

Abstract
Transposable elements have been used in Drosophila to detect gene expression, inactivate gene function, and induce ectopic expression or overexpression. We have combined all of these features in a single construct. A promoterless GAL4 cDNA is expressed when the construct inserts within a transcriptional unit, and GAL4 activates a GFP-encoding gene present in the same transposon. In a primary screen, patterned gene expression is detected as GFP fluorescence in the live progeny of dysgenic males. Many animals expressing GFP in distinct patterns can be recovered with relatively little effort. As expected, many insertions cause loss of function. After insertion at a genomic location, specific parts of the transposon can be excised by FLP recombinase, thus allowing it to induce conditional misexpression of the tagged gene. Therefore, both gain- and loss-of-function studies can be carried out with a single insertion in a gene identified by virtue of its expression pattern. Using this promoter trap approach, we have identified a group of cells that innervate the calyx of the mushroom body and could thus define a previously unrecognized memory circuit.
Keywords: gene function, GFP, memory, gene trap, misexpression
Determining the function of most genes is a long-term goal in the postgenomic era. This enterprise was initiated many decades ago, much before DNA sequencing, with the numerous forward genetic screens that have been carried out in Drosophila (1) and in other model organisms (2). Such screens have attained an exquisite degree of sophistication, allowing very specific biological functions to be probed. However, forward genetic screens are unlikely to uncover the function of all genes because their activity could be masked by redundancy. Moreover, the function of many genes might be overlooked if they serve a subtle function not needed for viability but essential for fitness in the wild. This is likely to be the case for many brain functions. Homologous recombination technology has the potential to knock out every gene, although this technology is still very laborious (3). Transgenic RNAi is another reverse genetic approach that has a place in the postgenomic era (4), but it is limited by the fact that it usually causes incomplete knock down and that it is still relatively laborious because it requires the construction and validation of individual transgenic strains. As a complement to the loss-of-function assays, misexpression screens based on the GAL4 system (5) have also been very successful in uncovering the activity of many genes in specific tissues (6). Ideally, however, gain-of-function analysis should always be complemented by the loss-of-function phenotype.
The pattern of expression can be an alternative starting point for a genetic screen. For example, our work on embryonic boundaries in Drosophila suggests that segmentally expressed genes involved in segmental groove formation remain to be discovered (7). Presumably, these genes have not been identified in the past because of redundancy. A screen based on expression patterns could identify these genes as long as subsequent analysis can probe the functional significance of such expression. Such an approach could also be particularly suited to identify genes involved in brain functions and/or to uncover previously unrecognized cell types in the brain. Expression-based screens have previously been performed in Drosophila using LacZ-based enhancer trap vectors (8–10). By current standards, the usefulness of enhancer trap insertions is limited by the effort needed for subsequent functional and molecular characterization. The advent of GFP technology provides an opportunity for dramatically improving the efficiency and focus of expression-based screens. Moreover, additional technological developments allow functional assays to dovetail readily on an expression-based screen. We report here on the design and activity of a transposon that achieves these aims. Using this approach, we identify a previously unrecognized group of cells that innervate the calyx of mushroom bodies.
Results and Discussion
Design and Features of the Promoter Trap.
A transposon carrying a promoterless cDNA accurately reflects endogenous gene expression when integrated downstream of a genomic transcription start site (11). However, flies carrying this construct have to be crossed to a GFP expressing reporter line to reveal the expression pattern in live animals. To allow the screening of new patterns in the first generation, we included UAS-GFP within an analogous GAL4-based construct (Fig. 1). Because the original construct by Lukacsovich et al. (11) was shown to trap promoters, the sequences upstream of GAL4 were kept the same, including a splice acceptor site (SA) and a so-called stop-start site (one small variation was added; see below). Because GFP and GAL4 are both present in our construct, the activity of endogenous promoters should be detectable in the first generation progeny of dysgenic animals. Moreover, because insertion of the transposon introduces three transcription termination sites (one downstream of GAL4, one in white, and one following GFP), it is expected that transcription of the endogenous gene would be prematurely terminated, thus leading to loss of function. To enable gain-of-function experiments, we introduced sequences that allow easy conversion to an inducible misexpression construct (after insertion at a specific genomic location). Conversion was achieved by introducing FLP recombination target (FRT) variants at suitable positions such that both GAL4 and GFP could be excised. A pair of mutated FRTs (called FRT2 here), which are incompatible with the wild-type FRT but pair with each other in vitro (12), were introduced on both sides of the coding sequence. Another pair of FRTs (FRT1), also incompatible with the wild-type FRT as well as with FRT2, was placed on both sides of the GFP coding sequence (including the polyadenylation site). In theory, FLP expression should excise both GAL4 and GFP while leaving in place the intervening sequence, which includes a miniwhite gene (as a marker) and the UAS-promoter cassette. The latter, which drives GFP expression before excision, should now point downstream into nearby genomic sequences. Excision should allow expression of the downstream gene in the presence of exogenous GAL4 (which would be brought in by a genetic cross). Overall, we expect the transposon to reveal active promoters by triggering transcription and hence GFP expression. By design, therefore, only insertions downstream of active endogenous promoters will be detected, and sequencing of flanking sequences after inverse PCR should unambiguously identify the tagged gene. In the cases in which the transposon inserts upstream of the translation start, it should be readily convertible into a misexpression construct after induction of GAL4 expression.
