

Abstract
The ability of the C3d component of complement to enhance antibody responses and protective immunity to influenza virus challenges was evaluated using a DNA vaccine encoding a C3d fusion of the hemagglutinin (HA) from influenza virus. Plasmids were generated that encoded a transmembrane HA (tmHA), a secreted form of HA (sHA), or a sHA fused to three tandem copies of the murine homologue of the C3d (sHA-3C3d). Analysis of the titers, avidity maturation, and hemagglutinin-inhibition activity of raised antibody revealed that immunizations with sHA-3C3d DNA accelerated both the avidity maturation of antibody to HA and the appearance of hemagglutinin-inhibition activity. These accelerated antibody responses correlated to a more rapid appearance of protective immunity. They also correlated to complete protection from live virus challenge by a single vaccination at a dose ten times lower than the protective dose for non-C3d forms of HA.
One of the challenges faced when developing influenza virus vaccines is the problem of how to protect populations in the face of spreading pandemics. With the advent of air travel, this readily transmitted virus can circumnavigate the globe within days. Each year influenza virus infection causes severe morbidity and even mortality, especially in immunocompromised individuals and the elderly1.
Protection against influenza virus is primarily mediated by antibodies to the hemagglutinin (HA) glycoprotein, which is responsible for the attachment and penetration of virus into cells during the initial stages of infection1–3. Variation in the HA glycoprotein caused by antigenic drift (mutations within a HA subtype) or antigenic shift (the change to another HA subtype) are responsible for the recurring outbreaks of influenza and poor ability to control these recurring infections by immunization1,4. HAs from different subtypes can have as little as 20% homology in their amino acid sequences. Within strains of a single subtype, the deduced amino acid sequences generally have differences of less than 10%5. Thus one solution to the control of emerging influenza infections is to immunize populations rapidly with HAs of the subtype that is currently spreading.
A new approach to the development of influenza vaccines has recently emerged with the advent of DNA use in immunization6–8. Theoretically, cDNA for an emergent virus could be rapidly cloned, introduced into a eukaryotic expression vector, amplified in bacteria and used for protective immunizations. This theoretical scenario has been limited by the reality that DNA-raised antibody responses typically require 2–3 months to reach maximal titers3. Compared to protein or attenuated viral vaccines10,11, the slow rise in antibody elicited by DNA vaccines is likely due to the raising of antibody responses by very low levels of antigen8. In the race against a pandemic, this relatively long period between immunization and protection could compromise the utility of a DNA-based anti-HA vaccine.
We examined whether a DNA vaccine expressing a fusion of HA and the C3d component of complement could achieve an earlier and more efficient anti-HA B cell response. In previous studies in mice, the fusion of two or three copies of C3d to a model antigen—hen egg lysozyme—increased the efficiency of immunizations by more than 1000-fold12. In the human immune system, one consequence of complement activation is the covalent attachment of the C3d fragment of the third complement protein to the activating protein. C3d in turn binds to CD21, a molecule with B cell stimulatory functions that amplify B lymphocyte activation, on B lymphocytes13. In a HA-C3d fusion protein, the HA moiety of the fusion would bind to anti-HA immunoglobulin receptors on B cells and signal through the B cell receptor, while the C3d moiety of the fusion would bind to CD21 and signal through CD19. According to this hypothesis, a B cell responding to a HA-C3d fusion protein would undergo more effective signaling than a B cell responding to HA alone. Our results demonstrate that mice vaccinated with DNA expressing a secreted HA fused to three copies of C3d- (sHA-3C3d) generated antibody that underwent more rapid avidity maturation than antibody generated by secreted or transmembrane forms of HA. This resulted in more rapid appearance of hemagglutination inhibition (HI) activity and protective immunity.
Results
Expression of plasmids
Three HA plasmids were constructed in the pGA vector to express either the transmembrane form of HA (tmHA), a secreted form of HA (sHA), or sHA-3C3d (Fig. 1). The tmHA represents the entire cDNA-coding region including the cytoplasmic region. The sHA represents the entire ectodomain of HA including the oligomerization domain but excluding the transmembrane and cytoplasmic region. The sHA-3C3d fusion protein was generated by cloning three tandem repeats of the mouse homologue of C3d12 in-frame with the gene encoding secreted HA (Fig. 1b). The proteolytic cleavage sites, found at the junction between each C3d molecule as well as the junction between the HA protein and the first C3d coding region, were destroyed by mutagenesis.
