

Abstract
erbB2/Her2, a ligandless receptor kinase, has pleiotropic effects on mammalian development and human disease. The absence of erbB2 signaling in cardiac myocytes results in dilated cardiomyopathy in mice, resembling the cardiotoxic effects observed in a subset of breast cancer patients treated with the anti-Her2 antibody herceptin. Emerging evidence suggests that erbB2 is pivotal for integrating signaling networks involving multiple classes of extracellular signals. However, its role in G protein-coupled receptor (GPCR) signaling remains undefined. Because the activation of the MAPK pathway through GPCR signaling is important for cardiac homeostasis, we investigated whether erbB2 is required for GPCR-mediated MAPK signaling in wild-type and heart-specific erbB2 mutant mice. Here we demonstrate that erbB2, but not EGF receptor, is essential for MAPK activation induced by multiple GPCR agonists in cardiac myocytes. erbB2 is immunocomplexed with a GPCR in vivo and is transactivated after ligand treatment in vitro. Coexpression of erbB2 with GPCRs in heterologous cells results in ligand-dependent complex formation and MAPK activation. Furthermore, MAPK activation and cardiac contractility are markedly impaired in heart-specific erbB2 mutants infused with a GPCR agonist. These results reveal an essential mechanism requiring erbB2 as a coreceptor for GPCR signaling in the heart. The obligatory role of erbB2 in GPCR-dependent signaling may also be important in other cellular systems.
Keywords: MAPK, cardiac myocytes, erbB2 mutants
The erbB signaling network is a key regulator of multiple developmental and physiological processes (1, 2). Of the four members of the EGF receptor (EGFR)/erbB family, erbB2 is the preferred and potent heterodimerization partner for all erbB receptors to elicit signaling pathways, including those induced by neuregulin 1 (NRG1) (3). Consistent with this model, the extracellular domain of the erbB2 receptor that is required for receptor heterodimerization is in a ligand-independent, activated configuration (4, 5). Thus, erbB2 is “primed” to couple with other erbB receptors for signaling. Emerging evidence suggests that it plays a pivotal role in integrating signaling networks involving other classes of extracellular signals. erbB2 is required, for example, by IL-6 for signaling through the gp130 receptor in prostate carcinoma cells (6). erbB2 may also function as a transcriptional factor (7). Hence, genetic loss of erbB2 or blocking of erbB2 by herceptin may result in an impairment of multiple signaling cascades impinging on diverse cellular and molecular activities.
Several lines of evidence led us to investigate the role of erbB2, through G protein-coupled receptor (GPCR) signaling, in MAPK Erk1/2 activation in the heart. First, EGFR/erbB1 has been shown to be essential in Erk1/2 activation induced by GPCR ligands in cell lines (8, 9). However, fibroblast cells from EGFR mutant mice remain responsive to several GPCR ligands for Erk1/2 activation (10), raising the possibility that another tyrosine kinase receptor may be required. Interestingly, low levels of erbB2 are expressed in some of these cell lines that were previously used for assessing the role of EGFR in GPCR signaling [see Daub et al. (8)]. Second, Erk1/2 plays a cardioprotective role against impaired erbB2 signaling and the cardiotoxicity observed with the chemotherapeutic agent doxorubicin, which is frequently used in conjunction with herceptin in breast cancer patients (11, 12). We have previously shown that erbB2-deficient cardiac myocytes are highly sensitive to doxorubicin (13). Heterozygous NRG1 (NRG1+/−) mutant mice are more susceptible to doxorubicin-induced heart failure. Doxorubicin treatment results in decreased phosphorylated Erk1/2 levels in NRG1+/− mice compared with controls (14). Finally, GPCR agonists are important in cardiac homeostasis (15). For example, impaired β-AR stimulation with decreased expression and coupling of β-AR subtypes is a hallmark of heart failure (15). Doxorubicin administration to β2-AR mutant mice results in altered Erk1/2 activation and decreased contractile function (16). Mice overexpressing MEK1, an upstream activator of Erk1/2, display enhanced cardiac contractility (17). Urocortin (Ucn) 2, acting through its GPCR, corticotropin-releasing factor receptor (CRFR) 2β, is cardioprotective against ischemia by activation of the Erk1/2 (18) and enhances cardiac contractility in a heart failure model (muscle-specific LIM protein-deficient mice) (19).
