maintenance of basal activity. In addition, this mutation led to an increase in PRL-stimulated activity in both CHO and MCF-10A cells, indicating that Oct-1 plays a negative regulatory role for PRL action in this region of the wild-type cyclin D1 promoter. One model that can be derived from these data is that Oct-1 is constitutively bound to the promoter, keeping it in a state of decreased activity. PRL treatment decreases the binding of Oct-1 to its site within the promoter, which in turn increases promoter activity. Since PRL does not appear to affect Oct-1 expression, it must post-translationally alter Oct-1 to decrease its interaction at this site. One possibility for this is phosphorylation of Oct-1 at Ser-385 (Segil et al., 1991). Another possible mechanism is that Oct-1 is involved in cross-talk with another PRL-dependent process within the cell. Upon PRL treatment, Oct-1 may be preferentially shuttled from its negative regulatory role at −234 in the cyclin D1 promoter to another site, thereby releasing an inhibitory mechanism.
Oct-1 has been shown to act at a number of sites within the cyclin D1 promoter in other cellular systems, through both direct DNA binding and protein–protein interactions. In MCF-7 and ZR-75.1 breast cancer cell lines, Oct-1, together with AP-1 and ERα, composes an activating complex that displaces YY1 at a distal regulatory region approximately 940 bp upstream of the cyclin D1 start site (Cicatiello et al., 2004). In CHO cells, PRL does not activate this region in the cyclin D1 promoter; other data from our lab indicate that the ability of PRL to activate AP-1 is highly variable between different cell types (Gutzman, Arendt, Rugowski, Rui and Schuler, submitted for publication). Others have shown that in MCF-7 cells, Oct-1 can also form a complex with CREB that then binds to the CRE site near the transcriptional start site of the cyclin D1 promoter (Boulon et al., 2002). In contrast, PRL does not appear to act through this mechanism in CHO cells, since a cyclin D1 promoter deletion mutant (D1Δ-71) containing this region does not show PRL-induced activity. In some cell contexts, however, these alternative sites of Oct-1 action within the cyclin D1 promoter may be areas of increased Oct-1 activity following a PRL-induced decrease in binding at the Oct-GAS2 site.
Interestingly, it has recently been shown that thrombopoietin (TPO)-activated Stat5 directly binds Oct-1 in a hematopoietic cell line (Magne et al., 2003). This complex binds to the Oct-GAS2 site in the cyclin D1 promoter examined here, leading to enhanced promoter activity. However, TPO action differed from PRL action at this promoter in that GAS2, and not GAS1, was critical for activity. They also showed that PRL treatment leads to an increase in Oct-1/Stat5 binding to an OCT-GAS2 EMSA probe in 293T cells, in contrast to our observed PRL-dependent decrease in Oct-1 binding to this region in CHO cells. These disparate data are not inconsistent with the diverse functions of Oct-1 that have been shown in multiple cellular contexts. Since Oct-1 function is, in part, controlled by alternative splicing of its pre-mRNA, with the resulting mRNA isoforms including both ubiquitous forms, and tissue-specific forms (Phillips and Luisi, 2000; Sytina and Pankratova, 2003), it is possible that different Oct-1 isoforms are expressed in 293T and CHO cells. In this way, the actions of a single transcription factor could direct a variety of cellular processes, and lead to diverse functional endpoints.
We also demonstrated that Oct-1 action at this site is important for both basal and PRL-stimulated cyclin D1 promoter activity in MCF-10A human breast cells. Since the mammary phenotype of the cyclin D1−/−(Sicinski and Weinberg, 1997) and PRLR−/− (Ormandy et al., 1997) mice are similar, and mammary carcinomas develop in mice overexpressing cyclin D1 within the mammary gland (Wang et al., 1994), this, along with our data, suggests a role for Oct-1 in PRL-dependent control of cyclin D1 in normal mammary development and potentially in mammary carcinoma as well.
Acknowledgments
The authors wish to thank Dr. Paul Bertics, Dr. C. Thomas Caskey, Dr. Charles Clevenger, Dr. James Darnell, Dr. Robert Ilaria, Dr. Rolf Müller and Dr. Jeff Rosen for the generous gift of constructs and antibodies, and Debra Rugowski for technical assistance. This work was supported by NIH grant R01 CA78312.
