

Abstract
The availability of choline during critical periods of fetal development alters hippocampal development and affects memory function throughout life. Choline deficiency during fetal development reduces proliferation and migration of neuronal precursor cells in the mouse fetal hippocampus and these changes are associated with modifications in the protein levels of some cell cycle regulators and early differentiation markers. We fed C57 BL/6 mouse dams diets deficient or normal in choline content from days 12 to 17 of pregnancy, and then collected fetal brains on embryonic day 17. Using laser-capture micro-dissection we harvested cells from the ventricular and subventricular zones of Ammon’s horn and from the prime germinal zone of the dentate gyrus (hippocampus). In the ventricular and subventricular zones from the choline deficient group, we observed increased protein levels for kinase-associated phosphatase (Kap) and for p15INK4b (two cell cycle inhibitors). In the dentate gyrus, we observed increased levels of calretinin (an early marker of neuronal differentiation). In fetal brain from mothers fed a choline deficient diet, DNA global methylation was decreased in the ventricular and subventricular zones of Ammon’s horn. We also observed decreased gene-specific DNA methylation of the gene (Cdkn3) that encodes for Kap, correlating with increased expression of this protein. This was not the case for p15INK4b or calretinin (Cdkn2b and Calb2, respectively). These data suggest that choline deficiency-induced changes in gene methylation could mediate the expression of a cell cycle regulator and thereby alter brain development.
Keywords: Cdkn3, Kap, Cdkn2b, p15INK4b, Calb2, calretinin, fetal brain, DNA methylation, choline deficiency, hippocampus
INTRODUCTION
Choline, an essential nutrient (1), is a required substrate for various molecules needed for the structural integrity and signaling functions of cell membranes, acetylcholine synthesis and methyl-group metabolism (2). Choline, via its metabolite betaine, methylates homocysteine to form methionine (3). Methionine is the precursor for S-adenosylmethionine (AdoMet), which is the physiological methyl-group donor for protein, RNA and DNA methylation (4).
Prenatal or postnatal dietary choline supplementation enhanced memory in rodents and these changes lasted throughout their lives (5-10). Conversely, dietary choline deprivation during pregnancy decreased memory performance in offspring (10). We previously reported that these changes were related to neuro-anatomical changes in the hippocampal areas that are known to regulate memory (11-13) and that feeding mothers choline deficient diets during pregnancy decreased the concentrations of phosphocholine and phosphatidylcholine in fetal mouse hippocampus (13). Choline-deprived primary neurons from cortex and hippocampus, as well as rat PC12 (pheochromocytoma) and human IMR-32 (neuroblastoma) cells, had increased apoptosis and decreased cell proliferation compared to cells grown in a control medium with choline (14-16). In rodents exposed to choline deficiency during their fetal development, we previously reported diminished cell division and increased apoptosis in the neuroepithelial layer of the fetal septum (11) and fetal hippocampus (11, 13). Choline deficient diets fed to dams also increased the expression, in fetal brain, of some markers and regulators of neural proliferation and differentiation such as TOAD-64 (a marker for neuronal differentiation), p27Kip1 (a cyclin-dependent kinase inhibitor), TGFβ1 (a growth factor) and calretinin (calcium binding protein expressed in GABA-ergic neurons, and a marker for early neuronal differentiation) (12, 17-19).
Methylation of the cytosines within cytosine-guanosine sites (CpG sites) of the promoter region of genes regulates gene expression (epigenetic regulation) (4). Often this methylation occurs in CpG sites within regions that have a higher incidence of CpG repeats than expected (CpG islands) (20). Changes in dietary availability of methyl-groups can induce stabile changes in gene expression and resulting phenotype (21, 22).
Based on our previous studies in neuroblastoma and pheochromocytoma cells, we suggested that, in part, the mechanism mediating the effects of choline on neuronal proliferation involves altered global DNA methylation and specifically hypomethylation of the promoter region of the cyclin-dependent kinase inhibitor 3 (Cdkn3) gene, with subsequent modulation of the expression of this gene, the kinase-associated phosphatase (Kap) (16). In order to determine whether choline-mediated changes in gene methylation observed in vitro had relevance for effects of dietary choline in fetal brain, we isolated the hippocampus from fetal mice (embryonic day 17, E17) whose dams were dietary choline-deprived versus from controls. Cells obtained by laser-capture microdissection (LCM) from the main proliferation area (Ammon’s horn ventricular and subventricular zones), and from the dentate gyrus of the hippocampus were used to study changes in protein levels of two cyclin-dependent kinase inhibitors (Kap and p15INK4b) and in calretinin (a calcium-binding protein expressed as neurons differentiate) and related these to changes in DNA methylation of their respective genes (Cdkn3, Cdkn2b and Calb2).
MATERIALS AND METHODS
All reagents and materials were purchased from Sigma-Aldrich, St. Louis, MO if not otherwise specified.
Animals
Timed-pregnant C57 BL/6 mice (obtained on pregnancy day 6-9 from Jackson Laboratory, Bar Harbor, ME) were used in all experiments according to a protocol described elsewhere (13). The mice consumed water ad libitum and AIN-76A purified diet (containing 1.1 g/kg choline chloride; Dyets, Bethlehem, PA) until embryonic day 12 (E12) when they were randomly assigned to one of the two feeding groups (n = 5/group). The choline-deficient group received AIN-76A diet containing no choline chloride, and the control group received AIN-76A diet containing 1.1 g/kg choline chloride. All pregnant mice received the special diets from the morning of day E12 until the morning of day E17.