Results
To determine the role of erbB2 in GPCR-mediated Erk1/2 activation, adult cardiac myocytes were prepared from control and heart-specific erbB2 mutant mice. Consistent with the idea that erbB2 is required for NRG1 signaling, we found that NRG1-induced Erk1/2 activation is abrogated in erbB2-deficient cardiac myocytes as compared with controls (Fig. 1a). Interestingly, EGF induced a marked Erk1/2 activation in both control and erbB2-deficient myocytes (Fig. 1b). We next tested the ability of Ucn 2 to activate Erk1/2. As shown in Fig. 1c, Ucn 2 activated Erk1/2 in controls but not in erbB2-deficient myocytes (Fig. 1c). This finding prompted us to compare the ability of three class A [angiotensin II, isoproterenol (ISO), and phenylephrine] and three class B (Ucn 2, glucagon, and vasoactive intestinal peptide) GPCR agonists to activate Erk1/2 in control versus erbB2-deficient myocytes. The stimulation of Erk1/2 observed in control myocytes by all six ligands is absent in erbB2-deficient myocytes (Fig. 1d). These results demonstrate that erbB2 is required for Erk1/2 activation by multiple GPCR agonists.
Fig. 1.

erbB2 is required for GPCR ligand-induced Erk1/2 activation in adult mouse cardiac myocytes. (a–c) Adult cardiac myocytes were isolated from control and heart-specific erbB2 mutant mice and stimulated with 5 ng/ml NRG (n = 10 controls; n = 6 mutants) (a), 5 ng/ml EGF (n = 8 controls; n = 9 mutants) (b), or 10 nM Ucn 2 (n = 10 controls; n = 9 mutants) (c). (d) Control and erbB2-deficient cardiac myocytes were untreated or were stimulated with Ucn 2 (100 nM), glucagon (Glu, 100 nM), vasoactive intestinal peptide (VIP, 100 nM), angiotensin II (Ang II, 100 nM), ISO (10 μM), and phenylephrine (PE, 50 μM). Fold of p-Erk1/2 induction was determined as described in Methods. (e) Cardiac myocytes were treated with ISO (10 μM) or Ucn 2 (100 nM) followed by measurement of cAMP levels. Fold of elevation was determined (n = 5 controls; n = 5 mutants). ∗, P < 0.05; ∗∗∗, P < 0.005.
We then focused our subsequent studies on the interaction of erbB2 with β2-AR or CRFR2β. To verify that the loss of Erk1/2 activation in response to the β2-AR agonist ISO and the CRFR2β agonist Ucn 2 was not due to a loss in receptor expression, we tested the ability of ISO and Ucn 2 to elevate cAMP levels in myocytes. Both ISO and Ucn 2 increased cAMP levels in control and erbB2-deficient myocytes (Fig. 1e), suggesting that erbB2-deficient myocytes do express functional β2-AR and CRFR2β. RT-PCR confirmed that β2-AR and CRFR2β levels are not significantly different between wild-type and erbB2-deficient myocytes (Fig. 6, which is published as supporting information on the PNAS web site). In addition, both NRG1 and EGF stimulated Erk1/2 activation in CRFR2β-deficient myocytes. In contrast, Ucn 2 fails to stimulate Erk1/2 activation in cardiac myocytes lacking CRFR2β (Fig. 7, which is published as supporting information on the PNAS web site). These results indicate that both CRFR2β and erbB2 are required for Ucn 2-induced Erk1/2 activation.
The requirement of erbB2 for GPCR agonist-induced Erk1/2 activation in myocytes led us to determine whether erbB2 forms a complex with GPCRs in vivo. We demonstrate that erbB2 and β2-AR form a complex in both whole heart and brain lysates (Fig. 2a). These results suggest a potential mechanism for the requirement of erbB2 in GPCR-dependent Erk1/2 activation via complex formation of these receptors. The lack of suitable antibodies to detect endogenous CRFR2β prevented us from examining the interaction of erbB2 with CRFR2β in myocytes. However, we show that erbB2 is transactivated in response to Ucn 2 stimulation in control myocytes (Fig. 2b).