Fig. 1.

Schematic representation of the promoter trap after it has inserted into an individual gene. Flanking genomic regions are shown in red with an arrow marking the endogenous start of transcription. The ends of the P element are indicated by black triangles. After insertion of the transposon, the endogenous promoter (UAS, upstream activating sequence) triggers transcription of and the subsequent production of GFP. A splice acceptor site (SA; AATTCTTATCCTTTCCTTTAGGCTAACGCCGAGGCCCAGAA) and a stop/start (TGATTGAATAAACATG) precede GAL4 as in the construct of Lukacsovich et al. (11). Both GAL4 and GFP are individually flanked by modified 35-bp FRTs (FRT2 and FRT1, respectively). The central core sequence (shown in the figure for FRT2 and FRT1), which determines specificity, was modified from the wild type (TCTAGAAA) to prevent cross-reactivity while still allowing self-pairing. After FLP expression, both GAL4 and GFP are expected to be excised, leaving all other sequences intact, including the miniwhite gene.
Testing the Conversion from Promoter Trap to Misexpression Construct.
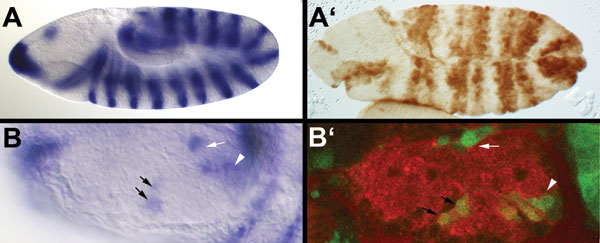
Because the specificity of the mutated FRT pairs present in our construct had not been tested previously in a heterologous system, we assessed the effect of expressing FLP in three lines picked from a small pilot screen. These represented insertions in Mhc (Myosin heavy chain), elav, and CG6301. Recombination was assessed by PCR in the progeny of males carrying both the transposon and a FLP-encoding transgene expressed from a testis-specific promoter (13). Primers were designed to amplify either the GAL4- or GFP-encoding sequences along with nearby flanking sequences (Fig. 2A). PCR amplification of the GAL4 region generated an expected band of 3.5 kb in the parental flies, whereas a 200-bp fragment was amplified with the same primers after FLP expression. Likewise, a region encoding GFP was amplified as a 2.7-kb fragment in the parental flies, and this fragment was reduced to 1 kb after crossing to the FLP-encoding transgene. All three lines retained the red eye color, indicating that the white gene, which is positioned between GAL4 and GFP in the parental stock, was not excised by FLP (Fig. 2B). This finding demonstrates that no cross-reactivity occurs between the two pairs of FRTs. Because white is not excised, the UAS promoter cassette is also expected to be retained after excision of GAL4 and GFP. This, according to our expectation, should allow expression of the downstream gene if the transposon is inserted upstream of the endogenous start of translation. We tested this directly for one of our line, which is inserted in the elav gene. This line was chosen because anti-Elav antibodies are readily available. Elav is normally expressed in the nervous system (shown at stage 14 in Fig. 2D). GAL4 and GFP were excised by crossing to flies expressing FLP as described above. The resulting flies, which we refer to as “flipped-out,” were crossed to engrailed-GAL4 and embryos were stained with anti-Elav. As can be seen, Elav is expressed in segmentally repeated stripe, mirroring the domain of engrailed-GAL4 activity (Fig. 2F). This finding demonstrates that the transposon can indeed be converted to a misexpression construct upon exposure to FLP.