Tissue collection
On gestational day 17 pregnant mice were anesthetized with a single injection of 0.03 mL ketamine (100 mg/mL) and 0.02 mL xylazine (20 mg/mL) (Henry Schein Inc., Melville, NY, USA) subcutaneously and the mice were kept on a heating pad to maintain body temperature. The uterine horns were exposed by a midline abdominal incision and the fetuses were removed individually for perfusion. The chest cavity of the fetus was opened and approximately 2 mL of perfusion fixative containing 4% formaldehyde and 0.2 % glutaraldehyde (Polysciences, Inc., Warrington, PA, USA) were injected into the fetal heart. The fetuses were decapitated and the fetal skull was opened for post fixation overnight in the perfusion fixative. Fetal brains were then stored in 0.1 mol/L phosphate buffer, pH 7.4. The fetal brains were embedded in paraffin and 5 μm coronal serial sections were cut and applied on glass slides for histological and immunohistochemical assays. Since there is a posterior to anterior gradient of neurogenesis in fetal mouse brain, the paraffin sections were reviewed at the time of sectioning to ensure that they included anatomically reproducible areas of the hippocampus as defined by a standard atlas of the developing brain (23) (Figure 2D). For each condition (choline deficiency and control), five randomly selected male fetal brains, each from a different dam, were used. Specimens from the same fetal brains were used for both immunohistochemical analysis and for the assessment of DNA methylation status.
Figure 2.
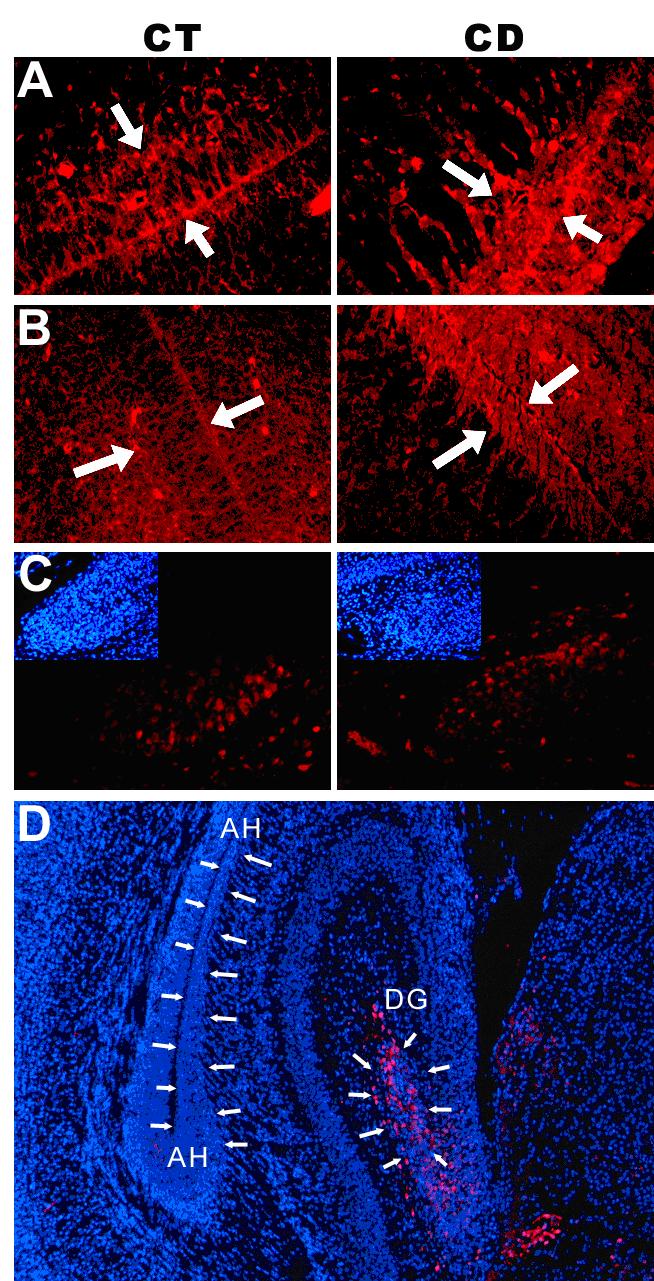
Maternal choline deficiency increased p15INK4b, Kap and calretinin protein levels in the ventricular and subventricular zones of Ammon’s horn and the dentate gyrus of the hippocampus in fetal brain.On embryonic day 12 (E12), pregnant C57 BL/6 mice were randomly assigned to one of the two feeding groups (n = 5/group). The choline-deficient group (CD) received AIN-76A diet containing no choline chloride, and the control group (CT) received AIN-76A diet containing 1.1 g/kg choline chloride. All pregnant mice received the special diets from the morning of day E12 until the morning of day E17 when brains were harvested and prepared as described in Methods. Fluorescent staining of specific proteins in CT and CD ventricular and subventricular zones and dentate gyrus was performed as described in Methods. In CD brains we observed Increased p15INK4b (A) and Kap (B) in ventricular and subventricular zones (bounded by white arrows). Panel C indicates the distribution of calretinin within the dentate gyrus (DG), with increased calretinin OD/cell in CD brains. The small inserts show the nuclear staining for the same areas. Panel D indicates the main areas that were selected for both protein assessment and gene-specific methylation (delineated by white arrows). AH denotes the Ammon’s horn ventricular and subventricular zones. DG denotes the prime germinal zone of the dentate gyrus The calretinin-positive cells were confined to the dentate-gyrus (red fluorescent staining) within the hippocampus. Optical density per cell (OD/cell) were determined by measuring the OD within selected areas, from which background was subtracted (using the same measurement on correspondent negative slides – see Materials and Methods). Finally, the computed values were divided by the total number of cells within the same area, as counted using the images with nuclear staining.