References
- Bole-Feysot C, Goffin V, Edery M, Binart N, Kelly PA. Prolactin (PRL) and its receptor: actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocr Rev. 1998;19:225–268. doi: 10.1210/edrv.19.3.0334. [DOI] [PubMed] [Google Scholar]
- Boulon S, Dantonel JC, Binet V, Vie A, Blanchard JM, Hipskind RA, Philips A. Oct-1 potentiates CREB-driven cyclin D1 promoter activation via a phospho-CREB- and CREB binding protein-independent mechanism. Mol Cell Biol. 2002;22:7769–7779. doi: 10.1128/MCB.22.22.7769-7779.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brivanlou AH, Darnell JE., Jr Signal transduction and the control of gene expression. Science. 2002;295:813–818. doi: 10.1126/science.1066355. [DOI] [PubMed] [Google Scholar]
- Brockman JL, Schroeder MD, Schuler LA. Prolactin activates the cyclin D1 promoter via the JAK2-STAT pathway. Mol Endocrinol. 2002;16:774–784. doi: 10.1210/mend.16.4.0817. [DOI] [PubMed] [Google Scholar]
- Buckley AR. Prolactin regulation of cell proliferation and apoptosis. In: Horseman ND, editor. Prolactin. Kluwer Academic Publishers; Boston: 2001. pp. 247–264. [Google Scholar]
- Cicatiello L, Addeo R, Sasso A, Altucci L, Petrizzi VB, Borgo R, Cancemi M, Caporali S, Caristi S, Scafoglio C, Teti D, Bresciani F, Perillo B, Weisz A. Estrogens and progesterone promote persistent CCND1 gene activation during G1 by inducing transcriptional derepression via c-Jun/c-Fos/estrogen receptor (progesterone receptor) complex assembly to a distal regulatory element and recruitment of cyclin D1 to its own gene promoter. Mol Cell Biol. 2004;24:7260–7274. doi: 10.1128/MCB.24.16.7260-7274.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevenger CV, Furth PA, Hankinson SE, Schuler LA. Role of prolactin in mammary carcinoma. Endocr Rev. 2003;24:1–27. doi: 10.1210/er.2001-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman ME, Kanyicska S, Lerant A, Nagy G. Prolactin: structure, function, and regulation of secretion. Physiol Rev. 2000;80:1523–1631. doi: 10.1152/physrev.2000.80.4.1523. [DOI] [PubMed] [Google Scholar]
- Friedrichsen BN, Richter HE, Hansen JA, Rhodes CJ, Nielsen JH, Billestrup N, Moldrup A. Signal transducer and activator of transcription 5 activation is sufficient to drive transcriptional induction of cyclin D2 gene and proliferation of rat pancreatic beta-cells. Mol Endocrinol. 2003;17:945–958. doi: 10.1210/me.2002-0356. [DOI] [PubMed] [Google Scholar]
- Gao J, Hughes JP, Auperin B, Buteau H, Edery M, Zhuang HM, Wojchowski DM, Horseman ND. Interactions among JANUS kinases and the prolactin (PRL) receptor in the regulation of a PRL response element. Mol Endocrinol. 1996;10:847–856. doi: 10.1210/mend.10.7.8813725. [DOI] [PubMed] [Google Scholar]
- Goffin V, Binart N, Touraine P, Kelly PA. Prolactin: the new biology of an old hormone. Annu Rev Physiol. 2002;64:47–67. doi: 10.1146/annurev.physiol.64.081501.131049. [DOI] [PubMed] [Google Scholar]
- Herber B, Truss M, Beato M, Muller R. Inducible regulatory elements in the human cyclin D1 promoter. Oncogene. 1994;9:1295–1304. [PubMed] [Google Scholar]
- Ilaria RL, Jr, Hawley RG, Van Etten RA. Dominant negative mutants implicate STAT5 in myeloid cell proliferation and neutrophil differentiation. Blood. 1999;93:4154–4166. [PubMed] [Google Scholar]
- Kabotyanski EB, Rosen JM. Signal transduction pathways regulated by prolactin and Src result in different conformations of activated Stat5b. J Biol Chem. 2003;278:17218–17227. doi: 10.1074/jbc.M301578200. [DOI] [PubMed] [Google Scholar]
- Kline JB, Roehrs H, Clevenger CV. Functional characterization of the intermediate isoform of the human prolactin receptor. J Biol Chem. 1999;274:35461–35468. doi: 10.1074/jbc.274.50.35461. [DOI] [PubMed] [Google Scholar]
- Luo GY, Yu-Lee LY. Transcriptional inhibition by Stat5—differential activities at growth-related versus differentiation-specific promoters. J Biol Chem. 1997;272:26841–26849. doi: 10.1074/jbc.272.43.26841. [DOI] [PubMed] [Google Scholar]