Figure 1.

Schematic representation of vector DNA vaccine constructs. (a) The pGA vector contains the cytomegalovirus immediate-early promoter (CMV-IE) plus intron A (IA) for initiating transcription of eukaryotic inserts and bovine growth hormone polyadenylation signal (BGH poly A) for termination of transcription. The vector also contains the Col E1 origin of replication for prokaryotic replication as well as the Kanamycin resistance gene (Kanr) for selection in antibiotic media. The λ T0 terminator has been placed 3′ to Kanr to increase the stability of eukaryotic inserts. Inserts were cloned into the vector using the Hind III and Bam HI restriction endonuclease sites. (b) Top, the wild-type transmembrane form of the HA protein used as a vaccine insert; middle, secreted form of the HA; bottom, the sHA-3C3d construct used as a vaccine insert. Linkers composed of two repeats of four glycines and a serine (G4S)2 were fused at the junctures of HA and C3d and between each C3d repeat. Rectangles indicate domains. 1, HA1; 2, HA2;TM, transmembrane.
HA was expressed at similar concentrations by plasmids containing either the secreted or transmembrane-associated forms of the antigen, but at a 50% lower concentration by the sHA-C3d–expressing plasmids (Table 1). Human 293T cells were transiently transfected with 2 μg of plasmid and both supernatants and cell lysates were assayed for HA using an antigen capture enzyme-linked immunosorbent assay (ELISA). Approximately 80% of the HA protein was secreted into the supernatant for both sHA DNA– and sHA-3C3d DNA–transfected cells (Table 1). As expected, ∼99% of the HA antigen was detected in the lysate of cells transfected with plasmids expressing tmHA (Table 1). The small amount of sHA detected in the cell lysate could have been antigen in the process of transport or sHA that had been reabsorbed onto the cell surface via binding to sialic acid14.
Table 1.
In vitro expression levels of HA vaccine plasmids
| Relative HA Unitsa | |||
|---|---|---|---|
| DNA vaccine | Supernatant | Cell lysate | % Supernatantb |
| sHA | 4.3 ± 0.3 | 1.3 ± 0.2 | 77 |
| tmHA | 0.1 ± 0.1 | 5.7 ± 0.6 | 1 |
| sHA-3C3d | 2.2 ± 0.4 | 0.5 ± 0.1 | 83 |
| Vector | 0.0 ± 0.0 | 0.2 ± 0.1 | N.A. |
Data are expressed as mean ± s.e.m.
Data are expressed as a percent of the total HA antigen secreted into the supernatant compared to the amount in the cell lysate fraction.
Western blot analyses revealed sHA, tmHA and sHA-3C3d proteins of expected sizes. Using a polyclonal rabbit antibody to HA, western blot analysis showed a broad band of 65–70 kD corresponding to HA (Fig. 2). A slightly lower molecular weight band was seen for the sHA molecules. This band size was consistent with truncation of the 33 amino acids corresponding to the transmembrane and cytoplasmic domains of HA (Fig. 2). A higher molecular weight band at ∼120 kD was consistent with the projected size of the sHA-3C3d fusion protein (Fig. 2). Strong protein bands were present in the supernatants of cells transfected with sHA DNA or sHA-3C3d DNA, whereas less intense bands were present in the cell lysates. No evidence for the proteolytic cleavage of the sHA-C3d fusion protein was seen by western analysis. As expected, a strong band was present in lysates from cells transfected with tmHA DNA and no protein was detected in the supernatant from these same transfected cells.
Figure 2.