Immunohistochemistry
Selected slides containing sections including the hippocampus were specifically labeled for global DNA methylation, p15INK4b, calretinin or Kap protein expression. Paraffin-embedded slides were first incubated at 60°C for 10 minutes, then deparaffinized in xylene 3 times for 25 minutes each. The sections were gradually rehydrated in absolute ethanol, 70% ethanol with 0.25% NH4OH for 1 hr, and then 50 % ethanol. Global DNA methylation was assessed by immunostaining for 5-methyl cytosine content of the DNA, according to a protocol described elsewhere (24), using a purified rabbit Anti-5mCytidine Polyclonal Antibody (cat# CP 50250, Megabase Research Products, Lincoln, NE) and a secondary Alexa-Fluor donkey anti-rabbit antibody (cat# A31573, Molecular Probes, Eugene, OR). For calretinin, p15INK4b and Kap staining, the following primary antibodies were used: Calretinin Cat.# sc-11644 (Santa Cruz Laboratories; Santa Cruz, CA), p15 Cat. #sc-1429 (Santa Cruz) and Kap Cat. # K32120 (Transduction Laboratories; San Diego, CA), diluted 1/200 in Blocking Buffer (Sigma, Cat. # B-6429). Slides were incubated overnight at 4°C. The following fluorescent secondary antibodies were used: Alexa-Fluor Cat. #A21468 and Cat.#A31573 (Molecular Probes) at a 1/200 dilution. All staining procedures were done according to protocols described elsewhere (12, 13, 19). Negative controls consisted of slides from the same specimens, where the incubation with the primary antibody was omitted. Slides were counterstained for nuclear DNA with diamino-phenylindole (DAPI), 0.1 μg/mL in phosphate-buffered saline (PBS) for nuclear DNA.
Image analysis
Images were acquired with an Olympus BX50 microscope using 10x or 20x objectives, with optical filters for UV light and red channel (above 600 nm). The protein levels were measured by assessing the Optical Density (OD) of each selected area (on images collected with the 20x objective) followed by background subtraction, and divided by the number of cells for each selected area, using the ScionImage software (Scion Corporation). The background OD was determined using similar areas on slides used as negative controls. Finally, the average value of OD/cell for each image from the same sample was computed. Five different fetuses were used for each condition.
Laser-Capture Microdissection and Bisulfite modification
In order to select a specific homogenous population of cells, we used Laser-Capture Microdissection (LCM; PixCell LCM II system, Arcturus Engineering, Mountain View, CA). We collected cells in the Ammon’s horn ventricular and subventricular zones (as anatomically defined in a neuroanatomical atlas (25)), and cells in the prime germinal zone of the dentate gyrus (as anatomically defined (26)). These areas were selected because they are the germinal zones for cells that will eventually form the hippocampus. The anatomical areas subjected to laser-capture microdissection (LCM) were the same as the areas used for the assessment of the protein levels (Figures 1 and 2). For the Ammon’s horn ventricular and subventricular zones, cells were captured along the basal layers of the hippocampus, facing the lateral ventricles. Paraffin-embedded sections from the same fetal brains used for the immunohistochemistry study were used (five fetuses for each condition, CD and CT). Cells were captured on CapSure HS LCM caps (Arcturus) (Figure 1) and genomic DNA subsequently extracted using a Pico Pure DNA Extraction Kit (KIT0103, Arcturus) according to the manufacturer’s protocol (version B), where the incubation with proteinase K was prolonged to 24 hours. For each sample, 8 captures were performed (2 captures per slide – right and left hippocampus), using 4 consecutive sections. DNA extracted from the captured cells was pooled on a per sample basis for subsequent bisulfite treatment using an EZ DNA Methylation kit (D5001, Zymo Research, Orange, CA), according to the manufacturer’s protocol.
Figure 1.