Fig. 2.

Postinsertion conversion to a misexpression construct. (A) Genomic DNA was isolated from various lines, and DNA fragments were amplified by PCR with primers indicated by red arrows in the diagram. Three insertions (in mhc, elav, and CG6301) were analyzed this way. (B) Fragments obtained by PCR. After excision (see lanes marked FO for flipped-out; two independent excisions), a 200-bp fragment was detected with the GAL4-specific primers, whereas a 3.5-kb fragment was produced in the parental flies. C, control; MHC, myosin heavy chain. Likewise, the flipped-out flies (three independent excisions) gave a 1-kb fragment with the GFP-specific primers, whereas a 2.7-kb fragment was obtained from the parental flies. (C) Diagram of the insertion site of the promoter trap (yellow triangle) in the 5′ end of the elav gene (transcription is from right to left). (D) GFP expression in the parental stock (promoter trap in elav). GFP is detected in the CNS as expected from a reporter of elav expression. (E) Immunocytochemical detection of Elav after FLP-mediated excision of GAL4 and GFP (PelavFO). (F) Elav expression in an embryo from a cross between engrailed-GAL4 and PelavFO. Note the expression in stripes in addition to the normal CNS expression.
A Pilot Screen.
To assess the activity and efficiency of our promoter trap, we mobilized it and screened for GFP expression in embryos and larvae. A silent insertion (no GFP expression) located on the third chromosome was used as a transposon source. To achieve good gamete representation, one dysgenic male was mated to 10 wild-type females. Depending on capacity, embryos from ≈3,000 females were screened each day, and a new GFP expression pattern is seen in 1 male out of 10. Thus, ≈30 new expression patterns per 3,000 females were identified. Approximately 100 lines were established from single fluorescent animals isolated during a pilot screen. Various expression patterns were selected (examples are shown in Fig. 3). In some lines, GFP expression was restricted, e.g., to the ring gland as in line 71. Other lines have a broad expression profile such as in line 50, which expresses GFP in many tissues such as the gut, trachea, and epidermis. Although the screen was carried out with embryos and early larvae, many insertions produce fluorescence throughout the life of the fly. GFP expression was recorded and is summarily described in Table 2, which is published as supporting information on the PNAS web site. Of particular significance are the patterns of expression detected in the brain (see below).
Fig. 3.

Some examples of GFP patterns. The number at the bottom of each panel refers to individual insertion lines. B, D, and G show photographs of live embryos, and the remaining panels depict live larvae. (A) Line 34 shows expression in the CNS and peripheral nervous system at larval stages. (B) Embryonic expression in segmentally arranged clusters of cells of the epidermis. (C) Line 50 shows expression in the tracheal system, intestinal tract, and the epidermis. (D) Line 4 is expressed in the CNS and the oenocytes. (E) Line 56 shows expression in the tracheal system. (F) Line 86 shows GFP expression in the fat bodies and somatic musculature. (G) In embryos from line 98, GFP is seen in narrow stripes of segmentally repeated epidermal cells. There is also expression in scattered cells throughout the embryo. (H) Line 35 exhibits exclusive expression in the ring gland. (I) Line 90 has GFP expression in segmentally repeated cells of the embryonic epidermis.