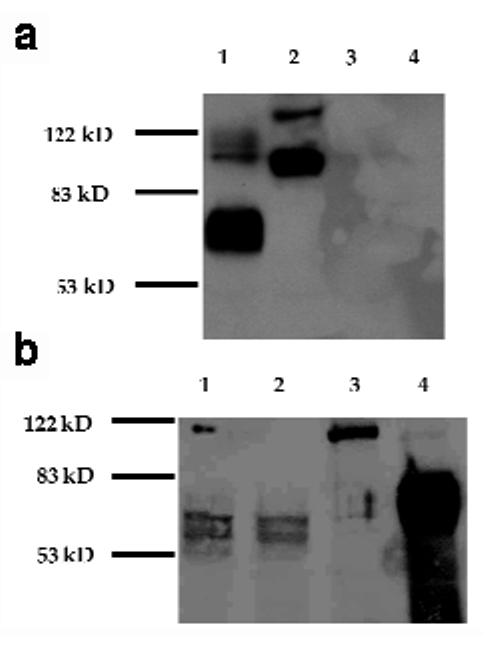
Expression of vaccine constructs in vitro. Human embryonic kidney cells, 293T, were transfected with 2 μg of each vaccine plasmid. (a) Supernatant was collected and 1.5% of total volume was electrophoresis on a 10% polyacrylamide gel. Lane 1, sHA DNA; lane 2, sHA-3C3d DNA; lane 3, tmHA DNA; lane 4, pGA vector. (b) Cells were lysed and total protein was extracted in 300 μl volume of RIPA lysis buffer and 5% of the protein was electrophoresis on a 10% polyacrylamide gel. Lane 1, pGA vector; lane 2, sHA DNA; lane 3, sHA-3C3d DNA; lane 4, tmHA DNA.
Antibody response to HA DNA immunizations
The tmHA– and sHA-3C3d–expressing DNA plasmids raised higher titers of ELISA antibody than the sHA DNA–expressing plasmids. BALB/c mice were vaccinated with DNA-coated gold particles via gene gun with a 0.1 μg or 1 μg inoculum. Four weeks after vaccination half of the mice in each group were boosted with an identical dose of DNA to that initially given. Total anti-HA IgG induced by the sHA-3C3d– and tmHA–expressing plasmids were similar in the different experimental mouse groups and three to fivefold higher than the amount raised by the sHA–expressing plasmids (Fig. 3a–d). In addition, the amount of anti-HA elicited increased relative to DNA used for vaccination, in a dose-dependent manner (Fig 3e,f). Overall, the dose response curves and temporal pattern for the appearance of anti-HA were similar in the mice vaccinated with tmHA DNA or sHA-3C3d DNA, but lower and slower in the mice vaccinated with sHA DNA. As expected, the booster immunization both accelerated and increased the titers of anti-HA.
Figure 3.

Anti-HA IgG raised by gene gun inoculation of DNAs expressing HA proteins. Mice were immunized with different doses of vaccine plasmid. Half of the mice were primed at day 0 and boosted at week 4 (a,b) and half were given a single vaccination at day 0 (c,d). A ratio of the dose of DNA to specific IgG concentrations was obtained at week 14 (e,f). Sera were obtained from mice with vector (filled squares), sHA (open circles), tmHA (open squares) or sHA-3C3d (filled circles). Sera collected at the indicated times from each group were pooled for determination of specific IgG levels by ELISA. Data are represented as the average of three individual assays. Preimmune sera from mice had no detectable specific IgG.
Avidity of mouse HA antiserum
Sodium thiocyanate (NaSCN)–displacement ELISA demonstrated that the avidity of the HA-specific antibody generated with sHA-3C3d–expressing DNA was consistently higher than antibodies from sHA DNA– or tmHA DNA–vaccinated mice (Fig. 4). The avidities of specific antibodies to HA were compared by using graded concentrations NaSCN, a chaotropic agent, to disrupt antigen-antibody interactions15. The binding of antibodies with less avidity to the antigen is disrupted at lower concentrations of NaSCN than that of antibodies with greater avidity to the antigen. The effective concentration of NaSCN required to release 50% of antiserum (ED50) collected at 8 weeks after vaccination from sHA DNA– or tmHA DNA–boosted mice (0.1 μg or 1 μg) was∼1.20 M (Fig. 4a). In contrast, antiserum from mice vaccinated and boosted with sHA-3C3d DNA had an ED50 of ∼1.75 M (Fig. 4b). At the time of challenge (14 weeks after vaccination), the ED50 had increased to ∼1.8 M for antibodies from both sHA DNA– and tmHA DNA–vaccinated mice (Fig. 4c). Antibodies from mice vaccinated with sHA-3C3d DNA had increased to an ED50 of ∼2.0 M (Fig 4d). These results suggest that the antibody from sHA-3C3d DNA–vaccinated mice had undergone more rapid affinity maturation than antibody from either sHA DNA– or tmHA DNA–vaccinated mice. The difference between the temporal avidity maturation of antibody for sHA-3C3d and tmHA was independent of the level of the raised antibody. Both of these plasmids had similar temporal patterns for the appearance of antibody and dose response curves for the ability to raise antibody (Fig. 3).