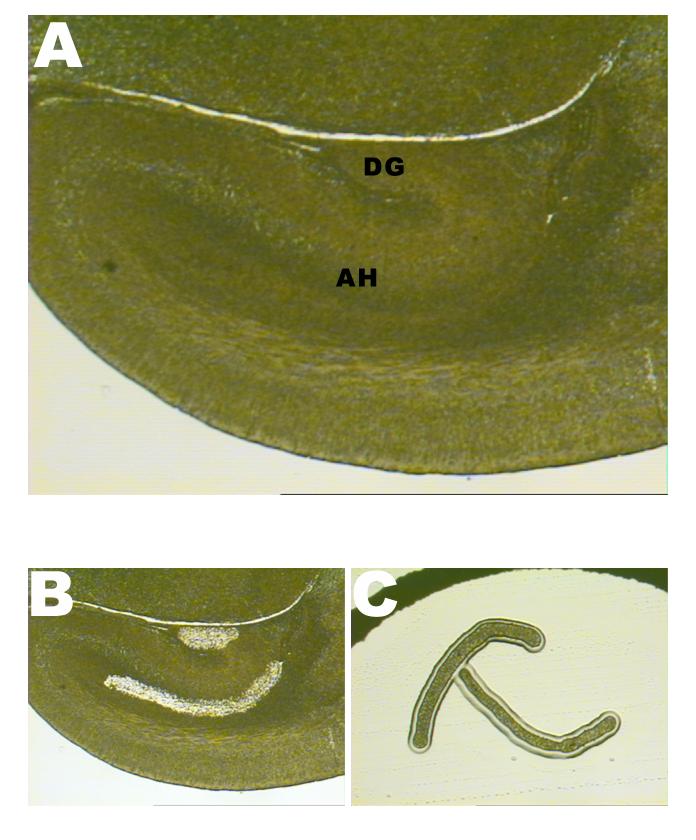
- A general view of the E17 mouse fetal hippocampus. AH denotes the Ammon’s horn ventricular and subventricular zones; DG denotes the prime germinal zone of the dentate gyrus.
- The same section of the hippocampus after cell removal using the Laser Capture Microdissection (LCM) technique.
- Two captured areas of Ammon’s horn ventricular and subventricular zones (right and left, from the same paraffin-embedded section), used for subsequent DNA extraction and bisulfite sequencing.
CpG island identification, PCR and bisulfite sequencing
The following DNA accession numbers were used to identify possible CpG islands in the promoter regions of the genes (start and end points of sequences given in brackets, position 1 is the first base of exon 1) : NT_039599 (Cdkn3, -1000 to +288); U66084.1 (promoter region and exon 1 for Cdkn2b); NT_078575 (Calb2, -1428 to +377). We used on-line MethPrimer software to identify possible CpG islands and to design specific bisulfite PCR primers (27). The following CpG islands were identified (start and end points of sequences where position 1 is the first nucleotide of exon 1): Cdkn3, -270 to +232 (502 bp length with 50 CpG sites); Cdkn2b, 3 consecutive CpG islands with 55 CpG sites (sites 289 to 912 within the U66084.1 sequence); Calb2, -163 to +79 (242 bp length with 29 CpG sites). We numbered CpG sites sequentially from the 5’ end of the CpG island. The primers used to amplify the bisulfite-treated DNA were: forward 5’-AGGGATATTTTTTATGTAAGGTTTTTTTT-3’ and reverse 5’-ACCACAAATCTACCACTCAACCTCC-3’ for Cdkn3, forward 5’-GGTTTGGGTTAAATAAAGATTTTTGTT-3’ and reverse 5’-CACACAACCAACACCAAAAAACTACC-3’ for Cdkn2b, forward 5’-GAGGGAAGGGAAGGTAGGAGTTGTTTT-3’ and reverse 5’-CCATACCCCTAACCCCTTTACCAAACT-3’ for Calb2. The PCR conditions were: initial denaturation at 95°C (2 min), 40 cycles of 95°C (40 sec), 57°C (or 64°C for CALB2) for 40 sec, and 68°C (60 sec), and a final extension step at 68°C. The PCR products were purified using a PCR Purification Kit (Qiagen, Valencia, CA) and sequenced using an ABI 3100 DNA Sequencer (Applied Biosystems, Foster City, CA, USA) at the University of North Carolina at Chapel Hill Genome Analysis Facility (http://152.19.68.152/gafsite/Main.asp). The products were sequenced in both directions using both primers for each PCR product. The sequences were analyzed using Chromas2.23 software (Technelysium Ptd Ltd, Tewantin, Qld, Australia). The chromatograms were exported into raw data files where, for each position, the peak height for each nucleotide is given.
Complete conversion of unmethylated cytosine to thymidine was confirmed as all cytosines from non-CpG sites were converted to thymidine. We identified unmethylated, methylated and incompletely methylated CpG sites. The latter sites were identified using the raw data files where, while the sequence for the wild-type DNA was unequivocally determined, the bisulfite-modified DNA PCR products presented mixed signals for C and T nucleotides, which indicated that in a given sample, a certain CpG site was methylated on some strands but not on others. For each site in a given sample, we used a modified qualitative scoring system (28) to assess the extent of cytosine methylation (methylation index) (29): 100 if completely methylated (T relative peak height < 25% of the C peak height), 50 for sites with mixed signals (T relative peak height more than 25% of the C peak, and 0 for unmethylated sites (C relative peak height < 25 % of the T peak height).
Statistics
Student’s t-test (JMP 3.2.6, SAS Institute INC, Cary, NC, USA) was used to determine the statistical significance of changes between the choline deficient group and controls for immunohistochemistry studies. Epi Info(TM) 3.3.2 software (http://www.cdc.gov/epiinfo/, Centers for Disease Control and Prevention, Atlanta, GA) was used for statistical tests involving the methylation status of CpG islands and for individual CpG sites across treatments (Bartlett’s chi square test and Kruskal-Wallis test for two groups). To test whether CpG islands and individual CpG sites were differently methylated between choline deficient and control samples, we used the Kruskal-Wallis test for two groups. The reasons for using this non-parametric test were that the outcome variable is a rank or score with only a few categories (0, 50, or 100) and does not allow us to assume a Gaussian distribution for the average of the score; in addition, Bartlett’s test for inequality of population variances revealed that, for many CpG sites, the variances were not homogeneous.