Fig. 2.

erbB forms a complex with β2-AR in whole heart and brain lysates and is transactivated by Ucn 2 in cardiac myocytes. (a) Adult mouse whole heart and brain lysates were incubated with either anti-erbB2 or anti-β2-AR antibodies. The resulting immune complexes were subjected to immunoblotting analysis with either anti-erbB2 or anti-β2-AR antibodies. (b) Control cardiac myocytes were treated with serum-free DMEM (−) or Ucn 2 (100 nM). Cell lysates were immunoprecipitated (IP) with anti-erbB2 antibodies and immunoblotted (IB) with antiphosphotyrosine antibodies PY-20 or anti-erbB2 antibody.
To further elucidate the mechanisms through which erbB2 and GPCRs interact to mediate Erk1/2 activation, we used heterologous cell culture systems. When coexpressed in COS7 cells, erbB2 forms a complex with either Flag-tagged β2-AR (Flag-β2-AR) (Fig. 3a) or Flag-tagged CRFR2β (Flag-CRFR2β) (Fig. 3b). Ligand-dependent Erk1/2 activation is observed in cotransfected cells. To determine the region(s) of erbB2 required for GPCR agonist-induced Erk1/2 activation, we generated tagged mutant constructs lacking either the extracellular domain (HA-erbB2ΔECD), with an HA tag, or the intracellular domain (erbB2ΔICD-Flag), with a Flag tag, of erbB2, as well as a kinase dead point mutant (erbB2-KD). Heterologous cells were cotransfected with the individual erbB2 mutant constructs, together with wild-type β2-AR or CRFR2β constructs, and analyzed for their ability to activate Erk1/2 in response to ligand stimulation. All three erbB2 mutants were detected on the cell surface by immunostaining (data not shown). As shown in Fig. 3, a functional kinase domain was required for both ISO- and Ucn 2-induced Erk1/2 activation (Fig. 3 c and d). In addition, both the ECD (Fig. 3e) and the ICD (Fig. 3f) were required for ISO-induced Erk1/2 activation and Ucn 2-induced Erk1/2 activation (data not shown).
Fig. 3.

Ligand-dependent complex formation and Erk1/2 activation after coexpression of erbB2 with β2-AR or CRFR2β in COS7 cells. (a and b) COS7 cells were transfected with erbB2 together with Flag-tagged β2-AR (Flag-β2-AR) (a) or Flag-tagged CRFR2β (Flag-CRFR2β) (b). Cells were treated with ISO or Ucn 2. Cell lysates were immunoprecipitated with anti-erbB2 or anti-Flag antibodies. The immunoblots (IBs) were probed with anti-erbB2. The levels of p-Erk1/2 and Erk1/2 were measured. (c and d) COS7 cells were transfected with Flag-β2-AR (c) or Flag-CRFR2β along with HA-erbB2ΔECD (d). (e and f) COS7 cells were transfected with Flag-β2-AR along with erbB2ΔICD-Flag (e) or erbB-kinase dead (erbB2-KD) (f).
To determine whether EGFR is required for GPCR-mediated Erk1/2 activation, we tested the ability of ISO, Ucn 1, and Ucn 2 to activate Erk1/2 in B82L cells that lack endogenous EGFR and express erbB2 (20), β2-AR, and CRFR2 (Fig. 8, which is published as supporting information on the PNAS web site). We show that ISO, Ucn 1, and Ucn 2 (Ucn 1 and Ucn 2 bind CRFR2) are capable of activating Erk1/2 in the absence of EGFR (Fig. 4a). In addition, clinical trials using human EGFR kinase inhibitors for treatment of cancer report no negative impact on cardiac performance, suggesting that EGFR-dependent signaling in the heart is not essential for cardiac homeostasis, at least for the duration of the trial (21). However, we cannot rule out the possibility that EGFR facilitates and/or modulates erbB2–GPCR cross-talk in response to GPCR agonist-induced Erk1/2 activation, including the pathway involving GPCR-mediated release of soluble ligands for EGFR (22, 23).
Fig. 4.