Figure 4.

Avidity of the anti-HA IgG raised by the three different HA DNA vaccines. Sera were analyzed from week 8 (a,b) and week 14 (c,d) in an A/PR/8/34 (H1N1)–specific NaSCN–displacement ELISA. Sera were obtained from mice inoculated. Sera were obtained from mice with sHA (open circles), tmHA (open squares) or sHA-3C3d (filled circles). Assays used pooled serum samples from each mouse group at a dilution of 1:300. Data are represented as the average of three independent experiments plus standard errors.
HA-inhibition titers
HI assays were performed to evaluate the ability of the raised antibody to block binding of A/PR/8/34 (H1N1) to sialic acid. The HI titers in serum samples collected from mice 8 and 14 weeks after vaccination were measured. All boosted mice had measurable HI titers at week 14 regardless of the dose or vaccine given (Fig. 5). The highest titers (up to 1:5280) were recorded for the sHA-3C3d DNA–vaccinated mice. Nonboosted mice showed more variation in HI titers (Fig. 5). Nonboosted mice vaccinated with a 0.1 μg dose of either sHA DNA– or tmHA DNA–expressing plasmids had low HI titers of 1:10. In contrast, mice vaccinated with sHA-3C3d DNA had titers greater than 1:640. The only vaccinated mice that had a measurable HI titer (1:160) at week 8 were boosted mice vaccinated with a 1 μg dose sHA-3C3d DNA. These results indicate that C3d, when fused to sHA, is able to stimulate specific B cells to increase the avidity maturation of antibody and thus the production of neutralizing antibodies to HA.
Figure 5.

Hemagglutinin-inhibition antibodies and protection generated by HA DNA vaccines. The HI assay was read as the endpoint dilution of serum that still inhibited hemagglutination. Data are represented as the Log2 titer plus standard error for three to five individual serum samples collected at week 8 (a) and five individual serum samples collected at week 14 (b) from each mouse group. The data is represented as a Log2 titer of antibody to HA. (Open bars, vector; shaded bars, sHA; hatched bars, tmHA; filled bars, sHA-3C3d.)
Protective efficacy to influenza challenge
Consistent with eliciting the highest titers of HI antibody, the sHA-3C3d DNA raised more effective protection than the sHA or tmHA DNAs. To test the protective efficacy of the various HA-DNA vaccines, mice were challenged with a lethal dose of A/PR/8/34 influenza virus (H1N1) and monitored daily for morbidity (as measured by weight loss) and mortality. Weight loss for each animal was plotted as a percentage of the average weight before challenge versus days after challenge (Fig. 6). Virus-challenged naïve mice and pGA vector only–vaccinated mice showed rapid weight loss, with all the mice losing >20% of their body weight by 8 days after challenge (Fig. 6). In contrast, PBS buffer mock-challenged mice showed no weight loss over the 14 days of observation. All boosted mice survived challenge, 14 weeks after vaccination, regardless of the dose of DNA plasmid administered. However, boosted mice vaccinated with a 0.1 μg dose of sHA DNA did drop to 92% of their initial body weight at 8 days after challenge before recovering (Fig. 6d). In contrast when 1 μg-dose–boosted mice were challenged 8 weeks after vaccination, the only mice to survive challenge were sHA-3C3d DNA– and tmHA DNA–vaccinated mice, albiet with greater weight loss than was observed from mice challenged 14 weeks after vaccination. The only 0.1 μg-dose–boosted mice to survive challenge 8 weeks after vaccination were the sHA-3C3d–vaccinated mice (Fig. 6b).
Figure 6.