RESULTS
Choline deficiency increased protein expression of p15INK4b and Kap cyclin-dependent kinase inhibitors in the hippocampal ventricular and subventricular zones of E17 fetal brains, while no changes were found in the dentate gyrus (Table 1 and Figure 2). Interestingly, the sub-cellular localization of p15INK4b also differed with treatment: control neuroepithelial cells had most of the p15INK4b protein in the cytoplasm, while protein localization in choline deficient cells also included the nuclear area (Figure 2). A similar localization pattern was detected for Kap, where choline deficient-mediated protein over-expression was associated with higher intra-nuclear distribution (Figure 2). Calretinin protein levels increased in the dentate gyrus of choline deficient brains compared to controls (Table 1), with calretinin positive cells restricted to the prime germinal zone and the subgranular zone (Figure 2). This is in agreement with our previous observations (19). Choline deficiency induced a significant decrease in global DNA methylation in the Ammon’s horn ventricular and in subventricular zones, whereas no significant changes were observed in the dentate gyrus (Table 1).
Table 1.
Maternal choline deficiency decreased DNA global methylation and increased expression of p15INK4b, Kap and calretinin in E17 fetal hippocampus.
| Ammon's horn ventricularand subventricular zones |
Dentate gyrus |
|||
|---|---|---|---|---|
| Control | Choline deficient | Control | Choline deficient | |
| Global DNA | 159.13 ± 6.7a | 109.9 ± 13.3b | 127.7 ± 20.3a | 117.2 ± 12.4a |
| methylation | ||||
| p15INK4b | 166.1 ± 7.9a | 220.9 ± 14.9b | 151.1 ± 32.5a | 171.1 ± 29.2a |
| KAP | 137.8 ± 16.2a | 290.3 ± 25.4b | 114.3 ± 11.3a | 134.1 ± 27.2a |
| Calretinin | ND | ND | 104.02 ± 4.34a | 195.55 ± 5.55b |
Pregnant mice were treated and fetal brains prepared as described in Figure 2 legend. DNA methylation and protein expression was assessed using immunohistochemical methods described in the Methods section. Optical density (OD) was measured by image analysis in selected areas of hippocampus: Ammon's horn ventricular and subventricular zones and the prime germinal zone of the dentate gyrus (for localization, see Figure 1). The computed values were divided by the number of cells in the selected areas. Results are expressed as OD mean values ± standard error (SE) of choline deficient and control samples. ND = not detected.
Values with different superscript letters are significantly different (p<0.05, Students t-test) within each assay.
The analysis of CpG islands’ methylation by bisulfite sequencing revealed different patterns of methylation between ventricular and subventricular zones and dentate gyrus, some of these being modulated by dietary choline availability (Table 2). We excluded some of the CpG sites within the Cdkn3 (sites 39 to 50) and Cdkn2b CpG islands (sites 1-8; 33-55) due to the low reliability of the sequencing. Therefore, the scoring system (see Materials and Methods) was applied to 38 CpG sites (out of 50) for Cdkn3 and to 24 CpG sites (out of 55) for Cdkn2b. The entire CpG island of the calretinin (Calb2) promoter was scored. The CpG island within the Cdkn3 promoter was significantly hypomethylated in the choline deficient ventricular and subventricular zones, and this change was confined to the first half of the island (Table 2 and Figure 3). Within the Cdkn3 CpG island, sites 5 to 10, and 18 were significantly hypomethylated in choline deficient brain from the ventricular and subventricular zones, while site 24 was significantly hyper-methylated in choline deficient brain from these areas. In the dentate gyrus there were no changes in DNA methylation due to the dietary treatment (map not shown).
Table 2.
Effects of maternal choline deficiency on qualitative scores for methylation of CpG islands from gene promoters Cdkn2b, Cdkn3, and Calb2 in fetal brain.
| Ammon's horn ventricular and subventricular zones |
Dentate gyrus |
|||
|---|---|---|---|---|
| CpG island | Control | Choline deficient | Control | Choline deficient |
| Cdkn3 | 35a | 20b | 13c | 147c |
| (CpG sites 1-38) | ||||
| Cdkn2b | 19a | 13a | 28b | 27b |
| (CpG sites 9-32) | ||||
| Calb2 | 14a | 10b | 28c | 28c |
| (CpG sites 1-29) | ||||
In the same brain areas used for studies in Table 1, cells were captured using laser capture microdissection and, using bisulfite sequencing, CpG sites within the listed gene promoters were scored according to their methylation status as described in the Methods section. The qualitative methylation score of the Cdkn3 promoter is significantly lower in choline deficient Ammon's horn compared to control samples. Mean scores (n=5 independent samples) across the entire sequences are presented. Values with different superscript letters are significantly different (p<0.05, Kruskal-Wallis test for two groups) within each gene.
Figure 3.