EGFR and nonreceptor tyrosine kinases Src, Yes, and Fyn are not required for ISO- and Ucn 2-induced Erk1/2 activation. (a) B82L EGFR-deficient fibroblasts were stimulated with serum-free DMEM, NRG (5 ng/ml), EGF (5 ng/ml), ISO (10 μM), Ucn 1 (10 nM), or Ucn 2 (10 nM). Phosphorylated Erk1/2 (p-Erk1/2) and total Erk1/2 levels were determined. (b) SYF cells were stimulated with serum-free DMEM, NRG (5 ng/ml), EGF (5 ng/ml), ISO (10 μM), Ucn 1 (10 nM), or Ucn 2 (10 nM). (c) SYF cells were transfected with pcDNA3, pcDNA3 plus erbB2, pcDNA3 plus Flag-CRFR2β, or erbB2 and Flag-CRFR2β. Cells were stimulated with serum-free DMEM and Ucn 2 (10 nM).
The Src family of nonreceptor tyrosine kinases, which includes Src, Lyn, Fyn, Yes, Lck, Blk, and Hyc, are important mediators of multiple physiological processes, such as cell proliferation, survival, and adhesion (24). Members of the Src nonreceptor tyrosine kinase family have also been shown to be important for GPCR agonist-induced Erk1/2 activation via EGFR (9). To test this possibility, we used a mouse embryonic fibroblast cell line lacking three nonreceptor tyrosine kinases, Src, Yes, and Fyn (SYF) (10), that also express β2-AR (Fig. 8). We found that ISO activates Erk1/2 in nontransfected SYF cells (Fig. 4b), consistent with data from Huang et al. (25). Ucn 2 does not activate Erk1/2 in nontransfected SYF cells because they do not express CRFR2β (Fig. 8). Cotransfection of erbB2 and CRFR2β in SYF cells resulted in Erk1/2 activation in response to Ucn 2 (Fig. 4c). Taken together, these results indicate that Src, Yes, and Fyn are not required for GPCR agonist-induced Erk1/2 phosphorylation via erbB2. These results suggest that other cellular kinases and/or scaffolding molecules are required for erbB2 to mediate GPCR ligand-induced Erk1/2 activation (26, 27).
To determine whether erbB2 is required for the in vivo activation of Erk1/2 in the heart, wild-type and heart-specific erbB2 mutants were studied before and after infusion with Ucn 2. There were no significant differences between basal levels of Erk1/2 in wild-type and heart-specific erbB2 mutants. Administration of Ucn 2 to control mice resulted in a significant increase in Erk1/2 activation. In contrast, infusion of Ucn 2 to heart-specific erbB2 mutant mice did not result in an elevation of Erk1/2 phosphorylation (Fig. 5a). To determine the effects of the loss of erbB2 in cardiac myocytes, the physiological effects of Ucn 2 were assessed by cardiac catheterization in both wild-type and heart-specific erbB2 mutant mice. In comparison with wild-type littermates, the responsiveness of left ventricular peak dP/dt (a relatively specific measure of contractility), ejection fraction, stroke work, and cardiac output after Ucn 2 infusion were all significantly decreased in the heart-specific erbB2 mutant mice (Fig. 5 b–e). There were no differences in the response to Ucn 2 in heart rate or aortic elastance in wild-type versus heart-specific erbB2 mutant mice (data not shown). Overall, our in vivo physiologic measurements suggest that the absence of erbB2 abrogates the activation of Erk1/2 by Ucn 2 and mitigates the potent enhancement of left ventricular function by this GPCR agonist. These results suggest that erbB2 is required for mediating the contractile response of a GPCR agonist, such as Ucn 2, in vivo, thus providing an in vivo correlate to the in vitro observations described above.
Fig. 5.