Protection from weight loss after virus challenge. At week 8 (a,b) or week 14 (c–f) mice were challenged intranasally with a lethal dose of influenza virus, A/PR/8/34 (H1N1), and monitored daily for weight loss. The data are plotted as percentage of the average initial weight. (a,c) Mice were primed and boosted with a 1 μg dose of DNA vaccine. (b,d) Mice were primed and boosted with a 0.1 μg dose of DNA vaccine. (e) Mice were given a single 1 μg dose of DNA vaccine. (f) Mice were given a single 0.1 μg dose inoculum of DNA vaccine. Sera were obtained from mice with vector (filled squares), sHA (open circles), tmHA (open squares), sHA-3C3d (filled circles), naïve-mock (open triangles) or naïve-virus (filled triangles). The open cross indicates the time-point at which all five mice in a group succumbed to disease.
Among the nonboosted 0.1 μg-dose immunizations, only the sHA-3C3d DNA–vaccinated mice survived challenge at 14 weeks after vaccination (Fig. 6f). All mice administered a single DNA vaccination lost weight. However, of these, the sHA-3C3d DNA–vaccinated mice lost the least weight and these mice were the only mice to survive the lethal challenge (Fig. 6). These results demonstrate the that 3C3d protein, when fused to HA, increased the efficiency of a DNA vaccine allowing for a reduction in dose of DNA and the number of vaccinations needed to afford protection to a lethal influenza virus challenge.
Discussion
The most likely mechanism by which C3d serves as an adjuvant for sHA is the binding of C3d to CD21 on the surface of B cells. CD21 and CD19 have been shown to activate B cells after receptor interaction13,16. C3d could act by one or more different mechanisms that include directly stimulating antibody-producing B cells or by providing antigen depots on follicular dendritic cells. One hypothesis for the induction of increased antibody responses by C3d is that C3d fusion proteins stimulate and expand the pool of anti-HA–specific B cells. The sHA-3C3d fusion protein allows for a cross-linking of HA-specific cell surface immunoglobulin and CD21, thereby leading to the expansion of B cells specific for HA and not just a generic stimulation of all B cells by nonfused C3d. A second hypothesis is that the fused C3d facilitates binding of the HA antigen to the follicular dendritic cells in germinal centers where affinity maturation occurs17,18. Once the HA is trapped in the germinal center it can both attract and stimulate affinity maturation in responding B cells. A role for complement receptors, such as CD21, has been shown to be important in responses of the B cells to protein antigens within germinal centers19. C3d could operate by either of these methods.
One of the more interesting features of the antibody raised by sHA-3C3d DNA was its avidity maturation. Avidity is a term used to describe the strength of interaction between multivalent antigens and antibodies in serum and is dependent on the individual affinities of the polyclonal antibodies in question20. After immunization, strong proliferation of B cells occurs in the follicles of peripheral lymphatic tissues leading to the formation of germinal centers. Within germinal centers, diversification of B cells, predominately by somatic hypermutations, results in a large repertoire of antigen-specific B cells with high affinity B cell receptors17,18. Eight weeks after vaccination, the avidity of the antibody to HA in boosted mice was stronger in sera from mice inoculated with sHA-3C3d than in sera from sHA DNA– or tmHA DNA–inoculated mice. By 14 weeks after vaccination, the avidity of the sera from all vaccinated mouse groups was more closely related than was observed at 8 weeks after vaccination. Differences in survival correlated with differences in the amount of HA and HI activity, which in turn correlated with the avidity maturation of antibody. For example, differences in survival of the tmHA– and the sHA-3C3d–vaccinated mice occurred despite the presence of similar ELISA titers of antibody to hemagglutinin.
Much effort is being made to improve the efficacy of influenza vaccines. One approach, which was tried in this study, is the use of a C3d fusion protein to reduce the dose and decrease the time to protection of the vaccine. The results demonstrate the effectiveness of C3d-fusions as a molecular adjuvant in enhancing antibody production, enhancing antibody maturation and leading to protective immunity in the influenza mouse model. Using sHA-3C3d DNA, we were able to drop the dose of DNA by 90% compared to wild-type tmHA and achieve both protection and a more rapid onset of protective immunity. This is remarkable in that in vitro, the plasmids expressing sHA-3C3d produced half as much protein as plasmids expressing sHA or tmHA. These results underscore the importance of increasing our understanding of the relationship between innate and acquired immunity. As one of the first molecularly defined adjuvants, C3d could be a major advance in vaccine technology. The results presented demonstrate the applicability of C3d using an antigen from a virus that caused an actual dip in the human population in the 20th century.