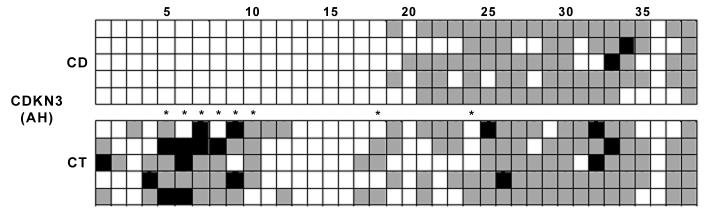
DNA methylation qualitative scores of the CpG islandd within the Cdkn3, Cdkn2b and Calb2 promoters in fetal brain.Animals were treated as described in figure legend 2 and laser capture microdissection (LCM) performed as described in methods. Bisulfite sequencing of DNA extracted from LCM samples was used to map the methylation status of CpG islands in Cdkn3, -270 to +232 (502 bp length with 50 CpG sites); Cdkn2b, 3 consecutive CpG islands with 55 CpG sites (sites 289 to 912 within the U66084.1 sequence); Calb2, -163 to +79 (242 bp length with 29 CpG sites). We numbered CpG sites sequentially from the 5’ end of the CpG island. Five independent samples were used for each treatment and each hippocampal area. Each square represents one CpG site within the respective CpG island.Black squares indicate CpG sites with methylation score of 100; grey squares indicate CpG sites with methylation score of 50; white squares indicate CpG sites with methylation score of 0 (see Materials and Methods); AH, Ammon’s horn ventricular and subventricular zones; CDKN3, cyclin-dependent kinase inhibitor 3 (encodes the Kap). An asterisk at a particular CpG site denotes a p value of less than 0.05 and statistical significance, between control (CT) and choline deficient (CD) samples, as determined by the Kruskal-Wallis test for two groups (n=5). AH denotes the Ammon’s horn ventricular and subventricular zones; DG denotes the prime germinal zone of the dentate gyrus.
The overall methylation score of the Cdkn2b promoter did not differ significantly within treatments (Table 2). The methylation of the same promoter in the dentate gyrus had higher scores than in the Ammon’s horn ventricular and subventricular zones, regardless of dietary treatment (Table 2). Conversely, the Calb2 methylation scores for the dentate gyrus were significantly lower than those for the Ammon’s horn ventricular and subventricular zones, regardless of dietary treatment (Table 2).
DISCUSSION
We report that maternal dietary choline deficiency induced significant changes in the mouse E17 fetal hippocampus. In the neuroepithelial layer, choline deficiency significantly increased the protein expression of p15INK4b and Kap, two important cyclin-dependent kinase inhibitors. Previous studies performed in various models have indicated that these cell-cycle regulators inhibit cell proliferation by inhibiting the phosphorylation of cyclin-dependent kinases Cdk2 and Cdk4 (30-32). Previously, we reported that choline deficiency decreased cell proliferation in neuronal progenitor cells within the Ammon’s horn ventricular and subventricular zones (11, 13). This reduction of cell proliferation was associated with increased apoptosis (13). Similar results were also reported using primary neurons or neuroblastoma/pheochromocytoma cells in culture (14, 16). Studies performed using cell culture models suggested that one mechanism responsible for reduced cell proliferation could be increased expression of cyclin-dependent kinase inhibitors p15Ink4B, p27Kip1 and Kap and subsequent activation of the retinoblastoma inhibitory pathway (12, 16). The studies using LCM captured cells from fetal brains that we now describe are consistent with this hypothesis. Choline deficient neuroepithelial cells had more total and nuclear staining for these cell cycle inhibitors (Figure 2A and 2B); it is the nuclear-distributed cyclin-dependent kinase inhibitors that inhibit activation of cyclin-dependent kinases and correspondent cyclins (33).
Global DNA methylation was decreased in neuroepithelium of fetuses from choline deficient mothers, but not in the dentate gyrus. As in the rat (on embryonic day 18) (25), the mouse neuroepithelia of the hippocampal ventricular and subventricular zones (on embryonic day 17) are the main sources for cells that proliferate and migrate(34). Thus, our data suggest that cells that have the potential to proliferate are the most sensitive to maternal dietary intake of methyl-groups.
Mouse Cdkn3 promoter has a CpG island highly similar to the human CpG island(16). Choline deficiency induced significant Cdkn3 hypo-methylation in the Ammon’s horn ventricular and subventricular zones, associated with increased Kap protein expression. A similar pattern exists for the Cdkn2b promoter, although these changes were not statistically significant.
We report a significant increase of calretinin staining in the dentate gyrus of CD fetuses. These findings confirm the changes previously reported in choline-deprived neural precursor cells (35), where the expression of Calb2 was increased. This calcium binding protein is expressed in mature neuronal cells, thus, this change suggests that choline deficiency increased neuronal differentiation (19, 36). The methylation status of the Calb2 (the gene coding for calretinin) promoter was higher in the dentate gyrus than in the ventricular and subventricular zones and this difference was inversely related to protein expression, as no calretinin-positive cells could be found in the ventricular and subventricular zones (Figure 2). This observation is consistent with the expected inhibition of gene expression by methylation of CpG islands in its promoter region. However, we did not observe changes in methylation of CpG islands in Calb2 associated with manipulating dietary choline. Thus, we hypothesize that some other mechanism is likely responsible for the increase in calretinin protein expression in brains of fetuses from choline deficient mothers.