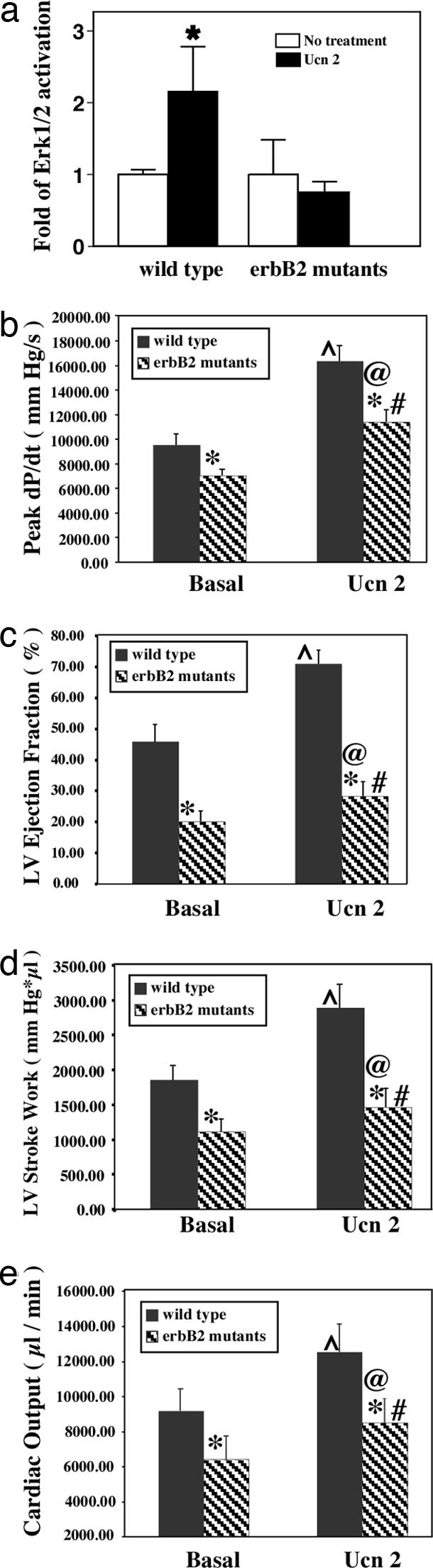
erbB2 is required for Ucn 2-induced Erk1/2 activation in vivo and left ventricular responsiveness. (a) Controls and heart-specific erbB2 mutants were untreated or infused with Ucn 2 (1.0 μg/kg). Levels of p-Erk1/2 and total Erk in heart lysates were determined. (b–e) Hemodynamic analysis of mice after Ucn 2 infusion. Peak dP/dt (b), left ventricle ejection fraction (c), left ventricle stroke work (d), and cardiac output (e) after administration of Ucn 2 to control and erbB2 mutant mice were determined by cardiac catheterization. Indices of left ventricular function (mean ± SEM) recorded in anesthetized basal state and in response to i.v. bolus administration of Ucn 2. Note that baseline values and responsiveness of these indices to Ucn 2 are significantly diminished in the erbB2 mutant mice as compared with their wild-type littermate controls. ∗, P < 0.05 wild type compared with erbB2 mutant basal or Ucn 2 responses; ⋀, P < 0.05 Ucn 2 response vs. basal wild type; #, P < 0.05 erbB2 mutant response to Ucn 2 compared with basal; @, P < 0.05 wild type response to Ucn 2 vs. erbB2 mutant response.
Discussion
In the present study we provide evidence that erbB2 is required for Erk1/2 activation induced by multiple GPCR ligands in the heart (Fig. 1e). We propose a model in which erbB2 forms a complex with GPCRs in a ligand-dependent fashion followed by transactivation of erbB2 and activation of downstream signaling cascade(s) leading to Erk1/2 activation.
To date, multiple mechanisms of GPCR-mediated activation of Erk1/2 have been proposed, including recruitment of β-arrestin, G protein-mediated activation of the nonreceptor tyrosine kinase (Src), and transactivation of tyrosine kinase receptors. Although a primary function of β-arrestins is to bind phosphorylated GPCRs and target these receptors for endocytosis, they have also been shown to be involved in Erk1/2 activation. Consistent with their established role in receptor endocytosis, genetic deletion of β-arrestin 2 or β-arrestin 1 and 2 in mouse embryonic fibroblasts results in the loss of β2-AR internalization in response to agonist stimulation (25). However, as in the case of the SYF-null cells (Fig. 4a), the loss of β-arrestin has no impact on ISO-induced Erk1/2 activation (data not shown), which is consistent with data from Huang et al. (25).