Methods
Plasmid DNA
pGA, a eukaryotic expression vector was constructed to contain CMV-IE plus IA for initiating transcription of eukaryotic inserts, and BGH poly A for termination of transcription (Fig 1a). The vector contains the Col E1 origin of replication for prokaryotic replication and Kanr for selection in antibiotic media. In addition, it contains the λ phage T0 terminator inserted 3′ to Kanr and in the same orientation as Kanr to limit prokaryotic transcriptional elongation into the vaccine insert and increase the stability of eukaryotic inserts during their amplification in bacteria21.
Influenza HA sequences from A/PR/8/34 (H1N1) and C3d sequences were cloned into the pGA vaccine vector using unique restriction endonuclease sites (Fig. 1b). The secreted version was generated by deleting the transmembrane and cytoplasmic domains of HA. This was accomplished using PCR to clone a fragment of the gene encoding HA from the ATG start site to nt163322. The vectors expressing sHA-C3d–fusion proteins were generated by cloning three tandem repeats of the mouse homologue of C3d in-frame with the gene encoding sHA. The construct design was based upon Dempsey et al. and used sequences from pSLG-C3d12. Linkers composed of (G4S)2 were fused at the junctures of HA and C3d and between each C3d repeat. Potential proteolytic cleavage sites between the junctions of C3d and the junction of sHA and 3C3d were mutated by using Bam HI and Bgl II fusion to mutate an arginine codon to a glycine codon12.
The plasmids were amplified in Escherichia coli strain, DH5α, purified using anion-exchange resin columns (Qiagen, Valencia, CA) and stored at -20 ° in dH20. Plasmids were verified by appropriate restriction enzyme digestion and gel electrophoresis. Purity of DNA preparations was determined by optical density reading at 260 nm and 280 nm.
Mice and DNA immunizations
BALB/c mice (Harlan Sprague Dawley, Indianapolis, IN) aged 6- to 8-weeks-old were used for inoculations. Mice, housed in microisolator units and allowed free access to food and water, were cared for under USDA guidelines for laboratory animals. Mice were anesthetized with 0.03–0.04 ml of a mixture of 5 ml ketamine HCl (100 mg/ml) and 1 ml xylazine (20 mg/ml). Gene gun immunizations were performed on shaved abdominal skin using the hand-held Accell gene delivery system as described previously23–25. Mice were immunized with doses containing 0.5 μg of DNA per 0.5 mg of ∼1 μm gold beads (DeGussa-Huls Corp., Ridgefield Park, NJ) at a helium pressure setting of 400 psi.
Transfections and expression analysis
The human embryonic kidney cell line 293T (5 × 105 cells per transfection) was transfected with 2 μg of DNA using 12% lipofectamine according to the manufacturer’s (Life Technologies, Grand Island, NY) guidelines. Supernatants were collected and stored at -20 °. Cell lysates were collected in 300 μl of RIPA lysis buffer (0.05M TRIS-HCl, pH 8.0, 0.1% SDS, 1.0% Triton-X-100, 2mM phenylmethylsulfonyl fluoride (PMSF), 0.15 M NaCl) and stored at -20 °. Quantitative antigen capture ELISAs for HA were conducted as described20.
For western hybridization analysis, 15 μl of supernatant or cell lysate was diluted 1:2 in SDS sample buffer (Bio-Rad, Hercules, CA) and loaded onto a 10% SDS–polyacrylamide gel. The resolved proteins were transferred onto a nitrocellulose membrane (Bio-Rad, Hercules, CA) and incubated with a 1:1000 dilution of polyclonal rabbit antibody to HA in PBS buffer containing 0.1% Tween 20 and 1% nonfat dry milk. After extensive washing, bound rabbit antibodies were detected using a 1:2000 dilution of horseradish peroxidase–conjugated goat antibody to rabbit, and enhanced chemiluminescence (Amersham).