In conclusion, we report that decreased choline availability to mother during development of fetal brain was associated with changes in DNA methylation that are specific to some CpG islands and even to specific CpG sites within genes that regulate cell cycling. Moreover, unknown mechanisms confined these changes to the main proliferation area of the hippocampus (Ammon’s horn ventricular and subventricular zones). These changes are associated with increased expression of cyclin-dependent kinase inhibitors. In the primordial dentate gyrus, decreased choline availability to mother during development of fetal brain was associated with changes in expression of a marker of cell differentiation, suggesting that timing of hippocampal development was accelerated. These results provide a reasonable molecular mechanism whereby decreased choline availability during development of fetal brain causes life-long changes in memory function.
ACKNOWLEDGEMENTS
This work was funded by grants from the National Institutes of Health (AG09525, ES012997, DK55865) and by the National Research Initiative of the USDA Cooperative State Research, Education and Extension Service, grant number 2005-35200-15247. Support for this work was also provided by grants from the NIH to the UNC Clinical Nutrition Research Unit (DK56350) and the Center for Environmental Health and Susceptibility (ES10126).
References
- 1.Institute of Medicine, and National Academy of Sciences USA . Dietary reference intakes for folate, thiamin, riboflavin, niacin, vitamin B12, panthothenic acid, biotin, and choline. Vol. 1. National Academy Press; Washington D.C: 1998. Choline; pp. 390–422. [PubMed] [Google Scholar]
- 2.Zeisel SH, Blusztajn JK. Choline and human nutrition. Ann. Rev. Nutr. 1994;14:269–296. doi: 10.1146/annurev.nu.14.070194.001413. [DOI] [PubMed] [Google Scholar]
- 3.Finkelstein JD. Pathways and regulation of homocysteine metabolism in mammals. Semin Thromb Hemost. 2000;26:219–225. doi: 10.1055/s-2000-8466. [DOI] [PubMed] [Google Scholar]
- 4.Jeltsch A. The Chemistry and Biology of DNA Methylation. ChemBioChem. 2002;3:274–293. doi: 10.1002/1439-7633(20020402)3:4<274::AID-CBIC274>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 5.Holmes GL, Yang Y, Liu Z, Cermak JM, Sarkisian MR, Stafstrom CE, Neill JC, Blusztajn JK. Seizure-induced memory impairment is reduced by choline supplementation before or after status epilepticus. Epilepsy Res. 2002;48:3–13. doi: 10.1016/s0920-1211(01)00321-7. [DOI] [PubMed] [Google Scholar]
- 6.Yang Y, Liu Z, Cermak JM, Tandon P, Sarkisian MR, Stafstrom CE, Neill JC, Blusztajn JK, Holmes GL. Protective effects of prenatal choline supplementation on seizure-induced memory impairment. J Neurosci. 2000;20:RC109. doi: 10.1523/JNEUROSCI.20-22-j0006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brandner C. Perinatal choline treatment modifies the effects of a visuo--spatial attractive cue upon spatial memory in naive adult rats. Brain Res. 2002;928:85–95. doi: 10.1016/s0006-8993(01)03363-7. [DOI] [PubMed] [Google Scholar]
- 8.Meck WH, Smith RA, Williams CL. Pre- and postnatal choline supplementation produces long-term facilitation of spatial memory. Dev. Psychobiol. 1988;21:339–353. doi: 10.1002/dev.420210405. [DOI] [PubMed] [Google Scholar]
- 9.Meck W, Williams C. Perinatal choline supplementation increases the threshold for chunking in spatial memory. Neuroreport. 1997;8:3053–3059. doi: 10.1097/00001756-199709290-00010. [DOI] [PubMed] [Google Scholar]
- 10.Meck WH, Williams CL. Metabolic imprinting of choline by its availability during gestation: implications for memory and attentional processing across the lifespan. Neurosci Biobehav Rev. 2003;27:385–399. doi: 10.1016/s0149-7634(03)00069-1. [DOI] [PubMed] [Google Scholar]
- 11.Albright CD, Tsai AY, Friedrich CB, Mar MH, Zeisel SH. Choline availability alters embryonic development of the hippocampus and septum in the rat. Brain Res Dev Brain Res. 1999;113:13–20. doi: 10.1016/s0165-3806(98)00183-7. [DOI] [PubMed] [Google Scholar]
- 12.Albright CD, Mar MH, Friedrich CB, Brown EC, Zeisel SH. Maternal choline availability alters the localization of p15Ink4B and p27Kip1 cyclin-dependent kinase inhibitors in the developing fetal rat brain hippocampus. Dev Neurosci. 2001;23:100–106. doi: 10.1159/000048701. [DOI] [PubMed] [Google Scholar]
- 13.Craciunescu CN, Albright CD, Mar MH, Song J, Zeisel SH. Choline availability during embryonic development alters progenitor cell mitosis in developing mouse hippocampus. J Nutr. 2003;133:3614–3618. doi: 10.1093/jn/133.11.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yen CL, Mar MH, Craciunescu CN, Edwards LJ, Zeisel SH. Deficiency in methionine, tryptophan, isoleucine, or choline induces apoptosis in cultured cells. J Nutr. 2002;132:1840–1847. doi: 10.1093/jn/132.7.1840. [DOI] [PubMed] [Google Scholar]