We show that EGF activates Erk1/2 in erbB2-deficient cardiac myocytes (Fig. 1b) and that EGFR is dispensable for GPCR agonist-induced Erk1/2 activation in B82l EGFR-null cells (Fig. 4a). The presence of EGFR signaling, together with the loss of GPCR ligand-induced Erk1/2 activation in erbB2-deficient myocytes, implicates erbB2 as the required receptor tyrosine kinase involved in a potential receptor tyrosine kinase/GPCR heterocomplex in cardiac myocytes. The use of cells isolated from genetic knockout mouse models of EGFR, Src, Yes, Fyn, and β-arrestin provide the strongest evidence that another tyrosine kinase receptor, likely erbB2, may be involved in mediating the Erk1/2 response in those models.
The molecular and cellular observations in erbB2-deficient cardiac myocytes were extended to an in vivo model in which multiple parameters of left ventricular function were shown to be impaired in heart-specific erbB2 mutant mice under basal and GPCR agonist (Ucn 2)-stimulated conditions (Fig. 5). These data provide an in vivo correlate to our in vitro observations demonstrating that erbB2 is required for mediating certain aspects of GPCR-dependent cardiac homeostasis.
It will be of interest to test our model with additional GPCR ligands in different cell types where erbB2 is known to be expressed. For example, we show that erbB2 forms a complex with β2-AR in the brain (Fig. 2a), where other classes of GPCR type neurotransmitter receptors and erbB2 are expressed and where the Erk1/2 signaling cascade is a key regulator of pathways for cell survival and synaptic function (28). The interaction of erbB2 and GPCRs may regulate cancer progression, including prostate, breast, and non-small cell lung cancers (29, 30).
Finally, for NRG1 (Fig. 1a) and GPCR ligands, we show that erbB2 is required for insulin (unpublished observation) and leukemia inhibitory factor/gp130 signaling pathways. For example, the signaling pathway mediated by the gp130 receptor complex is cardioprotective (31), and erbB2 forms a complex with gp130 (6). Therefore, we determined whether erbB2 is required for gp130-mediated signaling in adult cardiomyocytes. As shown in Fig. 9, which is published as supporting information on the PNAS web site, leukemia inhibitory factor-mediated Erk1/2 and STAT3 activation is impaired in erbB2-deficient cardiomyocytes. Taken together, these results suggest that erbB2 has distinct roles in mediating Erk1/2 activation in response to a variety of ligand and receptor partners. Our findings suggest that the potent antitumor effects of herceptin therapy are likely due to its ability to attenuate multiple oncogenic signaling pathways through inhibition of erbB2. In conclusion, we uncovered an essential role for erbB2 in cross-talk with GPCRs, consistent with the idea that erbB2 is required for the integration of diverse, key signaling pathways.
Methods
Adult Cardiac Myocyte Cultures and Stimulation.
Adult mouse cardiac myocytes were isolated from 3- to 6-month-old controls, heart-specific erbB2 mutants (13), or CRFR2β mutants (19) as described (18). Cells were stimulated with Ucn 2 (100 nM), glucagon (100 nM), vasoactive intestinal peptide (100 nM), angiotensin II (100 nM), ISO (10 μM), and phenylephrine (50 μM) for 5 min or 5 ng/ml NRG1 or EGF for 15 min. Levels of p-Erk1/2 and total Erk1/2 (Erk1/2) (Santa Cruz Biotechnology, Santa Cruz, CA) were measured by immunoblotting analysis using specific antibodies against p-Erk1/2 and Erk1/2, respectively. Fold increase of p-Erk1/2 was expressed as mean ± SEM. Single-factor one-way ANOVA was performed for each group of treatments. Differences among means were compared within the treatment groups by using Student's t test. Intracellular cAMP levels were measured by an RIA as described previously (18).
Transient Transfection and Immunoprecipitation.
COS7, B82L, and SYF cells were transfected with pcDNA3 or various expression plasmids. For immunoprecipitation, cells and tissues were lysed with RIPA buffer, precleared by incubation with protein A/G-agarose beads, and immunoprecipitated with anti-erbB2 [C-18 from Santa Cruz Biotechnology and AB3 and AB4 from Oncogene Science (Cambridge, MA)], anti-β2-AR (H-20; Santa Cruz Biotechnology), or anti-Flag antibodies. Tyrosine-phosphorylated erbB2 levels were determined by immunoblotting analysis using antiphosphotyrosine PY-20 antibodies.