ELISA, avidity and HI assays
A quantitative ELISA was performed to assess anti-HA–specific IgG in immune serum using purified influenza virus to coat plates as described3. Avidity ELISAs were performed similarly to serum antibody determination ELISAs up to the addition of samples and standards. Samples were diluted to give similar concentrations of IgG. Plates were washed three times with 0.05% PBS-Tween20. Different concentrations of the chaotropic agent, sodium thiocyanate (NaSCN) in PBS buffer, were then added (0, 0.5, 1, 1.5, 2, 2.5 and 3 M NaSCN). Plates were allowed to stand at room temperature for 15 min and then washed six times with PBS-Tween 20. Subsequent steps were performed similarly to the serum antibody determination ELISA. The HI assay was performed essentially as described20 using turkey erythrocytes (Lampiere Biologicals, Pipersville, PA) for agglutination and 4 hemagglutination units of the of A/PR/8/34 (H1N1) virus strain.
Influenza virus challenge
Challenge with live, mouse-adapted, influenza virus A/PR/8/34 (H1N1) was performed by intranasal instillation of 50 μl allantoic fluid, diluted in PBS buffer to contain three lethal doses of virus, into the nares of ketamine-anesthetized mice26. This method leads to rapid lung infections and is lethal to 100% of nonimmunized mice. Individual mice were challenge either 8 or 14 weeks after vaccination and monitored both for weight loss and survival. Data were plotted as the average individual weight in a group, as a percentage of prechallenge weight, versus days after challenge.
Acknowledgements
We thank D. Campbell and S. Winburn for technical assistance with serum collection, T. Tumpey from the Centers for Disease Control and Prevention for providing influenza A/PR/8/34–infected chicken lung homogenate and J. Katz for discussions. Supported by National Institute of Allergy and Infectious Diseases grant awards R21 AI44325 (T.M.R.) and R01 AI34946 (H.L.R.).
References
- 1.Subbarao K. Influenza vaccines: present and future. Adv. Virus Res. 1999;54:349–373. doi: 10.1016/s0065-3527(08)60371-1. [DOI] [PubMed] [Google Scholar]
- 2.Andrew ME, Coupar BEH, Boyle DB, Ada GL. The roles of influenza virus haemagglutinin and nucleoprotein in protection: analysis using vaccinia virus recombinants. Scand. J. Immunol. 1987;25:21–25. doi: 10.1111/j.1365-3083.1987.tb01042.x. [DOI] [PubMed] [Google Scholar]
- 3.Robinson HL, et al. DNA immunization for influenza virus: studies using hemagglutinin- and nucleoprotein-expressing DNAs. J. Infect. Dis. 1997;176:S50–55. doi: 10.1086/514176. [DOI] [PubMed] [Google Scholar]
- 4.Portela A, Zucker T, Nieto A, Ortin J. Replication of Orthomyxoviruses. Adv. Virus Res. 1999;54:319–348. doi: 10.1016/s0065-3527(08)60370-x. [DOI] [PubMed] [Google Scholar]
- 5.Murphy BR, Webster RG. In: Virology. 2nd edition Fields BN, et al., editors. Raven Press; New York: 1990. pp. 1091–1152. [Google Scholar]
- 6.Seder RA, Gurunathan S. DNA vaccines-designer vaccines for the 21st century. N. Engl. J. Med. 1999;341:277–278. doi: 10.1056/NEJM199907223410410. [DOI] [PubMed] [Google Scholar]
- 7.Donnelly JJ, Ulmer JB, Liu MA. DNA vaccines. Dev. Biol. Stand. 1998;95:43–53. [PubMed] [Google Scholar]
- 8.Robinson HL, Pertmer TM.DNA vaccines for viral infections: Basic studies and applications Adv. Virus Res 2000. in the press [DOI] [PubMed] [Google Scholar]
- 9.Robinson HL, Hunt LA, Webster RG. Protection against a lethal influenza virus challenge by immunization with a hemagglutinin-expressing plasmid DNA. Vaccine. 1993;11:957–960. doi: 10.1016/0264-410x(93)90385-b. [DOI] [PubMed] [Google Scholar]