More than one-half of the insertions were located on the third chromosome (Table 1), perhaps a consequence of the fact that our jumpstarter transposon was on the third chromosome and that local jumping is usually favored (14, 15). After our pilot screen, two silent insertions have been introduced on a marked second chromosome. Such a strain could be used as an alternative jumpstarter line that might favor insertions on the second chromosome. Twenty-five percent of our insertions were homozygous lethal (Table 1), indicating that integration of the P element can disrupt gene function as expected (16). However nonlethal insertions in essential genes were found (e.g., elav), maybe because a cryptic transcription start located downstream of the insertion site could become active. For these insertions, a loss-of-function mutation could be obtained by imprecise excision of the transposon. For 20 lines, the exact insertion site was determined by inverse PCR (Fig. 5 and Table 3, which are published as supporting information on the PNAS web site). Of these 20 gene traps, 11 were in an exon and 9 were in an intron. All but one were inserted before the start codon. For those, Flp-mediated conversion would allow inducible expression of the corresponding gene. Visual inspection showed that the embryonic expression of GFP driven by the promoter traps in cpo, vha68, m(2)21ab, mesk2, lobe, and odd-skipped fits with the corresponding in situ expression pattern posted on the Berkeley Drosophila Genome Project web site (http://www.fruitfly.org/cgi-bin/ex/insitu.pl). For two promoter traps, congruence between expression of GFP and that of the endogenous gene was verified by double immunofluorescence (line 106 in odd-skipped [data not shown] and line 95 in elav [Fig. 2 D and E]). Note that at any given stage, the GFP and in situ patterns were not always identical. However, the differences could be attributed to a delay in the appearance of GFP and to the perdurance of the GFP signal after the endogenous gene has turned off. This effect is illustrated for the odd gene in Fig. 6, which is published as supporting information on the PNAS web site. Overall, it appears that the promoter trap provides a good reporter of endogenous gene activity. The promoter trap vector was found to insert equally into introns and exons (Table 1). Interestingly, for 19 of the 20 lines, the promoter trap inserted upstream of the ATG (within the 5′ UTR). In all of these cases, conversion to a misexpression insertion should therefore be possible. Furthermore, some of these lines are homozygous lethal, indicating that insertion of the promoter trap into the 5′ UTR can cause disruption to gene expression. Because there is no obvious benefit from the splice acceptor site, an analogous construct was made without the splice acceptor. It is likely that this could lead to a higher proportion of GFP-expressing animals because out of 21 independent transformants, 6 expressed GFP in specific patterns (no GFP-producing lines were obtained from the original transformants carrying the original promoter trap). A large-scale mobilization experiment will be needed to confirm that efficiency is increased in the absence of a splice acceptor.
Table 1.
Insertion sites for some of the lines isolated in a pilot screen
|
P element insertions on |
Homozygotes lethal insertion | |||
|---|---|---|---|---|
| 3rd | 2nd | X | ||
| n = 110 | 62 | 27 | 21 | 27 |
A majority of insertions were found on the 3rd chromosome. Twenty-seven percent were homozygous lethal.
Identification of a Novel Group of Cells Innervating the Calyx.
As shown above, GFP expression from the promoter trap faithfully reports on the normal endogenous pattern. We expected faithfulness to exceed that of enhancer traps because, with the promoter trap, GAL4 is driven from a fusion transcript. GFP expression was also expected to be strong, thanks to the GAL4-mediated amplification step. The strength of the signal turned out to be particularly useful in cases in which expression is confined to a small number of cells. Strong expression can be seen, for example, in larvae carrying the promoter trap in odd-skipped, which is only expressed in ≈5–6 neurons in each brain lobe at the third instar (Fig. 4E). The ease of visualizing these groups of cells led us to postulate that genes expressed in restricted patterns in the larval brain could be screened for at the outset. The feasibility of this approach was tested by small-scale mobilization of the promoter trap and screening for GFP expression in the brain of larval progeny. Some examples are shown in Fig. 4 A–E. In these lines, neuronal processes can later be visualized by crossing the lines to flies carrying UAS-CD8-GFP. Fig. 4A shows the pattern from a promoter trap in vha16, a gene encoding a proton-transporting ATPase (http://flybase.bio.indiana.edu). As can be seen, the trap is expressed within the brain lobes in a restricted number of cells that project predominantly into the nerve cord. Another line (line 42) was found to be expressed exclusively in surface glia (Fig. 4B). To our knowledge, no such expression pattern has been reported previously. Fig. 4C shows the expression pattern from line 2 (B4), which is expressed almost exclusively in a group of cells located in an anterior-medial region of the brain lobe. Broader expression patterns were also identified in Fig. 4D.
Fig. 4.

Genes expressed in the brain. All images are of second instar larval brains with GFP in green. Anterior is left and medial is up. Each image is of one brain lobe. GFP is produced from the promoter trap itself as well as from a UAS-CD8-GFP transgene to highlight cell processes. In A–E, the preparations were stained with anti-N-cadherin (red) to visualize most neuronal processes. The site of insertion was determined for the lines shown in A, C, and E (hence the gene name is indicated above the corresponding panel). E shows the odd-skipped-expressing neurons (green) and their projection into the calyx of the mushroom body (white arrow) (for more details, see Movie 1). In F, the odd-skipped-expressing neurons (white arrow) are shown in a preparation stained with anti-Dachshund (red). The light blue arrow points to the Kenyon cells.