In Vivo Ucn 2 Signaling and Hemodynamic Analysis.
Left ventricular function in response to Ucn 2 (1 μg/kg i.v.) was assessed during general anesthesia by catheter micromanometry and volumetry in 9-month-old wild-type and heart-specific erbB2 mutant mRe. Hearts were frozen under liquid N2 and homogenized with 1 ml of ice-cold Tris·maleate (pH 7.0) buffer containing 0.2 M sucrose, 2 mM EDTA, 1 mM sodium orthovanadate, 10 mM sodium pyrophosphate, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 0.1 mM 3-isobutyl-1-methylxanthine. For measurement of cAMP, 100 μg of protein was precipitated with 6% (vol/vol) trichloroacteic acid (30 min, −20°C) and centrifuged at 10,000 rpm on an Eppendorf centrifuge (Model 5415R, rotor F-45-24-11; Eppendorf, Westbury, NY) (10 min, 4°C). The pellets were washed three times with 1 ml of acetone and dried under vacuum and intracellular cAMP levels were measured by an RIA, as previously described (12).
Supplementary Material
Acknowledgments
We thank R. Lefkowitz (Duke University, Durham, NC) for the β2-AR expression plasmid, J. Rivier (The Salk Institute) for peptides, T. Hunter (The Salk Institute) for PY-20 antibodies, and G. Gill (University of California at San Diego) for B82L cells. This work was supported by grants from the National Institutes of Health, the Robert J. and Helen C. Kleberg Foundation, the Adler Foundation, and the Foundation for Research. B.K.B. was partly supported by a British Heart Foundation International Research Fellowship. This work was supported in part by National Institutes of Health Grants HD034534 and DK26741 and the Clayton Medical Research Foundation.
Abbreviations
- Ucn
urocortin
- CRFR
corticotropin-releasing factor receptor
- GPCR
G protein-coupled receptor
- EGFR
EGF receptor
- NRG
neuregulin
- ISO
isoproterenol.
Footnotes
Conflict of interest statement: W.V. is a cofounder, consultant, equity holder, and member of the Board of Directors of Neurocrine Biosciences and Acceleron Pharma. CRF and CRF1 receptor have been licensed by the Salk Institute and/or Clayton Foundation (CRF to Ferring Pharmaceuticals and CRF1 receptor to Neurocrine Biosciences).
References
- 1.Yarden Y, Sliwkowski MX. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 2.Holbro T, Hynes NE. Annu Rev Pharmacol Toxicol. 2004;44:195–217. doi: 10.1146/annurev.pharmtox.44.101802.121440. [DOI] [PubMed] [Google Scholar]
- 3.Graus-Porta D, Beerli RR, Daly JM, Hynes NE. EMBO J. 1997;16:1647–1655. doi: 10.1093/emboj/16.7.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, Kofler M, Jorissen RN, Nice EC, Burgess AW, Ward CW. Mol Cell. 2003;11:495–505. doi: 10.1016/s1097-2765(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 5.Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW, Jr, Leahy DJ. Nature. 2003;421:756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 6.Qiu Y, Ravi L, Kung HJ. Nature. 1998;393:83–85. doi: 10.1038/30012. [DOI] [PubMed] [Google Scholar]
- 7.Wang SC, Lien HC, Xia W, Chen IF, Lo HW, Wang Z, Ali-Seyed M, Lee DF, Bartholomeusz G, Ou-Yang F, et al. Cancer Cell. 2004;6:251–261. doi: 10.1016/j.ccr.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 8.Daub H, Weiss FU, Wallasch C, Ullrich A. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 9.Luttrell LM, Daaka Y, Lefkowitz RJ. Curr Opin Cell Biol. 1999;11:177–183. doi: 10.1016/s0955-0674(99)80023-4. [DOI] [PubMed] [Google Scholar]