- 10.Zuckerman MA, et al. Serological response in volunteers to inactivated trivalent subunit influenza vaccine: antibody reactivity with epidemic influenza A and B strains and evidence of a rapid immune response. J. Med. Virol. 1991;33:133–137. doi: 10.1002/jmv.1890330213. [DOI] [PubMed] [Google Scholar]
- 11.Rimmelwaan GF, et al. Induction of protective immunity against influenza virus in a macaque model: comparison of conventional and iscom vaccines. J. Gen. Viral. 1997;78:757–765. doi: 10.1099/0022-1317-78-4-757. [DOI] [PubMed] [Google Scholar]
- 12.Dempsey PW, Allison MED, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: Bridging innate and acquired immunity. Science. 1996;271:348–350. doi: 10.1126/science.271.5247.348. [DOI] [PubMed] [Google Scholar]
- 13.Fearon DT, Carter RH. The CD19/CR2/TAPA-1 complex of B lymphocytes: linking natural to acquired immunity. Annu. Rev. Immunol. 1995;13:127–149. doi: 10.1146/annurev.iy.13.040195.001015. [DOI] [PubMed] [Google Scholar]
- 14.Gething MJ, Sambrook J. Construction of influenza hemagglutinin gene that code for intracellular and secreted forms of the protein. Nature. 1982;300:598–603. doi: 10.1038/300598a0. [DOI] [PubMed] [Google Scholar]
- 15.Pullen GR, Fitzgerald MG, Hosking CS. Antibody avidity determination by ELISA using thiocyanate elution. J. Immunol. Meth. 1986;86:83–87. doi: 10.1016/0022-1759(86)90268-1. [DOI] [PubMed] [Google Scholar]
- 16.Fearon DT. The complement system and adaptive immunity. Semin Immunol. 1998;10:355–361. doi: 10.1006/smim.1998.0137. [DOI] [PubMed] [Google Scholar]
- 17.Berek C, Berger A, Apel M. Maturation of the immune response in germinal centers. Cell. 1991;67:1121–1129. doi: 10.1016/0092-8674(91)90289-b. [DOI] [PubMed] [Google Scholar]
- 18.Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centres. Nature. 1991;354:389–392. doi: 10.1038/354389a0. [DOI] [PubMed] [Google Scholar]
- 19.Ahearn JM, et al. Disruption of the CR2 locus results in a reduction in B-1a cells and in a impaired B cell response to T-dependent antigen. Immunity. 1996;4:251–262. doi: 10.1016/s1074-7613(00)80433-1. [DOI] [PubMed] [Google Scholar]
- 20.Potter CW, Jennings R, Phair JP, Clarke A, Stuart-Harris CH. Dose response relationship after immunization of volunteers with a new surface-antigen-adsorbed influenza virus vaccine. J. Infect. Dis. 1977;155:423–431. doi: 10.1093/infdis/135.3.423. [DOI] [PubMed] [Google Scholar]
- 21.Scholtissek S, Grosse FA. cloning cartridge of λ t0 terminator. Nuc. Acids Res. 1987;15:3185. doi: 10.1093/nar/15.7.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Torres CAT, Yang K, Mustafa F, Robinson HL. DNA immunization: effect of secretion of DNA expressed hemagglutinins on antibody responses in preparation. Vaccine. 1999;18:805–814. doi: 10.1016/s0264-410x(99)00345-x. [DOI] [PubMed] [Google Scholar]
- 23.Pertmer TM, Robinson HL. Studies on antibody responses following neonatal immunization with influenza hemagglutinin DNA or protein. Virology. 1999;257:406–414. doi: 10.1006/viro.1999.9666. [DOI] [PubMed] [Google Scholar]
- 24.Haynes JR, Fuller DH, Eisenbraun MD, Ford MJ, Pertmer TM. Accell particle-mediated DNA immunization elicits humoral, cytotoxic, and protective immune responses. AIDS Res. Hum. Retroviruses. 1994;10:S43–45. [PubMed] [Google Scholar]
- 25.Pertmer TM, et al. Gene gun based nucleic acid immunization. eliciting of humoral and cytotoxic T lymphocyte response following epidermal delivery of nanogram quantitation of DNA. Vaccine. 1995;13:1427–1430. doi: 10.1016/0264-410x(95)00069-d. [DOI] [PubMed] [Google Scholar]
- 26.Montgomery DL, et al. Heterologous and homologous protection against influenza A by DNA vaccination: Optimization of DNA vectors. DNA Cell Biol. 1993;12:777–783. doi: 10.1089/dna.1993.12.777. [DOI] [PubMed] [Google Scholar]