- 15.Yen CL, Mar MH, Meeker RB, Fernandes A, Zeisel SH. Choline deficiency induces apoptosis in primary cultures of fetal neurons. FASEB J. 2001;15:1704–1710. doi: 10.1096/fj.00-0800com. [DOI] [PubMed] [Google Scholar]
- 16.Niculescu MD, Yamamuro Y, Zeisel SH. Choline availability modulates human neuroblastoma cell proliferation and alters the methylation of the promoter region of the cyclin-dependent kinase inhibitor 3 gene. J Neurochem. 2004;89:1252–1259. doi: 10.1111/j.1471-4159.2004.02414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Albright CD, Tsai AY, Mar M-H, Zeisel SH. Choline availability modulates the expression of TGFβ1 and cytoskeletal proteins in the hippocampus of developing rat brain. Neurochem. Res. 1998;23:751–758. doi: 10.1023/a:1022411510636. [DOI] [PubMed] [Google Scholar]
- 18.Albright CD, Friedrich CB, Brown EC, Mar MH, Zeisel SH. Maternal dietary choline availability alters mitosis, apoptosis and the localization of TOAD-64 protein in the developing fetal rat septum. Brain Res Dev Brain Res. 1999;115:123–129. doi: 10.1016/s0165-3806(99)00057-7. [DOI] [PubMed] [Google Scholar]
- 19.Albright CD, Siwek DF, Craciunescu CN, Mar MH, Kowall NW, Williams CL, Zeisel SH. Choline availability during embryonic development alters the localization of calretinin in developing and aging mouse hippocampus. Nutr Neurosci. 2003;6:129–134. doi: 10.1080/1028415031000084418. [DOI] [PubMed] [Google Scholar]
- 20.Robertson KD, Wolffe AP. DNA methylation in health and disease. Nat Rev Genet. 2000;1:11–19. doi: 10.1038/35049533. [DOI] [PubMed] [Google Scholar]
- 21.Cooney CA, Dave AA, Wolff GL. Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J Nutr. 2002;132:2393S–2400S. doi: 10.1093/jn/132.8.2393S. [DOI] [PubMed] [Google Scholar]
- 22.Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacobowitz D, Abbott L. Chemoarchitectonic atlas of the developing mouse brain. CRC Press; Boca Raton, FL: 1997. [Google Scholar]
- 24.Santos F, Hendrich B, Reik W, Dean W. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev Biol. 2002;241:172–182. doi: 10.1006/dbio.2001.0501. [DOI] [PubMed] [Google Scholar]
- 25.Altman J, Bayer SA. Migration and distribution of two populations of hippocampal granule cell precursors during the perinatal and prenatal periods. J. Comp. Neurol. 1990;301:365–381. doi: 10.1002/cne.903010304. [DOI] [PubMed] [Google Scholar]
- 26.Reznikov KY. Cell proliferation and cytogenesis in the mouse hippocampus. Adv Anat Embryol Cell Biol. 1991;122:1–74. doi: 10.1007/978-3-642-76447-9. [DOI] [PubMed] [Google Scholar]
- 27.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 28.Xu XL, Wu LC, Du F, Davis A, Peyton M, Tomizawa Y, Maitra A, Tomlinson G, Gazdar AF, Weissman BE, Bowcock AM, Baer R, Minna JD. Inactivation of human SRBC, located within the 11p15.5-p15.4 tumor suppressor region, in breast and lung cancers. Cancer Res. 2001;61:7943–7949. [PubMed] [Google Scholar]
- 29.Hamilton DW, Lusher ME, Lindsey JC, Ellison DW, Clifford SC. Epigenetic inactivation of the RASSF1A rumour suppresor gene in ependymoma. Cancer Letters. 2005:75–81. doi: 10.1016/j.canlet.2004.11.044. [DOI] [PubMed] [Google Scholar]
- 30.Davies T, Pratt D, Endicott J, Johnson L, Noble M. Structure-based design of cyclin-dependent kinase inhibitors. Pharmacol Ther. 2002;93:125–133. doi: 10.1016/s0163-7258(02)00182-1. [DOI] [PubMed] [Google Scholar]
- 31.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 32.Hannon GJ, Casso D, Beach D. KAP: a dual specificity phosphatase that interacts with cyclin-dependent kinases. Proc Natl Acad Sci U S A. 1994;91:1731–1735. doi: 10.1073/pnas.91.5.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Massague J. G1 cell-cycle control and cancer. Nature. 2004;432:298–306. doi: 10.1038/nature03094. [DOI] [PubMed] [Google Scholar]
- 34.Rodier PM. Chronology of neuron development: animal studies and their clinical implications. Dev Med Child Neurol. 1980;22:525–545. doi: 10.1111/j.1469-8749.1980.tb04363.x. [DOI] [PubMed] [Google Scholar]
- 35.Niculescu MD, Craciunescu CN, Zeisel SH. Gene expression profiling of choline-deprived neural precursor cells isolated from mouse brain. Brain Res Mol Brain Res. 2005;134:309–322. doi: 10.1016/j.molbrainres.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 36.Brandt MD, Jessberger S, Steiner B, Kronenberg G, Reuter K, Bick-Sander A, von der Behrens W, Kempermann G. Transient calretinin expression defines early postmitotic step of neuronal differentiation in adult hippocampal neurogenesis of mice. Mol Cell Neurosci. 2003;24:603–613. doi: 10.1016/s1044-7431(03)00207-0. [DOI] [PubMed] [Google Scholar]