The insertion into the odd-skipped gene is expressed at the second instar in a cluster of 5–6 neurons in each brain lobe. With CD8-GFP, these cells are seen to project into the calyx of the mushroom body (white arrow in Fig. 4E) (for 3D visualization, see Movie 1, which is published as supporting information on the PNAS web site). This finding is significant because all of the cells known so far to innervate the calyx are Kenyon cells, yet the odd-skipped cells (white arrow in Fig. 4F) are clearly distinct form the Kenyon cluster: As can be seen in Fig. 4F, odd-skipped-expressing cells (white arrow) do not colocalize with the Dachshund-positive Kenyon cell (light blue arrow). Therefore, the odd-skipped cells may define a previously unrecognized memory circuit. Not only was the promoter trap instrumental in identifying these cells, it also provides tools for future characterization. For example, the ontogeny of these cells could be uncovered by tracking them in live and fixed preparations. Our preliminary analysis suggests that these cells originate from a posterior cluster within the embryonic brain. In addition, GAL4 produced by the promoter trap could be used to drive the expression of additional markers such as CD2-HRP (7) to facilitate connectivity studies at the EM level. A toxin could also be expressed for cell ablation (17). Ablation could be done at a defined time if GAL80 [ts] is introduced to allow the control of GAL4 activity with temperature (18).
Conclusion
Our construct allows the efficient identification of genes that are expressed in specific patterns within a tissue. Morin et al. (19) reported a promoter trap vector that generates fusion proteins between endogenous gene products and GFP. GFP fusions can report on the subcellular localization of the endogenous product. Seeing the subcellular localization is a distinct advantage, but it comes at the cost of the need for three constructs (one for each frame). Because of its simplicity, our promoter trap allows numerous insertions (patterns) to be screened by a single human operator in a relatively short time, and this can be further increased by the use of a larva/embryo sorter. Efficiency and the ability to screen in the first generation after dysgenesis is such that one can afford to select only the desired patterns of expression for further analysis. Another benefit of our promoter trap is that, as a result of GAL4-mediated amplification, the GFP signal is readily detectable even if expression is restricted to a small number of cells. A further advantage of our construct is that it easily lends itself to gain- and loss-of-function analysis. Both gene and promoter traps allow the identification of the disrupted gene because the insertions occur within the transcription unit. All in all, the gene and promoter traps have distinct benefits (chiefly the creation of GFP fusions for the promoter trap and the bright signal and simplicity for the promoter trap) and hence should complement each other.
As we have shown, our promoter trap can be used to identify genes expressed in a particular tissue at any developmental stage of interest even if the cell population is very small. A proof-of-concept is provided by genes expressed in a subset of cells within the brain. Systematic screening for such expression patterns could provide a palette of tools to probe the development and function of various parts of the brain. Mutations in head gap genes such as orthodenticle, empty spiracles, tailless, and buttonhead (20, 21) have been known to cause large-scale deletions in the brain. However, relatively few mutations are known to affect restricted neuronal circuits in either embryos or larvae. The features of our promoter trap should help characterizing such circuits. As an assessment of this paradigm, it would be useful to test the role of the odd-skipped cluster and probing the role of odd-skipped in the development and function of mushroom bodies.
Materials and Methods
Construct of the Transposon.
Standard techniques of molecular biology were used. The full sequence of the transposon is available upon request.
Fly Stocks and Mating Crosses.
The jumpstarter stock used for our pilot screen carried a silent insertion on the third chromosomes. Mobilization was carried out by crossing flies containing the P element to flies carrying Δ2–3 as a source of transposase (20). Male progeny from this cross were mated to females from a white− virginizer stock containing heat shock-hid on the Y chromosome (a gift from Ruth Lehman, Skirball Institute, New York, NY). Progeny was screened by using a standard dissecting microscope with a fluorescence source or a COPAS embryo sorter (Union Biometrica, Holliston, Massachusetts). The following fly stocks were used for further experiments: engrailed-GAL4 (a gift from Andrea Brand, Cambridge University, Cambridge, U.K.) and a strain carrying FLP under the control of a testis-specific tubulin promoter (13).
Primers to Test FLP-Mediated Recombination.
The region was amplified with CGGACATTGACGCTAGGTAAC and GGATTTGCCATTGATCCTTCG, whereas GFP was amplified with CCGTTCGGAGTGATTAGGT and CACGTGCCGAAGTGTGCTATT.
Inverse PCR.