- Luo GY, Yu-Lee LY. Stat5b inhibits NFkappaB-mediated signaling. Mol Endocrinol. 2000;14:114–123. doi: 10.1210/mend.14.1.0399. [DOI] [PubMed] [Google Scholar]
- MacGregor GR, Caskey CT. Construction of plasmids that express E. coli beta-galactosidase in mammalian cells. Nucl Acids Res. 1989;17:2365. doi: 10.1093/nar/17.6.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey SL, Darlington GJ. CCAAT enhancer-binding protein alpha is required for interleukin-6 receptor alpha signaling in newborn hepatocytes. J Biol Chem. 2004;279:16206–16213. doi: 10.1074/jbc.M400737200. [DOI] [PubMed] [Google Scholar]
- Magne S, Caron S, Charon M, Rouyez MC, Dusanter-Fourt I. STAT5 and Oct-1 form a stable complex that modulates cyclin D1 expression. Mol Cell Biol. 2003;23:8934–8945. doi: 10.1128/MCB.23.24.8934-8945.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mynard V, Guignat L, Devin-Leclerc J, Bertagna X, Catelli MG. Different mechanisms for leukemia inhibitory factor-dependent activation of two proopiomelanocortin promoter regions. Endocrinology. 2002;143:3916–3924. doi: 10.1210/en.2002-220323. [DOI] [PubMed] [Google Scholar]
- Novaro V, Roskelley CD, Bissell MJ. Collagen-IV and laminin-1 regulate estrogen receptor a expression and function in mouse mammary epithelial cells. J Cell Sci. 2003;116:2975–2986. doi: 10.1242/jcs.00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormandy CJ, Binart N, Kelly PA. Mammary gland development in prolactin receptor knockout mice. J Mammary Gland Biol Ncoplasta. 1997;2:355–364. doi: 10.1023/a:1026395229025. [DOI] [PubMed] [Google Scholar]
- Pfitzner E, Jähne R, Wissler M, Stoecklin E, Groner B. P300/CREB-Binding protein enhances the prolactin-mediated transcriptional induction through direct interaction with the transactivation domain of Stat5, but does not participate in the Stat5-mediated suppression of the glucocorticoid response. Mol Endocrinol. 1998;12:1582–1593. doi: 10.1210/mend.12.10.0180. [DOI] [PubMed] [Google Scholar]
- Phillips K, Luisi B. The virtuoso of versatility: POU proteins that flex to fit. J Mol Biol. 2000;302:1023–1039. doi: 10.1006/jmbi.2000.4107. [DOI] [PubMed] [Google Scholar]
- Schmitt-Ney M, Happ B, Ball RK, Groner B. Developmental and environmental regulation of a mammary gland-specific nuclear factor essential for transcription of the gene encoding beta-casein. Proc Natl Acad Sci USA. 1992;89:3130–3134. doi: 10.1073/pnas.89.7.3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott P, Kessler MA, Schuler LA. Molecular cloning of the bovine prolactin receptor and distribution of prolactin and growth hormone receptor transcripts in fetal and utero-placental tissues. Mol Cell Endocrinol. 1992;89:47–58. doi: 10.1016/0303-7207(92)90210-w. [DOI] [PubMed] [Google Scholar]
- Segil N, Roberts SB, Heintz N. Mitotic phosphorylation of the Oct-1 homeodomain and regulation of Oct-1 DNA binding activity. Science. 1991;254:1814–1816. doi: 10.1126/science.1684878. [DOI] [PubMed] [Google Scholar]
- Sicinski P, Weinberg RA. A specific role for cyclin D1 in mammary gland development. J Mammary Gland Biol Neoplasia. 1997;2:335–342. doi: 10.1023/a:1026391128117. [DOI] [PubMed] [Google Scholar]
- Sytina EV, Pankratova EV. Oct-1 transcription factor-plasticity and polyfunctionality. Mol Biol (Mosk) 2003;37:755–767. [PubMed] [Google Scholar]
- Tseng YH, Schuler LA. Transcriptional regulation of interleukin-1β gene by interleukin-1β itself is mediated in part by Oct-1 in thymic stromal cells. J Biol Chem. 1998;273:12633–12641. doi: 10.1074/jbc.273.20.12633. [DOI] [PubMed] [Google Scholar]
- Wang TC, Cardiff RD, Zukerberg L, Lees M, Arnold A, Schmidt EV. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature. 1994;369:669–671. doi: 10.1038/369669a0. [DOI] [PubMed] [Google Scholar]
- Wen Z, Zhong Z, Darnell JE. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Wen Z, Darnell JE., Jr Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264:95–98. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]