- 10.Andreev J, Galisteo ML, Kranenburg O, Logan SK, Chiu ES, Okigaki M, Cary LA, Moolenaar WH, Schlessinger J. J Biol Chem. 2001;276:20130–20135. doi: 10.1074/jbc.M102307200. [DOI] [PubMed] [Google Scholar]
- 11.Klein PM, Dybdal N. Semin Oncol. 2003;30:49–53. doi: 10.1053/j.seminoncol.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 12.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, et al. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 13.Crone SA, Zhao YY, Fan L, Gu Y, Minamisawa S, Liu Y, Peterson KL, Chen J, Kahn R, Condorelli G, et al. Nat Med. 2002;8:459–465. doi: 10.1038/nm0502-459. [DOI] [PubMed] [Google Scholar]
- 14.Liu FF, Stone JR, Schuldt AJ, Okoshi K, Okoshi MP, Nakayama M, Ho KK, Manning WJ, Marchionni MA, Lorell BH, et al. Am J Physiol. 2005;289:H660–H666. doi: 10.1152/ajpheart.00268.2005. [DOI] [PubMed] [Google Scholar]
- 15.Rockman HA, Koch WJ, Lefkowitz RJ. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 16.Bernstein D, Fajardo G, Zhao M, Urashima T, Powers J, Berry G, Kobilka BK. Am J Physiol. 2005;289:H2441–H2449. doi: 10.1152/ajpheart.00005.2005. [DOI] [PubMed] [Google Scholar]
- 17.Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, Hewett TE, Jones SP, Lefer DJ, Peng CF, et al. EMBO J. 2000;19:6341–6350. doi: 10.1093/emboj/19.23.6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brar BK, Jonassen AK, Egorina EM, Chen A, Negro A, Perrin MH, Mjos OD, Latchman DS, Lee KF, Vale W. Endocrinology. 2004;145:24–35. doi: 10.1210/en.2003-0689. discussion 21–23. [DOI] [PubMed] [Google Scholar]
- 19.Bale TL, Hoshijima M, Gu Y, Dalton N, Anderson KR, Lee KF, Rivier J, Chien KR, Vale WW, Peterson KL. Proc Natl Acad Sci USA. 2004;101:3697–3702. doi: 10.1073/pnas.0307324101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wright JD, Reuter CW, Weber MJ. J Biol Chem. 1995;270:12085–12093. doi: 10.1074/jbc.270.20.12085. [DOI] [PubMed] [Google Scholar]
- 21.Tsuboi M, Kato H, Nagai K, Tsuchiya R, Wada H, Tada H, Ichinose Y, Fukuoka M, Jiang H. Anticancer Drugs. 2005;16:1123–1128. doi: 10.1097/00001813-200511000-00012. [DOI] [PubMed] [Google Scholar]
- 22.Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A. Nature. 1999;402:884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 23.Civenni G, Holbro T, Hynes NE. EMBO Rep. 2003;4:166–171. doi: 10.1038/sj.embor.embor735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luttrell DK, Luttrell LM. Oncogene. 2004;23:7969–7978. doi: 10.1038/sj.onc.1208162. [DOI] [PubMed] [Google Scholar]
- 25.Huang J, Sun Y, Huang XY. J Biol Chem. 2004;279:21637–21642. doi: 10.1074/jbc.M400956200. [DOI] [PubMed] [Google Scholar]
- 26.Lopez-Ilasaca M, Crespo P, Pellici PG, Gutkind JS, Wetzker R. Science. 1997;275:394–397. doi: 10.1126/science.275.5298.394. [DOI] [PubMed] [Google Scholar]
- 27.Kolch W. Nat Rev Mol Cell Biol. 2005;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 28.Adams JP, Sweatt JD. Annu Rev Pharmacol Toxicol. 2002;42:135–163. doi: 10.1146/annurev.pharmtox.42.082701.145401. [DOI] [PubMed] [Google Scholar]
- 29.Li YM, Pan Y, Wei Y, Cheng X, Zhou BP, Tan M, Zhou X, Xia W, Hortobagyi GN, Yu D, Hung MC. Cancer Cell. 2004;6:459–469. doi: 10.1016/j.ccr.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 30.Li S, Huang S, Peng SB. Int J Oncol. 2005;27:1329–1339. [PubMed] [Google Scholar]
- 31.Hirota H, Chen J, Betz UA, Rajewsky K, Gu Y, Ross J, Jr, Muller W, Chien KR. Cell. 1999;97:189–198. doi: 10.1016/s0092-8674(00)80729-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}