Inverse PCR was performed following the Berkeley Drosophila Genome Project standard protocol (as described in FlyBase) except that alternative primers were used in most cases: GGAGGCGACTCAACGCAGATG and CACCCAAGGCTCTGCCCCACAAT. In some cases, reliable PCR fragments were generated with the primers suggested by the Berkeley Drosophila Genome Project.
Supplementary Material
Abbreviation
- FRT
FLP recombination target.
Footnotes
The authors declare no conflict of interest.
References
- 1.Nüsslein-Volhard C, Wieschaus E. Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- 2.Jorgensen EM, Mango SE. Nat Rev Genet. 2002;3:356–369. doi: 10.1038/nrg794. [DOI] [PubMed] [Google Scholar]
- 3.Rong YS, Titen SW, Xie HB, Golic MM, Bastiani M, Bandyopadhyay P, Olivera BM, Brodsky M, Rubin GM, Golic KG. Genes Dev. 2002;16:1568–1581. doi: 10.1101/gad.986602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kennerdell JR, Carthew RW. Nat Biotechnol. 2000;18:896–898. doi: 10.1038/78531. [DOI] [PubMed] [Google Scholar]
- 5.Brand AH, Perrimon N. Development (Cambridge, UK) 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 6.Rorth P. Proc Natl Acad Sci USA. 1996;93:12418–12422. doi: 10.1073/pnas.93.22.12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larsen CW, Hirst E, Alexandre C, Vincent JP. Development (Cambridge, UK) 2003;130:5625–5635. doi: 10.1242/dev.00867. [DOI] [PubMed] [Google Scholar]
- 8.O'Kane CJ, Gehring WJ. Proc Natl Acad Sci USA. 1987;84:9123–9127. doi: 10.1073/pnas.84.24.9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bellen HJ, O'Kane CJ, Wilson C, Grossniklaus U, Pearson RK, Gehring WJ. Genes Dev. 1989;3:1288–1300. doi: 10.1101/gad.3.9.1288. [DOI] [PubMed] [Google Scholar]
- 10.Wilson C, Pearson RK, Bellen HJ, O'Kane CJ, Grossniklaus U, Gehring WJ. Genes Dev. 1989;3:1301–1313. doi: 10.1101/gad.3.9.1301. [DOI] [PubMed] [Google Scholar]
- 11.Lukacsovich T, Asztalos Z, Awano W, Baba K, Kondo S, Niwa S, Yamamoto D. Genetics. 2001;157:727–742. doi: 10.1093/genetics/157.2.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schlake T, Bode J. Biochemistry. 1994;33:12746–12751. doi: 10.1021/bi00209a003. [DOI] [PubMed] [Google Scholar]
- 13.Struhl G, Fitzgerald K, Greenwald I. Cell. 1993;74:331–345. doi: 10.1016/0092-8674(93)90424-o. [DOI] [PubMed] [Google Scholar]
- 14.Zhang P, Spradling AC. Proc Natl Acad Sci USA. 1994;91:3539–3543. doi: 10.1073/pnas.91.9.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bellen HJ, Levis RW, Liao G, He Y, Carlson JW, Tsang G, Evans-Holm M, Hiesinger PR, Schulze KL, Rubin GM, et al. Genetics. 2004;167:761–781. doi: 10.1534/genetics.104.026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsubota S, Schedl P. Genetics. 1986;114:165–182. doi: 10.1093/genetics/114.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allen MJ, O'Kane CJ, Moffat KG. Genesis. 2002;34:132–134. doi: 10.1002/gene.10129. [DOI] [PubMed] [Google Scholar]
- 18.McGuire SE, Mao Z, Davis RL. Sci STKE. 2004;2004:pl6. doi: 10.1126/stke.2202004pl6. [DOI] [PubMed] [Google Scholar]
- 19.Morin X, Daneman R, Zavortink M, Chia W. Proc Natl Acad Sci USA. 2001;98:15050–15055. doi: 10.1073/pnas.261408198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirth F, Therianos S, Loop T, Gehring WJ, Reichert H, Furukubo-Tokunaga K. Neuron. 1995;15:769–778. doi: 10.1016/0896-6273(95)90169-8. [DOI] [PubMed] [Google Scholar]
- 21.Younossi-Hartenstein A, Green P, Liaw GJ, Rudolph K, Lengyel J, Hartenstein V. Dev Biol. 1997;182:270–283. doi: 10.1006/dbio.1996.8475. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}